")
Back to Journals » International Journal of General Medicine » Volume 13
A Novel Missense Mutation of the CSF1R Gene Causes Incurable CSF1R-Related Leukoencephalopathy: Case Report and Review of Literature
Authors Chen J, Luo S, Li N, Li H, Han J , Ling L
Received 12 October 2020
Accepted for publication 7 December 2020
Published 22 December 2020 Volume 2020:13 Pages 1613—1620
DOI https://doi.org/10.2147/IJGM.S286421
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Jie Chen,1 Shiying Luo,1 Ning Li,1 Huimin Li,1 Jinming Han,2 Li Ling1
1Department of Neurology, Affiliated Hospital of Hebei University, Baoding, People’s Republic of China; 2Department of Clinical Neuroscience, Karolinska Institutet, Stockholm, Sweden
Correspondence: Li Ling
Department of Neurology, Affiliated Hospital of Hebei University, Baoding, People’s Republic of China
Email [email protected]
Jinming Han
Department of Clinical Neuroscience, Karolinska Institutet, Stockholm, Sweden
Email [email protected]
Abstract: CSF1R-related leukoencephalopathy, mainly caused by the mutation of the colony stimulating factor 1 receptor (CSF1R) gene on chromosome 5, is an underestimated neurological disease typically presenting as early-onset cognitive decline and personality changes. Currently, there is no specific treatment for CSF1R-related leukoencephalopathy. Most clinicians failed to recognize this disease during an early disease stage, leading to a high rate of misdiagnosis. Although rare, an increasing amount of CSF1R-related leukoencephalopathy cases have been reported recently. In this study, we first report a 35-year-old woman with CSF1R-related leukoencephalopathy carrying a novel missense mutation c.2463G >C (p.W821C) of CSF1R. An extensive literature research was performed in order to better understand the broader genetic and clinical characteristics of CSF1R-related leukoencephalopathy. A total of 147 patients with CSF1R-related leukoencephalopathy confirmed either by the genetic test or brain biopsy were identified. Among them, 49 patients were sporadic, and the rest of individuals had a family history originating from 46 different families. Our study indicated that the average age of CSF1R-related leukoencephalopathy onset was 41.4 years. Typical clinical symptoms of CSF1R-related leukoencephalopathy include cognitive decline, movement disorders, behavior changes and mental disorders. Genetic studies have reported 93 missense mutations, 13 splicing mutations, 6 deletion/insertion mutations, 1 code shift mutation and 1 nonsense mutation of the CSF1R gene in patients with CSF1R-related leukoencephalopathy. Early genetic detection and brain biopsy would be helpful for a confirmed diagnosis, and more translational studies are needed to combat this devastating disease.
Keywords: CSF1R-related leukoencephalopathy, clinical symptoms, CSF1R
Introduction
CSF1R-related leukoencephalopathy is a rapidly progressive neurodegenerative disease. This disease is mainly caused by the mutations of the colony stimulating factor 1 receptor (CSF1R) gene located in the chromosome 5q32 and now considered as a primary central nervous system (CNS) microgliopathy.1 Patients with CSF1R-related leukoencephalopathy present complex clinical symptoms which make this disease more difficult to recognize during an early disease stage and can often be misdiagnosed as multiple sclerosis,2 frontotemporal dementia, corticobasal syndrome, Alzheimer disease, atypical cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Unfortunately, there is no specific treatment for this disease yet and patients may gradually lose their motor function, become bedridden and eventually die from lung infections or other secondary infections.3–5 Thus, more effort is needed to understand this incurable disease and eventually improve the life quality of patients with CSF1R-related leukoencephalopathy.
Case Presentation
A 35-year-old woman was admitted to Department of Neurology at the affiliated hospital of Hebei University due to speech impairment and gait disorder for almost 3 months. She had no remarkable past medical history. This patient graduated from a junior college, while her speech was clumsy at admission. Specifically, she had difficulty in pronouncing certain words when she talked fast, often causing frustration. Cognitive function tests revealed that the Mini-Mental State Exam (MMSE) score was 29/30 and the Montreal Cognitive Assessment (MoCA) score was 28/30 at admission. Upon neurological evaluation, the pharyngeal reflex response was reduced and the uvula deviated toward the left side. The tendon reflexes of the limbs were slightly more active and other remarkable signs were not evident. Routine laboratory investigations indicated a sign of liver damage without apparent cause, evidenced by increased levels of alanine aminotransferase (52, normal range 7–40U/L) in the circulation. Sagittal T2-weighted fluid-attenuated inversion recovery (FLAIR) images showed periventricular white matter hyperintensities and abnormal signals on the bilateral semi-oval centers (Figure 1A). Ventricular enlargement, thinning of the corpus callosum and global brain atrophy were also noted on MRI scans (Figure 1A). Four months later, the patient experienced communication difficulties, dragging gait, posture instability, dysphagia, progressive cognitive decline and obvious tremors of the jaw and hands. Repeated routine laboratory investigations also showed an increased serum level of alanine aminotransferase (73, normal range 7–40U/L). Multiple patchy abnormal signals, global brain atrophy and thinning of corpus callosum were evident in bilateral lateral ventricles on MRI scans (Figure 1B). Glucocorticoids and therapeutic strategies controlling related clinical symptoms were then used. However, the patient continued to progressively deteriorate during follow-up period. Nine months after the first admission, the patient suffered from dysarthria, tongue muscle atrophy, upper body tremors and walking abnormalities. Most of the daily activities could not be performed by herself. Cognitive function, especially short-term memory, became worse with the MMSE score being 23/30 and the MoCA score being 22/30. Repeated MRI scans did not show any significant changes from the last time (Figure 1C). After 12 months, the patient could not walk and essentially restricted to bed much of day. Genetic testing results indicated that the CSF1R gene on chromosome 5 (exon 19) was reported as c.2463G >C (p.W821C) with a missense mutation (Figure 2A). The genetic code was mutated from GGA to GCA, leading to the changes of tryptophan number 821 to cysteine (Figure 2A). We confirmed this novel mutation c.2463G >C (p.W821C) by performing insilico analysis using different methods. The results are “probably damaging” (Polyphen); “deleterious” (Sorting Intolerant from Tolerant, SIFT) and “disease causing” (Mutation Taster) (Figure 2B). The predicted result using the Mutation Assessor is medium (Figure 2B and Supplementary Figure). The criterion is PM1 (the American College of Medical Genetics and Genomics, ACMG) since it is located at a mutational hot spot and/or critical and well-established functional domain. Whole exome sequencing was carried out with the consent of this patient’s parents, sister and daughters. Genetic tests of patient’s relatives did not show abnormalities. Our case is sporadic (de novo mutation), which has not been reported in genome databases. Pathological results of the brain tissue removed during biopsy reported that the structure of white matter was seriously damaged, in particular with axonal destruction. Numerous axonal spheroids, gliosis and proliferating phagocytic cells were also noted (pathological images were not shown since the brain biopsy was not performed in our hospital).
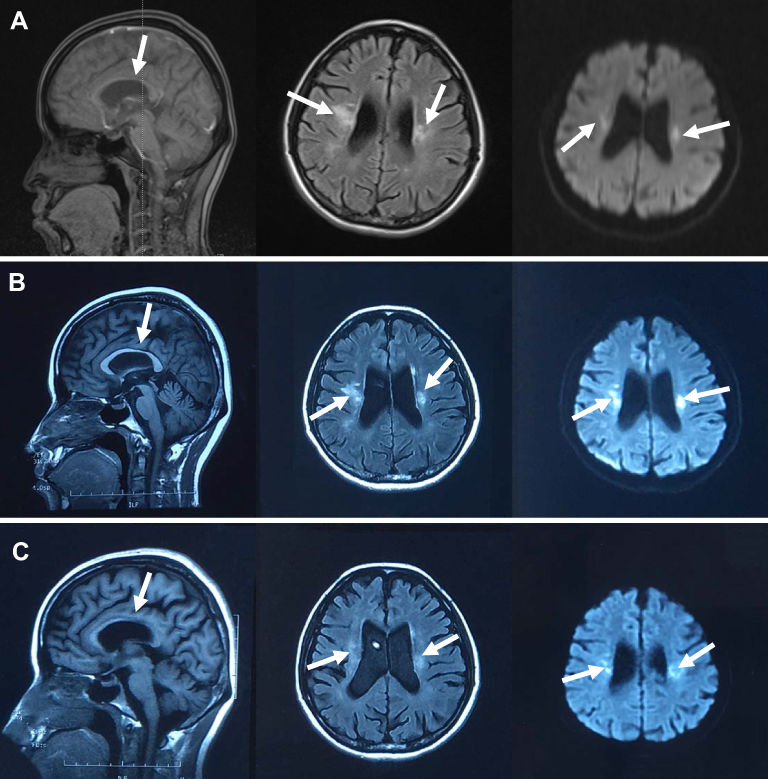 |
Figure 1 MRI features of CSF1R-related leukoencephalopathy. (A) T1-weighted sagittal images showed thinning of the corpus callosum (arrows). Fluid-attenuated inversion recovery (FLAIR) and diffusion weighted imaging (DWI) images showed periventricular white matter hyperintensities, abnormal signals on bilateral semi-oval centers (arrows). Global brain atrophy was noted. MRI was taken on 13th, June, 2019. (B) T1-weighted sagittal images showed that the corpus callosum is thinner than the last time (arrows). FLAIR images showed increased diffuse white matter lesions and the progression of global brain atrophy (arrows). DWI images revealed various isolated spots of hyperintense signals. MRI was taken on 08th, July, 2019. (C) Repeated MRI scans were taken on 10th, October, 2019. White arrows represent the brain lesions. |
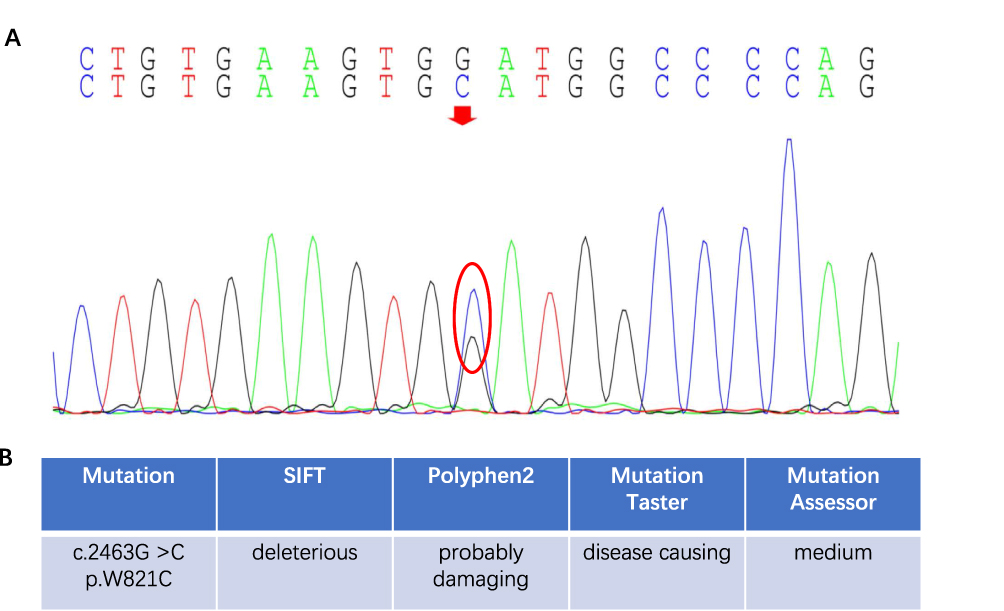 |
Figure 2 Genetic test results of CSF1R-related leukoencephalopathy. (A) The CSF1R gene on chromosome 5 (exon 19) was c.2463G >C (p.W821C) with the missense mutation. The genetic code was mutated from GGA to GCA, leading to the changes of tryptophan number 821 to cysteine. The red arrow and red circle mean the base mutation site. (B) Results of insilico analysis for the W821C mutations by SIFT, Polyphen2, Mutation Taster and Mutation Assessor. |
Discussion
We conducted a literature review by searching PubMed, WANFANG (old.g.wanfangdata.com.cn) and CNKI (www.cnki.net). The following keywords “axonal leukodystrophy”, “adult-onset leukodystrophy with axonal spheroids”, “CSF1R-related leukoencephalopathy” and “hereditary diffuse leukoencephalopathy with axonal spheroids” were used. Both English and Chinese articles were included (from 1987 to February 2020) in our study.3,5–66 All patients with CSF1R-related leukoencephalopathy were confirmed either by the genetic test or brain biopsy. Individuals who presented similar symptoms in the family without genetic tests or brain biopsy were excluded in our study. We extracted the following information: population, gender, age of onset, age of death, survival time, first symptoms, clinical features, neuroimaging findings, genetic testing, pathological results and family history.
A total of 120 patients with detailed MRI images were available. One of the most common imaging findings was brain white matter lesions (109/120, 90.8%). Specifically, they were mainly located in the periventricular areas (57/109, 52.3%), frontal lobes (43/109, 39.4%), corpus callosum (24/109, 22.0%) and parietal lobes (24/109, 22.0%). Brain lesions in some patients with CSF1R-related leukoencephalopathy can also be noted in the midbrain (3/109, 2.8%), temporal lobes (3/109, 2.8%), internal capsule hind limbs (2/109, 1.8%) and brain stem (2/109, 1.8%). Furthermore, atrophy (71/120, 59.2%, particularly frontal lobe atrophy) and thinning of the corpus callosum (48/120, 40.0%) were also frequently found (Figure 3A). In some patient, partial periventricular white matter lesions were oval in shape and perpendicular to the ventricles. Persistent limited diffusion on diffusion-weighted imaging (DWI) was also evident. In support of this, Onder et al recently proposed that the persistence of the diffusion restriction in deep white matter lesions may serves as a crucial sign for CSF1R-related leukoencephalopathy.67
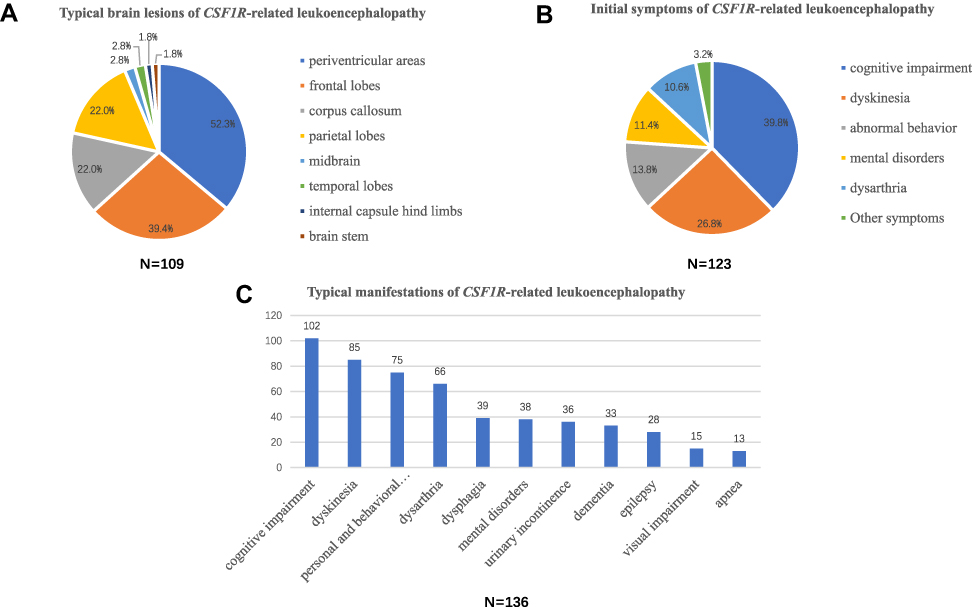 |
Figure 3 Clinical spectrum of CSF1R-related leukoencephalopathy. (A) Typical brain lesions of CSF1R-related leukoencephalopathy. White matter lesions were the most common lesions, which mainly located in the periventricular areas, frontal lobes, corpus callosum and parietal lobes. Atrophy and thinning of the corpus callosum were also noted. (B) Initial symptoms of CSF1R-related leukoencephalopathy. Cognitive impairment, parkinsonism, abnormal behaviors and mental disorders are the most common initial symptoms. (C) Typical manifestations of CSF1R-related leukoencephalopathy during the disease course. Cognitive impairment, parkinsonism, gait disorder, personal and behavioral abnormalities, and dysarthria are the most common typical manifestations during the disease course. |
A total of 147 patients with CSF1R-related leukoencephalopathy were included in our study. Specifically, 49 patients were sporadic and 98 individuals had a family history (from 46 families). The average age of onset was 41.4 years (15 of them were not included due to unknown age of onset), among which males were 43.6 years old and females 39.7 years old. The average course of disease was 5.2 years. The duration of disease ranging from 3 weeks to 11 years. The first symptoms were mainly cognitive impairment (49/123, 39.8%), parkinsonism (22/123, 17.9%), abnormal behaviors (17/123, 13.8%), mental disorders (14/123, 11.4%), dysarthria (13/123, 10.6%) and gait disorder (11/123, 8.9%). Other potential symptoms included visual impairment (2/123, 1.6%), progressive weight loss (1/123, 0.8%) and nominalization (1/123, 0.8%) (Figure 3B). Furthermore, typical clinical manifestations of CSF1R-related leukoencephalopathy during the disease course included cognitive impairment (102/136, 75.0%), parkinsonism (57/136, 41.9%), gait disorder (28/136, 20.6%), personal and behavioral abnormalities (75/136, 55.1%), dysarthria (66/136, 48.5%), dysphagia (39/136, 28.7%), mental disorders (38/136, 27.9%), urinary incontinence (36/136, 26.5%), dementia (33/136, 24.3%), epilepsy (28/136, 20.6%), visual impairment (15/136, 11.0%) and apnea (13/136, 9.6%) (Figure 3C). Some rare manifestations such as drinking cough and orthostatic hypotension have also been reported.
The clinical spectrum of CSF1R-related leukoencephalopathy is expanding. For example, the involvement of spinal cord in CSF1R-related leukoencephalopathy has been reported.68 Apart from typical CSF1R-related leukoencephalopathy clinical symptoms such as progressive motor impairment, hyperintense signals on DWI of this patient were noted from the subcortex white matter to the medulla, then extending to cervical and thoracic spinal cord.68 Some researchers proposed that brain and spinal cord microglia may differ regarding their development, phenotypes and even functions.69 Potential underlying mechanisms of microglial regional specificity in the condition of CSF1R-related leukoencephalopathy need to be further investigated. Furthermore, visual impairment had also been reported in a Chinese patient with CSF1R-related leukoencephalopathy.25 Bilateral optic nerves lesions were evident on optic nerve MRI scans, causing difficulties to differentiate from MS.
A total of 114 patients with detailed genetic information were included for analysis, including 93 with missense mutation, 13 with splicing mutation, 6 with deletion/insertion mutation, 1 with code shift mutation and 1 with nonsense mutation. The mutation site of one patient was located on exon 2, while the rest were located on exon 12–22 with exon 18 being the most common one. We summarized that 15 patients with CSF1R-related leukoencephalopathy from 11 unrelated families in 4 different countries had the same gene mutation: p.I794T, while their clinical manifestations were different. Furthermore, some family members who carried the mutations of CSF1R gene might not present obvious clinical symptoms. We suppose that it may be attributed to the changes of penetrance, making the mutation causing different degrees of disease severity. Meanwhile, this phenomenon also indicates that the dominant transmission pattern has familial phenotypic variation or incomplete penetrance.
Among 147 patients with CSF1R-related leukoencephalopathy, the brain biopsy was conducted in a total of 60 patients. Typical pathological features of CSF1R-related leukoencephalopathy are white matter pathologies with an enlargement of the frontal horn in the lateral ventricle. Microscopically, white matter damage presents as increased mitochondrial vacuolation and disorganized neurofilaments in ballooned axons or myelin loss.14 Histopathologic studies showed that axonal spheroids were positive for phosphorylated neurofilament, amyloid precursor protein (APP) and variably for ubiquitin.70 Macrophages with lipid-laden vacuoles and lipofuscin-like pigment of glial cells were visible. The formation of axonal spheroid is considered as a pathological manifestation of CSF1R-related leukoencephalopathy.71,72 Tada et al indicated that in layers 3 and 4 of the frontal cortex in CSF1R-related leukoencephalopathy fewer numbers of Iba1 positive microglia were recorded than healthy control brains. A high proliferation of microglia was noted in selective brain regions.73 Oosterhof et al also noted decreased microglial cell density and numbers in patients with CSF1R-related leukoencephalopathy.74 Deep cortical layers could be more vulnerable than superficial cortical layers in patients with CSF1R-related leukoencephalopathy.75
CSF1R-related leukoencephalopathy is a rare but rapidly progressive neurodegenerative disease. A clinical diagnosis of CSF1R-related leukoencephalopathy should be considered when the patients present cognitive impairment, movement disorders and personality changes. A combination of genetic testing and MRI imaging is needed for early accurate diagnosis and therapeutic decision making due to the complexity of CSF1R-related leukoencephalopathy. CSF1R-related leukoencephalopathy disease is relatively rare with unclear pathophysiological mechanisms and complex genetic mutation mechanisms. It is worth noting that with the popularization of genetic diagnostic methods, more and more mutations have been discovered. CSF1R gene mutation type has a dominant-negative effect,13 haploinsufficiency,74 biallelic variants76 and homozygous mutations.77 Hematopoietic stem cell transplantation (HSCT) is relatively effective in some patients,78 however, some patients did not show any improvements after HSCT,79 indicating that HSCT may not a specific treatment for CSF1R-related leukoencephalopathy. CSF1R-related leukoencephalopathy is considered as a primary CNS microgliopathy. Therefore, development of a potential microglia-based treatment is warranted.80 Only when we are sufficiently knowledgeable can we find potential translational cues and clinical interventions to combat CSF1R-related leukoencephalopathy.
Ethics and Consent
As this typical case was not involved in any experimental interventions, no formal research ethics approval was required. The relatives of this patient provided written informed consent to the publication of the information and images related to this manuscript.
Disclosure
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest and report no conflicts of interest for this work.
References
1. Rademakers R, Baker M, Nicholson AM, et al. Mutations in the colony stimulating factor 1 receptor (CSF1R) gene cause hereditary diffuse leukoencephalopathy with spheroids. Nat Genet. 2011;44:200–205. doi:10.1038/ng.1027
2. Sundal C, Baker M, Karrenbauer V, et al. Hereditary diffuse leukoencephalopathy with spheroids with phenotype of primary progressive multiple sclerosis. Eur J Neurol. 2015;22:328–333. doi:10.1111/ene.12572
3. Funayama M, Sugihara M, Takata T, Mimura M, Ikeuchi T. Remarkable behavioural signs and progressive non-fluent aphasia in a patient with adult-onset leucoencephalopathy with axonal spheroids and pigmented glia. Psychogeriatrics. 2019;19(3):282–285. doi:10.1111/psyg.12387
4. Hancock N, Poon M, Taylor B, McLean C. Hereditary diffuse leucoencephalopathy with spheroids. J Neurol Neurosurg Psychiatry. 2003;74:1345–1347. doi:10.1136/jnnp.74.9.1345
5. Spitzer P, Kohl Z, Golitz P, et al. Biochemical markers of neurodegeneration in hereditary diffuse leucoencephalopathy with spheroids. BMJ Case Rep. 2014;2014(jun02 1):bcr2012008510–bcr2012008510. doi:10.1136/bcr-2012-008510
6. Van Gerpen JA, Wider C, Broderick DF, Dickson DW, Brown LA, Wszolek ZK. Insights into the dynamics of hereditary diffuse leukoencephalopathy with axonal spheroids. Neurology. 2008;71(12):925–929. doi:10.1212/01.wnl.0000325916.30701.21
7. Shi T, Li J, Tan C, Chen J. Diagnosis of hereditary diffuse leukoencephalopathy with neuroaxonal spheroids based on next-generation sequencing in a family: case report and literature review. Medicine (Baltimore). 2019;98:e15802. doi:10.1097/MD.0000000000015802
8. Kawakami I, Iseki E, Kasanuki K, et al. A family with hereditary diffuse leukoencephalopathy with spheroids caused by a novel c.2442+2T>C mutation in the CSF1R gene. J Neurol Sci. 2016;367:349–355. doi:10.1016/j.jns.2016.06.013
9. Saitoh B-Y, Yamasaki R, Hayashi S, et al. A case of hereditary diffuse leukoencephalopathy with axonal spheroids caused by a de novo mutation in CSF1R masquerading as primary progressive multiple sclerosis. Mult Scler. 2013;19(10):1367–1370. doi:10.1177/1352458513489854
10. Zhuang LP, Liu CY, Li YX, Huang HP, Zou ZY. Clinical features and genetic characteristics of hereditary diffuse leukoencephalopathy with spheroids due to CSF1R mutation: a case report and literature review. Ann Transl Med. 2020;8:11. doi:10.21037/atm.2019.12.17
11. Lopes C, Duarte S, Santos E, Reis C, Pinto M. Intrafamilial heterogeneity in hereditary diffuse leukoencephalopathy with axonal spheroids. Neurol Clin Pract. 2019;9(6):500–502. doi:10.1212/CPJ.0000000000000647
12. Tian WT, Zhan FX, Liu Q, et al. Clinicopathologic characterization and abnormal autophagy of CSF1R-related leukoencephalopathy. Transl Neurodegener. 2019;8:32. doi:10.1186/s40035-019-0171-y
13. Leng C, Lu L, Wang G, et al. A novel dominant-negative mutation of the CSF1R gene causes adult-onset leukoencephalopathy with axonal spheroids and pigmented glia. Am J Transl Res. 2019;11:6093–6101.
14. Yang X, Huang P, Tan Y, Xiao Q. A novel splicing mutation in the CSF1R gene in a family with hereditary diffuse leukoencephalopathy with axonal spheroids. Front Genet. 2019;10:491. doi:10.3389/fgene.2019.00491
15. Sharma R, Graff-Radford J, Rademakers R, Boeve BF, Petersen RC, Jones DT. CSF1R mutation presenting as dementia with lewy bodies. Neurocase. 2019;25(1–2):17–20. doi:10.1080/13554794.2019.1601230
16. Kraya T, Quandt D, Pfirrmann T, et al. Functional characterization of a novel CSF1R mutation causing hereditary diffuse leukoencephalopathy with spheroids. Mol Genet Genomic Med. 2019;7:e00595. doi:10.1002/mgg3.595
17. Du Q, Chen H, Shi Z, Zhang Y, Wang J, Zhou H. A novel mutation in the CSF1R gene causes hereditary diffuse leukoencephalopathy with axonal spheroids. Neurol Sci. 2019;40(6):1287–1290. doi:10.1007/s10072-018-3693-7
18. Makary MS, Awan U, Kisanuki YY, Slone HW. Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia: clinical and imaging characteristics. Neuroradiol J. 2019;32(2):139–142. doi:10.1177/1971400918822136
19. Wang D, Yu M, Zhang W, Wang Z, Yuan Y. AARS2 compound heterozygous variants in a case of adult-onset leukoencephalopathy with axonal spheroids and pigmented glia. J Neuropathol Exp Neurol. 2018;77(11):997–1000. doi:10.1093/jnen/nly087
20. Coomans C, Sieben A, Lammens M, et al. Early-onset dementia, leukoencephalopathy and brain calcifications: a clinical, imaging and pathological comparison of ALSP and PLOSL/nasu hakola disease. Acta Neurol Belg. 2018;118(4):607–615. doi:10.1007/s13760-018-1023-8
21. Lan M-Y, Liu J-S, Chang -C-C, et al. Clinicopathologic and genetic studies of 2 patients with hereditary diffuse leukoencephalopathy with axonal spheroids. Alzheimer Dis Assoc Disord. 2016;30(1):73–76. doi:10.1097/WAD.0000000000000067
22. Wu L, Liu J, Sha L, et al. Sporadic cases with novel mutations and pedigree in hereditary leukoencephalopathy with axonal spheroids. J Alzheimers Dis. 2017;56:893–898.
23. Adams SJ, Kirk A, Auer RN. Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP): integrating the literature on hereditary diffuse leukoencephalopathy with spheroids (HDLS) and pigmentary orthochromatic leukodystrophy (POLD). J Clin Neurosci. 2018;48:42–49. doi:10.1016/j.jocn.2017.10.060
24. Blume J, Weissert R. Suspected perinatal depression revealed to be hereditary diffuse leukoencephalopathy with spheroids. J Mov Disord. 2017;10:59–61. doi:10.14802/jmd.16050
25. Shu Y, Long L, Liao S, et al. Involvement of the optic nerve in mutated CSF1R-induced hereditary diffuse leukoencephalopathy with axonal spheroids. BMC Neurol. 2016;16(1):171. doi:10.1186/s12883-016-0694-0
26. Eichler FS, Li J, Guo Y, et al. CSF1R mosaicism in a family with hereditary diffuse leukoencephalopathy with spheroids. Brain. 2016;139(6):1666–1672. doi:10.1093/brain/aww066
27. Ueda S, Yamashita H, Hikiami R, Sawamoto N, Yoshida K, Takahashi R. A novel A792D mutation in the CSF1R gene causes hereditary diffuse leukoencephalopathy with axonal spheroids characterized by slow progression. eNeurologicalSci. 2015;1(1):7–9. doi:10.1016/j.ensci.2015.07.001
28. Robinson JL, Suh E, Wood EM, et al. Common neuropathological features underlie distinct clinical presentations in three siblings with hereditary diffuse leukoencephalopathy with spheroids caused by CSF1R p. Arg782His. Acta Neuropathol Commun. 2015;3:42. doi:10.1186/s40478-015-0219-x
29. Meyer-Ohlendorf M, Braczynski A, Al-Qaisi O, et al. Comprehensive diagnostics in a case of hereditary diffuse leukodystrophy with spheroids. BMC Neurol. 2015;15(1):103. doi:10.1186/s12883-015-0368-3
30. Foulds N, Pengelly RJ, Hammans SR, et al. Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia caused by a novel R782G mutation in CSF1R. Sci Rep. 2015;5(1):10042. doi:10.1038/srep10042
31. Cheng X, Shen W, Zou H, et al. Analysis of CSF1R gene mutation in a Chinese family with hereditary diffuse leukoencephalopathy with neuroaxonal spheroids. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2015;32:208–212.
32. Kim E-J, Shin J-H, Lee JH, et al. Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia linked CSF1R mutation: report of four Korean cases. J Neurol Sci. 2015;349(1–2):232–238. doi:10.1016/j.jns.2014.12.021
33. Riku Y, Ando T, Goto Y, et al. Early pathologic changes in hereditary diffuse leukoencephalopathy with spheroids. J Neuropathol Exp Neurol. 2014;73(12):1183–1190. doi:10.1097/NEN.0000000000000139
34. Kitani-Morii F, Kasai T, Tomonaga K, et al. Hereditary diffuse leukoencephalopathy with spheroids characterized by spastic hemiplegia preceding mental impairment. Intern Med. 2014;53(12):1377–1380. doi:10.2169/internalmedicine.53.1932
35. Levin J, Tiedt S, Arzberger T, et al. Diffuse leukoencephalopathy with spheroids: biopsy findings and a novel mutation. Clin Neurol Neurosurg. 2014;122:113–115. doi:10.1016/j.clineuro.2014.04.022
36. Hoffmann S, Murrell J, Harms L, et al. Enlarging the nosological spectrum of hereditary diffuse leukoencephalopathy with axonal spheroids (HDLS). Brain Pathol. 2014;24(5):452–458. doi:10.1111/bpa.12120
37. Terasawa Y, Osaki Y, Kawarai T, et al. Increasing and persistent DWI changes in a patient with hereditary diffuse leukoencephalopathy with spheroids. J Neurol Sci. 2013;335(1–2):213–215. doi:10.1016/j.jns.2013.08.027
38. Inui T, Kawarai T, Fujita K, et al. A new CSF1R mutation presenting with an extensive white matter lesion mimicking primary progressive multiple sclerosis. J Neurol Sci. 2013;334(1–2):192–195. doi:10.1016/j.jns.2013.08.020
39. Ahmed R, Guerreiro R, Rohrer JD, et al. A novel A781V mutation in the CSF1R gene causes hereditary diffuse leucoencephalopathy with axonal spheroids. J Neurol Sci. 2013;332(1–2):141–144. doi:10.1016/j.jns.2013.06.007
40. Calatayud T, Turkalp ZT, Gonzales AA, Munoz DG. Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia: report on a case with morphometric studies. Clin Neuropathol. 2013;32(11):492–501. doi:10.5414/NP300603
41. Kleinfeld K, Mobley B, Hedera P, Wegner A, Sriram S, Pawate S. Adult-onset leukoencephalopathy with neuroaxonal spheroids and pigmented glia: report of five cases and a new mutation. J Neurol. 2013;260(2):558–571. doi:10.1007/s00415-012-6680-6
42. Kinoshita M, Yoshida K, Oyanagi K, Hashimoto T, Ikeda S. Hereditary diffuse leukoencephalopathy with axonal spheroids caused by R782H mutation in CSF1R: case report. J Neurol Sci. 2012;318:115–118.
43. Sundal C, Ekholm S, Nordborg C, et al. Update of the original HDLS kindred: divergent clinical courses. Acta Neurol Scand. 2012;126(1):67–75. doi:10.1111/j.1600-0404.2011.01624.x
44. Sundal C, Lash J, Aasly J, et al. Hereditary diffuse leukoencephalopathy with axonal spheroids (HDLS): a misdiagnosed disease entity. J Neurol Sci. 2012;314(1–2):130–137. doi:10.1016/j.jns.2011.10.006
45. Martinez-Saez E, Shah S, Costa C, Fleminger S, Connor S, Bodi I. Adult onset leukodystrophy with neuroaxonal spheroids and demyelinating plaque-like lesions. Neuropathology. 2012;32(3):285–292. doi:10.1111/j.1440-1789.2011.01257.x
46. Freeman SH, Hyman BT, Sims KB, et al. Adult onset leukodystrophy with neuroaxonal spheroids: clinical, neuroimaging and neuropathologic observations. Brain Pathol. 2009;19(1):39–47. doi:10.1111/j.1750-3639.2008.00163.x
47. Baba Y, Ghetti B, Baker MC, et al. Hereditary diffuse leukoencephalopathy with spheroids: clinical, pathologic and genetic studies of a new kindred. Acta Neuropathol. 2006;111:300–311.
48. Terada S, Ishizu H, Yokota O, et al. An autopsy case of hereditary diffuse leukoencephalopathy with spheroids, clinically suspected of alzheimer’s disease. Acta Neuropathol. 2004;108(6):538–545. doi:10.1007/s00401-004-0920-5
49. Marotti JD, Tobias S, Fratkin JD, Powers JM, Rhodes CH. Adult onset leukodystrophy with neuroaxonal spheroids and pigmented glia: report of a family, historical perspective, and review of the literature. Acta Neuropathol. 2004;107(6):481–488. doi:10.1007/s00401-004-0847-x
50. van der Knaap MS, Naidu S, Kleinschmidt-Demasters BK, Kamphorst W, Weinstein HC. Autosomal dominant diffuse leukoencephalopathy with neuroaxonal spheroids. Neurology. 2000;54(2):463–468. doi:10.1212/WNL.54.2.463
51. Jin C, Washimi Y, Yoshida K, Hashizume Y, Yazawa I. Characterization of spheroids in hereditary diffuse leukoencephalopathy with axonal spheroids. J Neurol Sci. 2015;352(1–2):74–78. doi:10.1016/j.jns.2015.03.033
52. Nicholson AM, Baker MC, Finch NA, et al. CSF1R mutations link POLD and HDLS as a single disease entity. Neurology. 2013;80(11):1033–1040. doi:10.1212/WNL.0b013e31828726a7
53. Daida K, Nishioka K, Li Y, Nakajima S, Tanaka R, Hattori N. CSF1R mutation p.G589R and the distribution pattern of brain calcification. Intern Med. 2017;56(18):2507–2512. doi:10.2169/internalmedicine.8462-16
54. Battisti C, Di Donato I, Bianchi S, et al. Hereditary diffuse leukoencephalopathy with axonal spheroids: three patients with stroke-like presentation carrying new mutations in the CSF1R gene. J Neurol. 2014;261(4):768–772. doi:10.1007/s00415-014-7257-3
55. La Piana R, Webber A, Guiot M-C, Del Pilar Cortes M, Brais B. A novel mutation in the CSF1R gene causes a variable leukoencephalopathy with spheroids. Neurogenetics. 2014;15(4):289–294. doi:10.1007/s10048-014-0413-1
56. Lynch DS, Jaunmuktane Z, Sheerin U-M, et al. Hereditary leukoencephalopathy with axonal spheroids: a spectrum of phenotypes from CNS vasculitis to parkinsonism in an adult onset leukodystrophy series. J Neurol Neurosurg Psychiatry. 2016;87(5):512–519. doi:10.1136/jnnp-2015-310788
57. Gore E, Manley A, Dees D, Appleby BS, Lerner AJ. A young-onset frontal dementia with dramatic calcifications due to a novel CSF1R mutation. Neurocase. 2016;22(3):257–262. doi:10.1080/13554794.2016.1175635
58. Granberg T, Hashim F, Andersen O, Sundal C, Karrenbauer VD. Hereditary diffuse leukoencephalopathy with spheroids - a volumetric and radiological comparison with multiple sclerosis patients and healthy controls. Eur J Neurol. 2016;23(4):817–822. doi:10.1111/ene.12948
59. Kortvelyessy P, Krageloh-Mann I, Mawrin C, et al. Hereditary diffuse leukoencephalopathy with spheroids (HDLS) with a novel CSF1R mutation and spinal cord involvement. J Neurol Sci. 2015;358(1–2):515–517. doi:10.1016/j.jns.2015.09.370
60. Di Donato I, Stabile C, Bianchi S, et al. A novel CSF1R mutation in a patient with clinical and neuroradiological features of hereditary diffuse leukoencephalopathy with axonal spheroids. J Alzheimers Dis. 2015;47(2):319–322. doi:10.3233/JAD-150097
61. Prieto-Morin C, Ayrignac X, Ellie E, Tournier-Lasserve E, Labauge P. CSF1R-related leukoencephalopathy mimicking primary progressive multiple sclerosis. J Neurol. 2016;263(9):1864–1865. doi:10.1007/s00415-016-8197-x
62. Traschutz A, Hattingen E, Klockgether T, Paus S. Mirror movements and blepharoclonus as novel phenomena in hereditary diffuse leukoencephalopathy with spheroids. Parkinsonism Relat Disord. 2019;58:83–84.
63. Mascalchi M, Gavazzi C, Morbin M, et al. CT and MR imaging of neuroaxonal leukodystrophy presenting as early-onset frontal dementia. AJNR Am J Neuroradiol. 2006;27:1037–1039.
64. Kondo Y, Kinoshita M, Fukushima K, Yoshida K, Ikeda S. Early involvement of the corpus callosum in a patient with hereditary diffuse leukoencephalopathy with spheroids carrying the de novo K793T mutation of CSF1R. Intern Med. 2013;52(4):503–506. doi:10.2169/internalmedicine.52.8879
65. Liu YQ, Zhang LX, Qiu ZH, Li J, Wang ZX. Report of a case of hereditary diffuse leukoencephalopathy complicated with axonal globular change and literature review. J Stroke Neurol Dis. 2020;37(01):55–57.
66. Yu XP, Cui RR, Sun J, Liu X, Li FJ, Zhou C. Clinical and imaging features of inherited diffuse leukoencephalopathy with axonal globular degeneration (report of 1 case). J Clin Neurol. 2017;30(06):464–467.
67. Onder H, Oguz KK, Soylemezoglu F, Varli K. Deep white matter lesions with persistent diffusion restriction on MRI as a diagnostic clue: neuroimaging of a turkish family with hereditary diffuse leukoencephalopathy with spheroids and literature review. Ann Indian Acad Neurol. 2020;23:280–288.
68. Li S, Zhu Y, Yao M. Spinal cord involvement in adult-onset leukoencephalopathy with axonal spheroids and pigmented glia. JAMA Neurol. 2020;77(9):1169. doi:10.1001/jamaneurol.2020.2204
69. Xuan FL, Chithanathan K, Lillevali K, Yuan X, Tian L. Differences of microglia in the brain and the spinal cord. Front Cell Neurosci. 2019;13:504. doi:10.3389/fncel.2019.00504
70. Lin W-L, Wszolek ZK, Dickson DW. Hereditary diffuse leukoencephalopathy with spheroids: ultrastructural and immunoelectron microscopic studies. Int J Clin Exp Pathol. 2010;3:665–674.
71. Miura T, Mezaki N, Konno T, et al. Identification and functional characterization of novel mutations including frameshift mutation in exon 4 of CSF1R in patients with adult-onset leukoencephalopathy with axonal spheroids and pigmented glia. J Neurol. 2018;265:2415–2424. doi:10.1007/s00415-018-9017-2
72. Fernández-Vega I, Pérez de Heredia-goñi K, Santos-Juanes J, et al. Sporadic adult-onset leucodystrophy with axonal spheroids and pigmented glia with no mutations in the known targeted genes. Histopathology. 2016;68(2):308–312. doi:10.1111/his.12741
73. Tada M, Konno T, Tada M, et al. Characteristic microglial features in patients with hereditary diffuse leukoencephalopathy with spheroids. Ann Neurol. 2016;80:554–565. doi:10.1002/ana.24754
74. Oosterhof N, Kuil LE, van der Linde HC, et al. Colony-Stimulating Factor 1 Receptor (CSF1R) regulates microglia density and distribution, but not microglia differentiation in vivo. Cell Rep. 2018;24(5):1203–1217 e6. doi:10.1016/j.celrep.2018.06.113
75. Kempthorne L, Yoon H, Madore C, et al. Loss of homeostatic microglial phenotype in CSF1R-related Leukoencephalopathy. Acta Neuropathol Commun. 2020;8(1):72. doi:10.1186/s40478-020-00947-0
76. Tamhankar PM, Zhu B, Tamhankar VP, et al. A novel hypomorphic CSF1R gene mutation in the biallelic state leading to fatal childhood neurodegeneration. Neuropediatrics. 2020;51(4):302–306. doi:10.1055/s-0040-1702161
77. Oosterhof N, Chang IJ, Karimiani EG, et al. Homozygous mutations in CSF1R cause a pediatric-onset leukoencephalopathy and can result in congenital absence of microglia. Am J Hum Genet. 2019;104(5):936–947. doi:10.1016/j.ajhg.2019.03.010
78. Gelfand JM, Greenfield AL, Barkovich M, et al. Allogeneic HSCT for adult-onset leukoencephalopathy with spheroids and pigmented glia. Brain. 2020;143(2):503–511. doi:10.1093/brain/awz390
79. Wang M, Zhang X. A novel CSF-1R mutation in a family with hereditary diffuse leukoencephalopathy with axonal spheroids misdiagnosed as hydrocephalus. Neurogenetics. 2019;20(3):155–160. doi:10.1007/s10048-019-00579-0
80. Han J, Sarlus H, Wszolek ZK, et al. Microglial replacement therapy: a potential therapeutic strategy for incurable CSF1R-related leukoencephalopathy. Acta Neuropathol Commun. 2020;8(1):217. doi:10.1186/s40478-020-01093-3
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.