")
Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 12
A Novel Mutation Of The EMD Gene In A Family With Cardiac Conduction Abnormalities And A High Incidence Of Sudden Cardiac Death
Authors Kong D, Zhan Y , Liu C, Hu Y, Zhou Y, Luo J, Gu L, Zhou X, Zhang Z
Received 1 July 2019
Accepted for publication 27 September 2019
Published 31 October 2019 Volume 2019:12 Pages 319—327
DOI https://doi.org/10.2147/PGPM.S221444
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Demiao Kong,1,2,* Yi Zhan,3,* Canzhao Liu,4 Yerong Hu,1 Yangzhao Zhou,1,4 Jiawen Luo,1 Lu Gu,1 Xinmin Zhou,1 Zhiwei Zhang1,4
1Department of Cardiovascular Surgery, The Second Xiangya Hospital, Central South University, Changsha, Hunan 410011, China; 2Department of Thoracic Surgery, Guizhou Provincial People’s Hospital, Guiyang, Guizhou 550002, China; 3Department of Dermatology, The Second Xiangya Hospital, Central South University, Changsha, Hunan 410011, China; 4Department of Medicine, University of California San Diego, La Jolla, CA 92093, USA
*These authors contributed equally to this work
Correspondence: Zhiwei Zhang
Department of Cardiovascular Surgery, The Second Xiangya Hospital, Central South University, No. 139 Middle Renmin Road, Changsha, Hunan 410011, People’s Republic of China
Tel +86 13787083210 Email [email protected]
Background: Emery-Dreifuss muscular dystrophy, caused by mutations in genes such as emerin (EMD) or lamin A/C (LMNA), is a disorder affecting the joints, muscles, and heart, with a wide spectrum of patient phenotypes including muscle wasting and cardiac conduction defects.
Methods and results: Here we report a multi-generation family from the Hunan Province of China. Affected family members displayed an uncommon clinical presentation of serious cardiac conduction abnormalities at an early age and a high incidence of sudden cardiac death along with mild skeletal muscular atrophy and joint contracture. Clinical analysis of affected members provided evidence of X-linked recessive inheritance. Consequently, using Sanger sequencing of X chromosome exomes, we identified a novel duplication mutation (c.405dup/p.Asp136X) in the EMD gene as the cause for the disease in this family. This variant is a novel mutation that has not been previously reported in Pubmed, Clinvar or other cases reported in the Human Gene Mutation Database.
Conclusion: Our finding expands the mutation spectrum of Emery-Dreifuss muscular dystrophy and provides a rationale for EMD mutation testing in cases of X-linked inherited cardiac conduction disease and sudden cardiac death, even in those lacking pathognomonic neuromuscular features.
Keywords: Emery-Dreifuss muscular dystrophy, emerin, sudden cardiac death, cardiac conduction abnormalities, mutation
Introduction
Emery-Dreifuss muscular dystrophy (EDMD) is a rare inherited disease characterized clinically by humero-peroneal muscle atrophy and weakness, multi-joint contractures, spine rigidity and cardiac insufficiency with conduction defects.1–3 EDMD is caused by mutations in different genes with approximately 50% of cases linked to mutations in EMD (STA), LMNA, FHL1, SYNE1, SYNE2, LUMA and SUN1.4
EDMD1, one common type of EDMD, has an X-linked recessive pattern of inheritance and is caused by mutations in the EMD gene (OMIM 310300) on chromosome Xq28.5 The EMD gene contains five introns and six exons that span 2.2 kb of genomic DNA. EMD encodes a protein known as emerin, which belongs to the family of type II integral membrane proteins anchored to the inner nuclear membrane via hydrophobic tails, with the remainder of the molecule projecting into the nucleoplasm.6 Emerin is involved in a number of biological processes, including direct and indirect regulation of transcription factor activity and localization, intra- and intercellular signaling, nucleo-cytoskeletal mechanotransduction, nuclear structure,7 chromatin condensation, as well as epigenetic modifications.8,9 Since most of the mutations in EMD are premature termination point or frame-shift mutations, they typically result in absence or decreased production of full-length emerin.
In EDMD1, the first symptoms of the disease are usually seen in the first decade of life and manifest as ankle and elbow contractures and spine rigidity. These symptoms precede muscle atrophy and weakness, which are typically visible in the second to third decades of life.10 Symptoms involving the skeletal muscles usually arise before cardiac disease, which initially includes sinus bradycardia, supraventricular tachyarrhythmia and paroxysmal atrial fibrillation.10 Atrial standstill and heart insufficiency emerge later in the fourth or fifth decades of life. The phenotypic variability in disease presentation among patients makes diagnosis relatively difficult, with physicians often relying on gene sequencing and duplication analysis. However, this type of diagnostic approach obviously necessitates the responsible mutation having already been identified.
In this study, we identified a Chinese family with EDMD1 associated with a novel duplication mutation (c.405dup/p. Asp136X) in the EMD gene. The affected family members displayed an uncommon clinical presentation of serious cardiac conduction abnormalities at an early age and a high incidence of sudden cardiac death (SCD) along with mild skeletal muscular atrophy and joint contracture. Our finding expands the mutation spectrum of EMD and provides a rationale for EMD mutation testing in cases of X-linked inherited cardiac conduction disease (CCD) and SCD, even in those lacking pathognomonic neuromuscular features.
Materials And Methods
Subjects
The proband, a 28-year-old man, was admitted to our clinical due to slight dyspnea. Electrocardiogram (ECG) showed sinus bradycardia and occasional sinus cardiac arrest. Upon further inquiry, the proband revealed that there were several cases of SCD in his family. Due to the family disease history of the proband, we decided to investigate his family carefully. Extensive genealogical records provided by the proband were utilized for pedigree construction. A questionnaire on health history was completed, medical records were collected, and electrocardiographic screening was performed for each individual. The available medical records for all deceased individuals were also reviewed.
The study was approved by the Ethics Committee of the Second Xiangya Hospital, Central South University in China. Written informed consent was obtained from all study participants or their legal representatives or guardians.
Clinical Evaluation
All subjects of the family underwent serial physical examinations, 24 hr ambulatory electrocardiogram, transthoracic echocardiography (TTE), electromyography muscle (EMG), magnetic resonance spectroscopy (MRI) and blood sampling for creatine kinase (CK) measurements.
Genetic Analysis
Genomic DNA was extracted from a peripheral venous blood sample of each subject using the QiAamp® DNA Blood Mini Kit (Qiagen Corp., USA). To complete the X chromosome exome sequencing (X-exome sequencing), genomic DNA (3mg) was sheared to produce 180bp fragments. Illumina adapters were added using SureSelect XT kit reagents (Agilent Technologies, Santa Clara, CA). The adapter-ligated DNA underwent hybridization with the X-exome targeted biotinylated RNA baits (SureSelect X chromosome demo kit, Agilent Technologies) for 24 hrs at 65°C. Hybridized DNA X-exome targets were captured using streptavidin-coated magnetic beads. The X-exome targets of interest were eluted and barcoded/indexed after a series of washes to remove the non-targeted, unbound genome fragments. The sample was indexed and sequenced to obtain approximately 50 average coverage using 2×100-base pair paired end reads (HiSeq2000, Illumina, San Diego, CA). SOAP aligner (soap2.21) and SOAP snp (v.1.03)11 were used for single nucleotide polymorphism (SNP) calling, whereas BWA (Burrows-Wheeler Aligner) and GATK (Genome-Analysis-Tool-Kit)11 were used for indel detection. The reads were aligned to the UCSC reference genome (Hg19) and variants were called with a probabilistic variant detection. All variants present in dbSNP137 (MAF> 0.01), the 1000 Genome variants database, HapMap exomes and an in-house database containing the WES data from 1000 normal individuals were then filtered out. The data interpretation rules were according to the guidelines of American College of Medical Genetics and Genomics (ACMG).12
Sanger sequencing was performed as follows: PCR primer sequences were designed through Ensembl (http://asia.ensembl.org/index.html). Mutation analysis of genes by direct sequencing was performed on the PCR products. The sequencing results were then analyzed using Chromas 2.33 and compared with the sequences in the NCBI GenBank database.
Immunoblotting Of Lymphocytes
Peripheral blood samples were collected from all participants and lymphocytes were separated using lymphocyte separation medium (MP, USA). The cells were washed twice with phosphate-buffered saline (PBS) solution and lysed with RIPA lysis buffer (Beyotime, China) containing a proteinase inhibitor (Beyotime, China). Protein concentrations were measured using the Pierce BCA Protein Assay Kit (Thermo-Fisher Scientific, USA) and samples were stored at −80°C until use. Total protein aliquots were loaded onto a 10% sodium dodecyl sulfate polyacrylamide gel for electrophoresis and were then transferred onto PVDF membranes. The membranes were blocked for in Tris-buffered saline with Tween-20 (TBS-T) containing 5% non-fat milk. Membranes were then incubated overnight at 4°C with a 1:500 dilution of the anti-emerin antibody (ab54996, Abcam, USA) followed by the horse radish peroxidase–conjugated goat anti-rabbit IgG secondary antibody (1:6000 dilution, BioTeke, China) and detection using the Enhanced Chemiluminescence Substrate Kit (Thermo-Fisher, USA).
Immunofluorescence Of Mucosa Exfoliating Cells
For all subjects, specimens of mucosa exfoliating cells from the cheek oral mucosa were used for immunofluorescence analysis. In brief, the inner surface of the cheek was first cleaned with a piece of gauze wetted with PBS and then gently scraped with a plastic spatula measuring 1.5×4cm. The collected cells were smeared on to a poly-lysine coated glass slide and left to dry. After fixation with cold methanol for 10 mins at room temperature and rinsing in PBS, the samples were incubated with an anti-emerin antibody (ab54996) (Abcam, USA, dilution 1:50) in Tris-HCl-phosphate buffered saline (TBS) containing 1% bovine serum albumin (Abcam, USA) overnight at 4°C. The samples were then incubated with Alexa Fluro®448 goat anti-mouse antibody (Life Technologies Corp. USA) for 1 hr in the dark at room temperature. After the samples were counterstained with Hoechst 33,258 to visualize nuclei (Sigma Corp. USA), a Nikon E600 fluorescent microscope was used for visualization photo acquisition.
Results
Clinical Evaluation Of Proband
A 28-year-old man came to our clinical complaining slight dyspnea. ECG and 24 hr ambulatory electrocardiogram of the man revealed sinus bradycardia, atrial escape rhythm and occasional sinus cardiac arrest (Figure 1A and B). Echocardiography displayed mild left ventricle dilation but normal systolic function (ejection fraction: 68%, left ventricular end diastolic diameter: 50 mm). He denied the use of alcohol, tobacco or drugs. The patient (proband) had a normal birth and developed without any obvious signs or symptoms of neuromuscular disease (Figure 1E). No obvious muscular atrophy was detected through visual inspection. However, serum creatine phosphokinase (CK) levels were high (CK = 1234.8 U/L; normal: 24–195 U/L). The electromyography (EMG) evaluation displayed chronic myogenic damage (Figure 1C), and muscular MRI showed mild fatty infiltration in the skeletal muscle (Figure 1D).
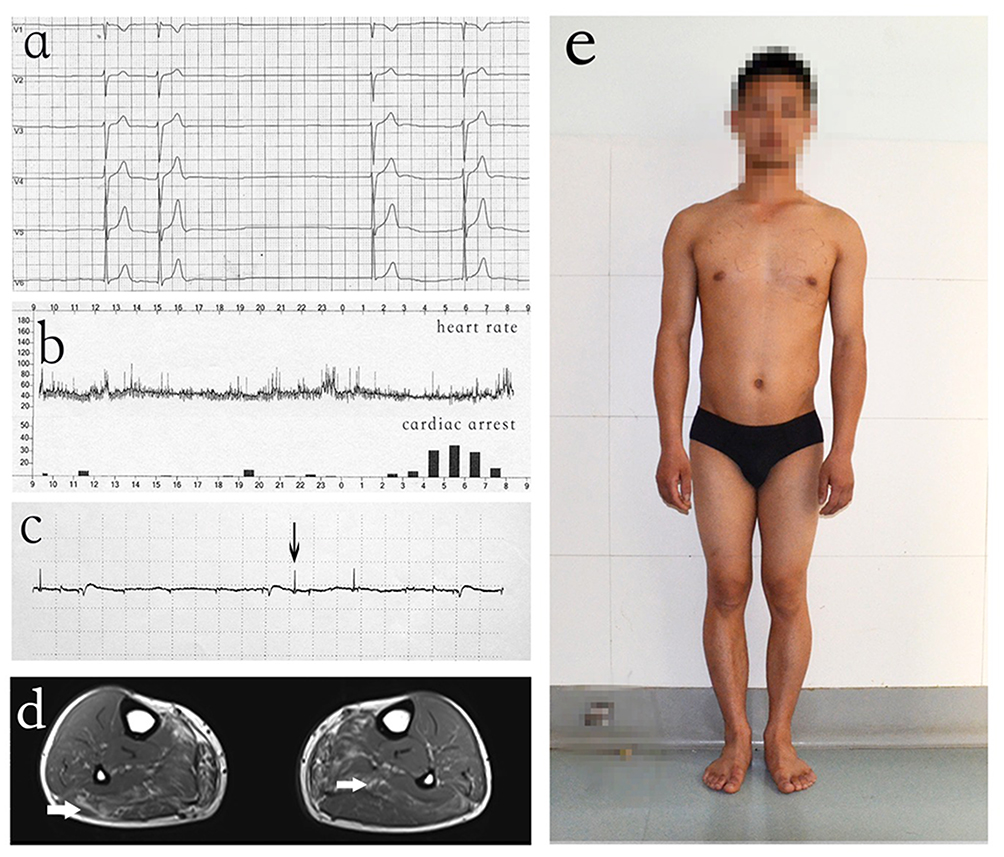 |
Figure 1 Clinical characteristics of proband (V-4). (A) ECG showed sinus bradycardia and occasional sinus cardiac arrest. (B) 24 hr ambulatory electrocardiogram showed the changes in heart rate and sinus cardiac arrest in a day. (C) EMG displayed spontaneous electromyographic activity in limb muscles. (D) Muscular MRI showed mild fatty infiltration in the skeletal muscle. (E) The patient (proband) developed without any obvious symptoms of neuromuscular disease. |
Cardiac Conduction Abnormalities Were Observed In Multiple Members Of The Proband Family In X-Linked Recessive Manner
The patient (proband) also mentioned that some of his family members died suddenly. We then decided to determine whether any other members of the family also display similar phenotypes. This pedigree, from the Hunan Province of China, consisted of 77 family members (38 males and 39 females) across six generations (Figure 2). In this family, ECG analysis showed serious cardiac conduction abnormalities in family members III-5,IV-4, IV-29,V3 and V4 (proband). In addition, III-3,III-7,III-8,IV-30 died from SCD. Other members had no obvious cardiac complications. The main clinical features of the other affected members were similar to those of the proband (Table 1). None of the female heterozygous carriers (ranging between 2–82 years of age) had skeletal muscular dystrophy, joint contracture or serious cardiac complications in our study. Clinical analysis of the pedigree provided evidence of X-linked recessive inheritance (Figure 2).
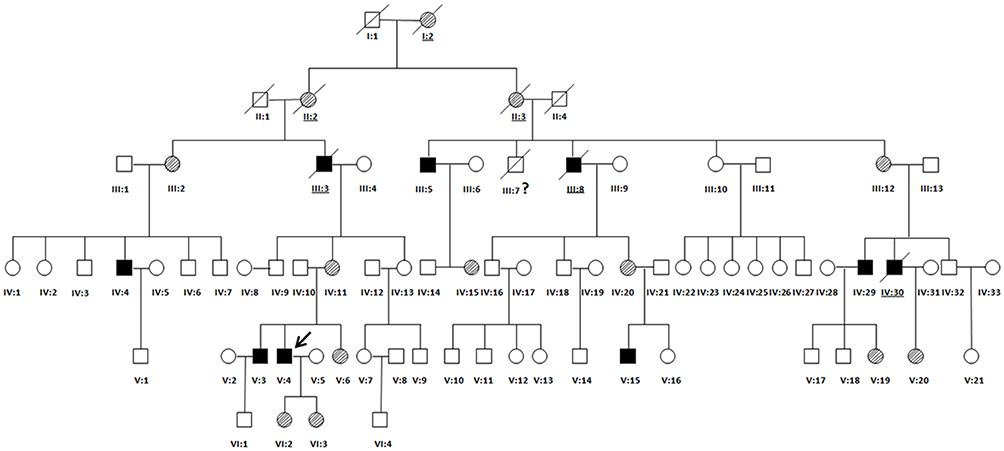 |
Figure 2 Family Pedigree. All individuals in the pedigree are identified by their Roman numerals below the symbol. Arabic numerals denote each individual in a generation. Squares represent males, and circles represent females. The proband (V-4) is designated with an arrow. Open symbols are unaffected individuals, filled symbols are hemizygous individuals, symbols with backslashes are heterozygous individuals, and symbols with diagonal lines indicate deceased subjects. Underlined numerals are obligate carriers who did not undergo molecular analysis. Numeral with a question mark (?) is an individual with an uncertain genetic status. |
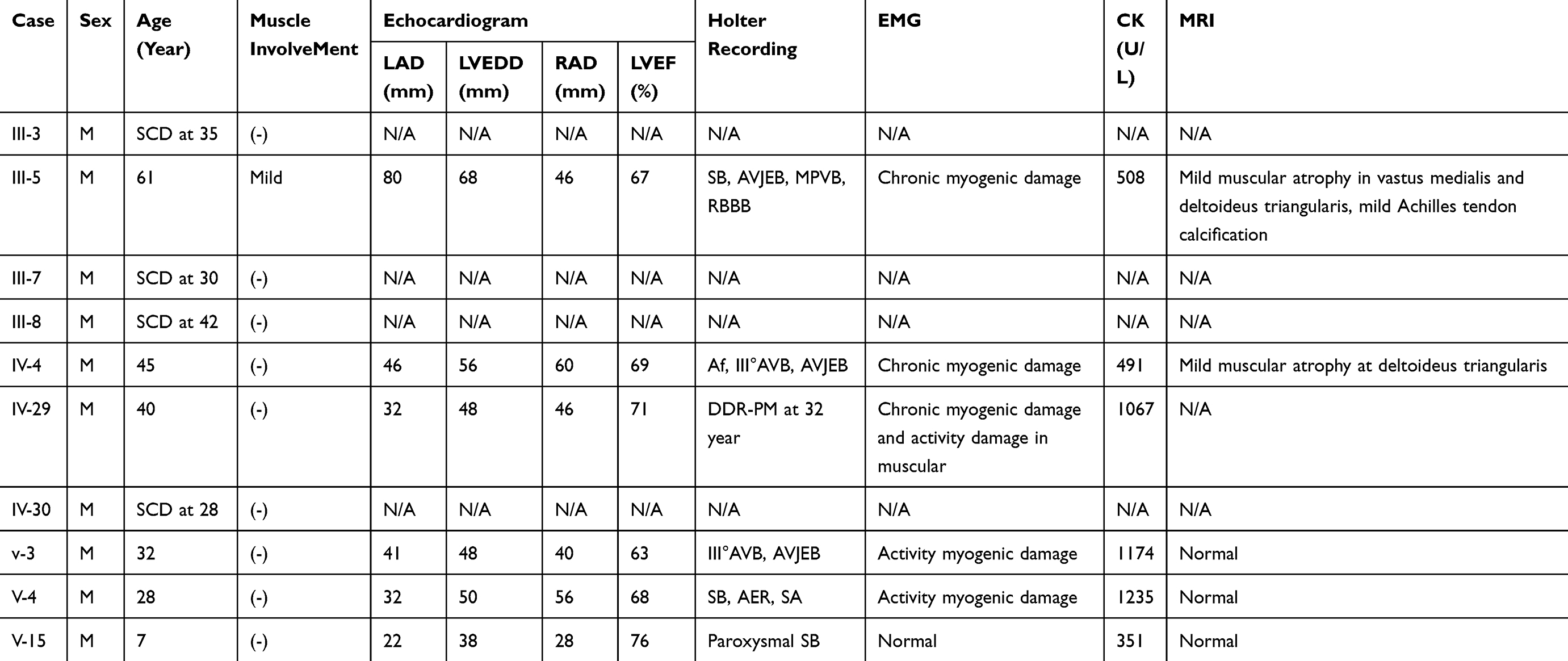 |
Table 1 Clinical Features Of The Proband And 9 Affected Family Members |
A Novel Duplication Mutation (c.405dup/p.Asp136X) Was Identified In The Patients
Since the pedigree (Figure 2) provided evidence of X-linked recessive inheritance, X-exome sequencing was performed for the proband. Approximately 7,653 exons on the X chromosome were captured and sequenced. The average base coverage depth of the X chromosome exome sequencing was 98.32 and 93.79% of genomic regions covered more than 20 times. According to the ACMG guidelines, we identified that the EMD mutation c.405dupin exon5 was a possible candidate mutation. Mutation screening for the candidate mutation was performed on all living individuals in the pedigree by PCR-Sanger sequencing. This variant was also identified by Sanger sequencing (Figure 3). The novel duplication mutation (c.405dup/p.Asp136X) resulted in a premature termination at position 136. This variant was a novel mutation and has not yet been reported in Pubmed, Clinvar or in any other previous cases of HGMD.
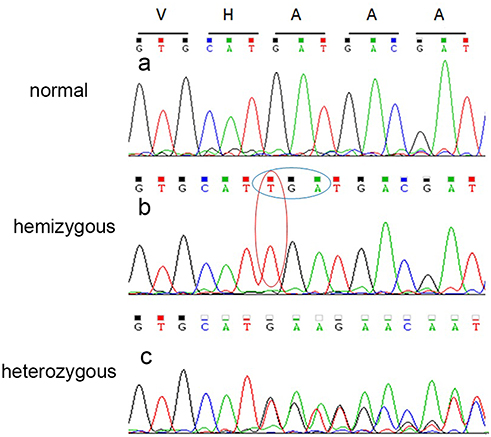 |
Figure 3 Sequencing results of EMD mutation. (A) Image a shows results for a normal individual. Sanger sequencing demonstrated a duplication mutation (c.405dup) in exon 5 of EMD in the hemizygous individual (B) and the overlapping peaks in the heterozygous individual (C). |
The Mutation Leads To Absent Of EDMD Proteins
Immunoblotting of peripheral blood lymphocytes revealed that emerin protein expression was present in the heterozygous individuals. The expression of emerin in hemizygous individuals was completely absent (Figure 4A).
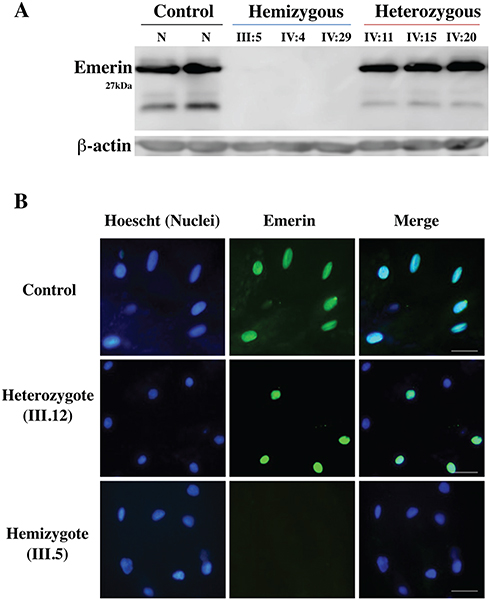 |
Figure 4 Immunoblot and immunohistochemistry results revealed that EMD hemizygous individuals do not express emerin protein. (A) Immunoblotting of emerin protein (29 kDa) expression from total protein extracts of lymphocytes from the hemizygotes (III5, IV4, IV29), heterozygotes (IV11, IV15, IV20) and a normal control, with use of the emerin monoclonal antibody. No emerin protein bands were detected in the hemizygous patients. (B) Left panels, nucleus (Hoechst) staining (blue); middle panels, emerin staining (green); right panels, merged emerin and nucleus staining. Patients include a normal male control, heterozygote female (III.12) and a male hemizygote (III.5). Scale bar represents 50 μm. |
The immunofluorescence results of mucosa exfoliating cells demonstrated an absence of emerin expression in nuclei of all hemizygous members, while about 50% of nuclei of heterozygous female carriers of the EMD mutation were positive for emerin staining (Figure 4B), indicating that emerin expression from the wild type allele occurs only when that allele is not inactivated by random X chromosome inactivation.
Discussion
In this current study discovered a novel duplication mutation in the EMD gene that led to the development of EDMD1 in a multi-generation Chinese family. The family consists of 77 members, of which all of the hemizygous members suffered from cardiac conduction disorder (Figure 2). This study is particularly novel since previous studies were limited in the amount of data from large families displaying similar cardiac phenotypes that we report here. Mutations in EMD are very rare, with an estimated incidence of 0.13/100,000.13 To date, approximately 150 different EMD mutations have been reported.10 In our study, we identified a duplication mutation (c.405dup/p.Asp136X) of EMD which is a novel variant that has not yet been reported in HMGD cases, Clinvar or Pubmed. We also revealed that patients with this mutation no longer express emerin protein, including any of the potential small isoforms (Figure 4). Ultimately, our study expands the genetic spectrum of the EDMD1 disease presentation and provides an additional genetic marker for diagnostic measures.
Thus far, the correlations between gene mutations and phenotypes have not been fully characterized. In EDMD1, symptoms involving the skeletal muscles usually arise before cardiac disease.10 However, the patients in our study were characterized by early serious cardiac conduction abnormalities accompanied by mild skeletal muscular dystrophy and joint contracture. To date, fewer than five EMD mutations have been reported in patients with predominant cardiac diseases and mild skeletal muscle disorders.14–16 These cases, together with our findings, may reveal that environmental or genetic modifications, such as functionally overlapping proteins, may contribute to the observed clinical variability caused by EMD mutations. Furthermore, female carriers of an EMD mutation have no muscular symptoms, but some may be at risk of cardiac arrhythmias and sudden death. Sakata K et al reported that approximately 20% of the female carriers in their study developed overt cardiac disease necessitating pacemaker implantation and pharmacotherapy.14 However, none of the female carriers in our study (range, 2–82 years of age) had serious cardiac complications.
In this pedigree, SCD occurred in 4 of 10 patients (40%). Of the other 6 surviving patients, 5 developed serious cardiac conduction abnormalities, and a pacemaker eventually needed to be implanted in a 35-year-old patient (IV:29). SCD is a serious cardiac complication and is an important cause of mortality in the EDMD population.17,18 Although the youngest patient did not have signs of cardiac conduction abnormality, paroxysmal bradyarrhythmia was observed in the ECG measurements. There is a general consensus regarding the efficacy of the implantable cardioverter-defibrillator (ICD) in primary prevention of SCD in EDMD patients with reduced ejection fraction values19 or with ventricular arrhythmias.20 However, reports thus far may have been biased by cases of autosomal dominant disease.21–23 In patients with Emery–Dreifuss and limb-girdle type 1B muscular dystrophies associated with LMNA mutations, sudden death is responsible for 30% of all deaths.23 However, the incidence rate of SCD in EDMD1 is unknown because of the scarcity cases.18,24–26 Therefore, the 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death20 considered that management of the rare X-linked recessive Emery–Dreifuss muscular dystrophy associated with mutations in the emerin gene is complicated by a lack of clinical data; in the absence of gene-specific information, it seems reasonable to adopt the management strategy used in the dominant form of Emery–Dreifuss.
SCD is a leading cause of mortality, with an annual incidence of one death per 1000 person-years, affecting all ages.27 However, no obvious extra-cardiac causes may be identified by post-mortem examination in some cases of SCD. Therefore, the identification of genetic factors that predispose these patients to SCD is important because this enables genetic testing that may contribute to diagnosis and risk stratification.28 Currently, most studies have focused on genes that encode ion channels (SCN5A, HCN4, KCNJ2),29,30 structural proteins (LMNA, DES, JUP), cardiac transcription factors (NKX2-5, TBX5), gap junctions (Cx40) and energy metabolism regulators (PRKAG2),28,31,32–34 but few studies have selected EMD as a positional candidate gene for sequencing. Our findings provide a rationale for EMD mutation testing in cases of X-linked inherited CCD or SCD, even if pathognomonic neuromuscular features may not be obvious. Once an EMD mutation is identified, genetic testing should be offered to at-risk men and women. The mechanism by which the emerin mutation causes sudden death is unclear. The presumed mechanism could be emerin regulation of megakaryoblastic leukaemia 1 (MKL1)–serum response factor (SRF) activity.7 MKL1 (also known as MAL or MRTF-A) is a mechano-sensitive transcription factor with important roles in the cardiovascular system.7
In conclusion, we identified a novel duplication mutation (c.405dup/p.Asp136X) in the EMD gene in a multi-generation Chinese family with EDMD1. The pedigree displayed an uncommon clinical presentation of serious cardiac conduction abnormalities at an early age and a high incidence of SCD. This discovery expands the mutation spectrum of EMD and indicates the importance of EMD in SCD. Moreover, our study provides a rationale for EMD mutation testing in cases of X-linked inherited CCD or SCD, even if there is a lack of other obvious pathologic markers. However, the correlations between the gene mutation type and phenotype or observed clinical variability need further research.
Statement Of Ethics
The patient (proband) provided written informed consent for the case details to be published. The study protocol has been approved by the research institute’s committee on human research.
Acknowledgments
We would like to thank the patients and their families for participating in this study. We would also like to thank Prof. Ju Chen and Dr. Christa Trexler of University of California, San Diego, for the help in preparing this manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Emery AE, Dreifuss FE. Unusual type of benign x-linked muscular dystrophy. J Neurol Neurosurg Psychiatry. 1966;29(4):338–342. doi:10.1136/jnnp.29.4.338
2. Dubowitz V, editor. Muscle disorders in childhood.
3. Emery AE. Emery-Dreifuss muscular dystrophy - a 40 year retrospective. Neuromuscul Disord. 2000;10(4–5):228–232.
4. Meinke P, Schneiderat P, Srsen V, et al. Abnormal proliferation and spontaneous differentiation of myoblasts from a symptomatic female carrier of X-linked Emery–Dreifuss muscular dystrophy. Neuromuscular Disord. 2015;25(2):127–136. doi:10.1016/j.nmd.2014.09.012
5. Bione S, Maestrini E, Rivella S, et al. Identification of a novel X-linked gene responsible for Emery-Dreifuss muscular dystrophy. Nat Genet. 1994;8(4):323–327. doi:10.1038/ng1294-323
6. Manilal S, Nguyen TM, Sewry CA, Morris GE. The Emery-Dreifuss muscular dystrophy protein, emerin, is a nuclear membrane protein. Hum Mol Genet. 1996;5(6):801–808. doi:10.1093/hmg/5.6.801
7. Ho CY, Jaalouk DE, Vartiainen MK, Lamin LJ. A/C and emerin regulate MKL1–SRF activity by modulating actin dynamics. Nature. 2013;497(7450):507–511. doi:10.1038/nature12105
8. Muchir A, Worman HJ. Emery-Dreifuss muscular dystrophy. Curr Neurol Neurosci Rep. 2007;7(1):78–83.
9. Koch AJ, Holaska JM. Emerin in health and disease. Semin Cell Dev Biol. 2014;29:95–106. doi:10.1016/j.semcdb.2013.12.008
10. Madej-Pilarczyk A, Kochanski A. Emery-Dreifuss muscular dystrophy: the most recognizable laminopathy. Folia Neuropathol. 2016;54(1):1–8. doi:10.5114/fn.2016.58910
11. Mondal K, Shetty AC, Patel V, Cutler DJ, Zwick ME. Targeted sequencing of the human X chromosome exome. Genomics. 2011;98(4):260–265. doi:10.1016/j.ygeno.2011.04.004
12. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi:10.1038/gim.2015.30
13. Norwood FL, Harling C, Chinnery PF, Eagle M, Bushby K, Straub V. Prevalence of genetic muscle disease in Northern England: in-depth analysis of a muscle clinic population. Brain. 2009;132(Pt 11):3175–3186. doi:10.1093/brain/awp236
14. Sakata K. High incidence of sudden cardiac death with conduction disturbances and atrial cardiomyopathy caused by a nonsense mutation in the STA gene. Circulation. 2005;111(25):3352–3358. doi:10.1161/CIRCULATIONAHA.104.527184
15. Karst ML, Herron KJ, Olson TM. X-Linked nonsyndromic sinus node dysfunction and atrial fibrillation caused by Emerin mutation. J Cardiovasc Electr. 2008;19(5):510–515. doi:10.1111/j.1540-8167.2007.01081.x
16. Zhang M, Chen J, Si D, et al. Whole exome sequencing identifies a novel EMD mutation in a Chinese family with dilated cardiomyopathy. BMC Med Genet. 2014;15:77. doi:10.1186/1471-2350-15-77
17. Chen W, Huo J, Ma A, Bai L, Liu P. A novel mutation of the LMNA gene in a family with dilated cardiomyopathy, conduction system disease, and sudden cardiac death of young females. Mol Cell Biochem. 2013;382(1–2):307–311. doi:10.1007/s11010-013-1734-3
18. Finsterer J, Stollberger C, Maeztu C. Sudden cardiac death in neuromuscular disorders. Int J Cardiol. 2016;203:508–515. doi:10.1016/j.ijcard.2015.10.176
19. Russo V, Nigro G. ICD role in preventing sudden cardiac death in Emery-Dreifuss muscular dystrophy with preserved myocardial function: 2013 ESC guidelines on cardiac pacing and cardiac resynchronization therapy. Europace. 2015;17(2):337. doi:10.1093/europace/euu146
20. Priori SG, Blomstrom-Lundqvist C, Mazzanti A, et al. ESC guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: the task force for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death of the European Society of Cardiology (ESC). Endorsed by: association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J. 2015;36(41):2793–2867.
21. van Berlo JH, de Voogt WG, van der Kooi AJ, et al. Meta-analysis of clinical characteristics of 299 carriers of LMNA gene mutations: do lamin A/C mutations portend a high risk of sudden death? J Mol Med (Berl). 2005;83(1):79–83. doi:10.1007/s00109-004-0589-1
22. Meune C, Van Berlo JH, Anselme F, Bonne G, Pinto YM, Duboc D. Primary prevention of sudden death in patients with lamin A/C gene mutations. N Engl J Med. 2006;354(2):209–210. doi:10.1056/NEJMc052632
23. van Rijsingen IA, Arbustini E, Elliott PM, et al. Risk factors for malignant ventricular arrhythmias in lamin a/c mutation carriers a European cohort study. J Am Coll Cardiol. 2012;59(5):493–500. doi:10.1016/j.jacc.2011.08.078
24. Fishbein MC, Siegel RJ, Thompson CE, Hopkins LC. Sudden death of a carrier of X-linked Emery-Dreifuss muscular dystrophy. Ann Intern Med. 1993;119(9):900–905. doi:10.7326/0003-4819-119-9-199311010-00006
25. Rakovec P, Zidar J, Sinkovec M, Zupan I, Brecelj A. Cardiac involvement in Emery-Dreifuss muscular dystrophy: role of a diagnostic pacemaker. Pacing Clin Electrophysiol. 1995;18(9 Pt 1):1721–1724. doi:10.1111/j.1540-8159.1995.tb06996.x
26. Ishikawa K, Mimuro M, Tanaka T. Ventricular arrhythmia in X-linked Emery-Dreifuss muscular dystrophy: a lesson from an autopsy case. Intern Med. 2011;50(5):459–462. doi:10.2169/internalmedicine.50.4598
27. Straus SM, Bleumink GS, Dieleman JP, van der Lei J, Stricker BHC, Sturkenboom MCJM. The incidence of sudden cardiac death in the general population. J Clin Epidemiol. 2004;57(1):98–102. doi:10.1016/S0895-4356(03)00210-5
28. Bezzina CR, Lahrouchi N, Priori SG. Genetics of sudden cardiac death. Circ Res. 2015;116(12):1919–1936. doi:10.1161/CIRCRESAHA.116.304030
29. Iio C, Ogimoto A, Nagai T, et al. Association between genetic variation in the SCN10A gene and cardiac conduction abnormalities in patients with hypertrophic cardiomyopathy. Int Heart J. 2015;56(4):421–427. doi:10.1536/ihj.14-411
30. Tzimas I, Zingraf JC, Bajanowski T, Poetsch M. The role of known variants of KCNQ1, KCNH2, KCNE1, SCN5A, and NOS1AP in water-related deaths. Int J Legal Med. 2016;130(6):1575–1579. doi:10.1007/s00414-016-1424-2
31. Hertz CL, Christiansen SL, Larsen MK, et al. Genetic investigations of sudden unexpected deaths in infancy using next-generation sequencing of 100 genes associated with cardiac diseases. Eur J Hum Genet. 2016;24(6):817–822. doi:10.1038/ejhg.2015.198
32. Christiansen SL, Hertz CL, Ferrero-Miliani L, et al. Genetic investigation of 100 heart genes in sudden unexplained death victims in a forensic setting. Eur J Hum Genet. 2016. doi:10.1038/ejhg.2016.118
33. Ackerman M, Atkins DL, Triedman JK. Sudden Cardiac Death in the Young. Circulation. 2016;133(10):1006–1026. doi:10.1161/CIRCULATIONAHA.115.020254
34. Miles CJ, Behr ER. The role of genetic testing in unexplained sudden death. Transl Res. 2016;168:59–73. doi:10.1016/j.trsl.2015.06.007
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.