")
Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 15
A Novel Missense Mutation in TWNK Gene Causing Perrault Syndrome Type 5 in a Chinese Family and Review of the Literature
Authors Wei L, Hou L, Ying YQ, Luo XP
Received 8 October 2021
Accepted for publication 3 December 2021
Published 8 January 2022 Volume 2022:15 Pages 1—8
DOI https://doi.org/10.2147/PGPM.S341172
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Lan Wei, Ling Hou, Yan-Qin Ying, Xiao-Ping Luo
Department of Pediatrics, Tongji Hospital, Tongji Medical College, Huazhong University of Science & Technology, Wuhan, Hubei, 430030, People’s Republic of China
Correspondence: Yan-Qin Ying
Department of Pediatrics, Tongji Hospital, Tongji Medical College, Huazhong University of Science & Technology, Jiefang Avenue 1095#, Wuhan, Hubei, 430030, People’s Republic of China
Tel/Fax +86 27 83662684
Email [email protected]
Background: Perrault syndrome (PRLTS) is a rare autosomal recessive disorder characterized by sensorineural hearing loss in both sexes and ovarian dysfunction in females. In some cases, patients present with a diversity of neurological signs. Six genes are known to cause Perrault syndrome.
Case Report: We report an 11-year-old Chinese girl with delayed gonadal development, sensorineural hearing loss, and neurologic manifestations. Genetic etiology was identified by whole-exome sequencing and confirmed via Sanger sequencing. Compound heterozygous variants with one novel variant c.1752C>A (p.D584E) and one known pathogenic variant c.1172G>A (p.R391H) in TWNK were discovered in the child and inherited from her parents, respectively.
Conclusion: The compound heterozygous variants c.1172G>A (p.R391H) and c.1752C>A (p.D584E) of the TWNK gene probably underlie PRLTS type 5 (PRLTS5). This study expands the mutation spectrum of TWNK pathogenicity in the PRLTS5 phenotype.
Keywords: Perrault syndrome, TWNK gene, Chinese, whole-exome sequencing
Background
Perrault syndrome (PRLTS) is a rare autosomal recessive disorder characterized by sensorineural hearing loss (SNHL) in both males and females and ovarian dysfunction in females with normal karyotypes.1 SNHL is bilateral, sometimes progressive, and ranges from profound with prelingual (congenital) onset to early childhood onset.2,3 Ovarian dysfunction in females ranges from absent or streak gonads manifesting as primary amenorrhea to primary ovarian insufficiency (POI) with raised gonadotropins (LH & FSH) and reduced estrogen.2,4 Affected males often show normal pubertal development.5 Some patients also have a diversity of neurological manifestations, including cerebellar ataxia, nystagmus, ophthalmoplegia, mild intellectual disability, muscle weakness, and peripheral neuropathy.3,5,6 According to the clinical features, Perrault syndrome has been classified as type I, which is static and without neurological disease, and type II, which is with progressive neurological disease.7
Perrault syndrome is also genetically heterogeneous. So far, six subtypes of Perrault syndrome (PRLTS 1~6) have been identified, corresponding to six pathogenic genes: HSD17B4 (MIM 601860), HARS2 (MIM 600783), CLPP (MIM 601119), LARS2 (MIM 604544), TWNK (MIM 606075), and ERAL1 (MIM 607435).2,7–11 However, the identified Perrault syndrome genes collectively explain only a fraction of cases, suggesting that additional disease-causing genes remain to be discovered. In addition, the number of reported cases and the spectrum of mutations are too small to establish conclusive genotype-phenotype correlations.
Here, we report a Chinese family with PRLTS5 carrying compound heterozygous variants in the TWNK gene and review the literature about PRLTS5 of the clinical and molecular investigations.
Case Report
Clinical Case
An 11-year-old girl was admitted to our hospital with delayed secondary sexual characteristics. She was the first baby of non-consanguineous parents and was a full-term birth. The birth weight was 2950 g, and the length was 51 cm. She started walking alone at 19 months. She was admitted to her doctor for unstable walking with an ataxic gait at four years. Abnormal hearing and nystagmus were first noted at the age of three. Bilateral severe sensorineural deafness was diagnosed at four years, and unilateral cochlear implantation was performed. Speech development was age-appropriate. The level of intelligence was normal. No epilepsy or unconsciousness seizures occurred.
Her height was 147.5 cm (+0.14 SD) and weight 38 kg (+0.29 SD). She had a highly arched palate and horizontal nystagmus. Pubic hair and bilateral breast development were all at stage 1, according to the Tanner staging. She had no scoliosis but had cubitus valgus. Her muscle strength was normal, and she had no muscle atrophy. Deep tendon reflexes were absent. The left Babinski reflex was suspicious positive, and the Romberg sign was positive.
Laboratory data were normal, including liver and renal function, serum electrolytes, blood glucose and lipids, myocardial enzyme profile, adrenal function, thyroid function, lactate, and pyruvate. Lactate was 1.41 mmol/L (normal range: 0.5–2.2 mmol/L) and pyruvate was 48.9 μmol/L (normal range: 20–100 μmol/L), Unfortunately we did not perform the aerobic exercise test to evaluate the levels of lactate and pyruvate during exercise. Sex hormone studies showed a low level of serum estradiol (E2) (<20 pg/mL) and raised levels of gonadotropins (LH 26.77 mIU/mL and FSH 78.27 mIU/mL). Serum anti-mullerian hormone (AMH) concentrations were 0.01 ng/mL, and Inhibin B was 11.5 pg/mL, indicating diminished ovarian reserves and poor ovarian response. Gonadotropic hormone stimulation tests showed high levels with LH peak 142.31 mIU/mL and FSH peak 141.78 mIU/mL. Bone age was ten years. The karyotype was normal (46, XX). Pelvic ultrasonography revealed the uterus was smaller than other children of the same age (1.7 × 0.5×1.0 cm), and there were no pubertal changes in the ovary (about 1.6×0.8 cm on the left side and 1.7×0.9 cm on the right side). Several small anechoic areas could be seen. The larger one, which was a follicle, had a diameter of about 0.2 cm. The electroencephalogram showed sharp waves and spike waves released in the left frontal and central areas during sleep.
Whole-Exome Sequencing
Genomic DNA was purified from peripheral blood using standard procedures. A library was prepared using a GenCap Whole-exome capture kit (MyGenostics GenCap Enrichment Technologies). Sequencing was performed on Illumina HiSeq X ten sequencer. After sequencing, the raw data were saved as a FASTQ format, then followed by the bioinformatics analysis. First, Illumina sequencing adapters and low-quality reads (<80 bp) were filtered by Cutadapt. After quality control, the clean reads were mapped to the UCSC hg19 human reference genome using Burrows–Wheeler Aligner (BWA). The average sequencing depth was more than 100×, and the targeted region was more than 20× for >90% and 10× for >95%. Duplicated reads were removed using Picard tools, and mapping reads were used for variation detection. Second, the variants of SNP and InDel were detected by Genome Analysis Toolkit (GATK) Haplotype Caller, then GATK Variant Filtration was used to filter variants. After these steps, the data were transformed to a VCF format. Variants were further annotated by ANNOVAR and associated with multiple databases, such as gnomAD, 1000 genomes, dbSNP, and predicted by SIFT, PolyPhen-2, MutationTaster, GERP++, phyloP, and phastCons. The American College of Medical Genetics and Genomics (ACMG) guidelines were employed for sequence variation interpretation.
Literature Searching
The literature was searched up to April 2021 from PubMed, Human Gene Mutation Database (HGMD), and some Chinese databases, such as CNKI, Wanfang, and China Science and Technology Journal Database with “Perrault syndrome”, “TWNK”, and the corresponding terms in Chinese. Clinical features and gene mutations were summarized.
Results
Genetic Analysis
By whole-exome sequencing (WES) and Sanger sequencing, we identified two heterozygous variants on the TWNK gene, corresponding to c.1172G>A (p.R391H) (NM_021830 GRch37/hg19; exon 1) on chr10:102749139, inherited from her father, and c.1752C>A (p.D584E) (NM_021830 GRch37/hg19; exon 5) on chr10:102752964, inherited from her mother (Figure 1). c.1172G>A (p.R391H) was a known missense variant,2 while c.1752C>A (p.D584E) was a novel missense variant (Figure 2). According to the gnomAD database, the allele frequency of the c.1172G>A (p.R391H) was 0.000064. It was considered as likely pathogenic according to the ACMG guidelines (PM1, PM2, PM3, PP3). The variant c.1752C>A (p.D584E) was absent in gnomAD, it was considered as uncertain according to the ACMG guidelines (PM2, PP3), which was predicted to be damaging by SIFT, probably damaging by PolyPhen-2, and disease-causing by MutationTaster. Conservation analysis indicated that c.1752C>A (p.D584E) was highly conserved across mammals (the larger the score, the more conserved the site) with a GERP++ score of 5.7 (scores ranged from −12.3 to 6.17), a phyloP conservation score of 0.935 (scores ranged from −13.282 to 1.199), and a phastCons conservation score of 0.997 (scores ranged from 0 to 1). The variant c.1752C>A (p.D584E), located in the helicase domain of the Twinkle protein, was expected to impair helicase activity and result in the stalling of mtDNA replication.
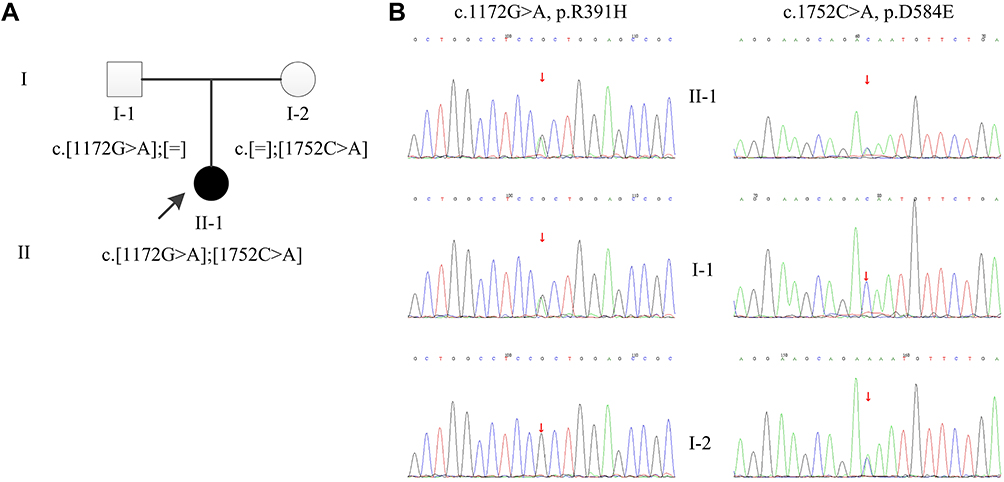 |
Figure 1 Pedigree and sequence analysis of the Chinese family. (A) Pedigree of the Chinese family. The filled circle indicates affected individual, and the open circle and box indicate non-affected individuals. The proband is indicated by an arrow. (B) Sanger sequencing confirmation of the Chinese family. The proband (II-1) carried compound heterozygous mutations: c.1172G>A inherited from his father (I-1) and c.1752C>A from his mother (I-2). Square: Male; Circle: Female. |
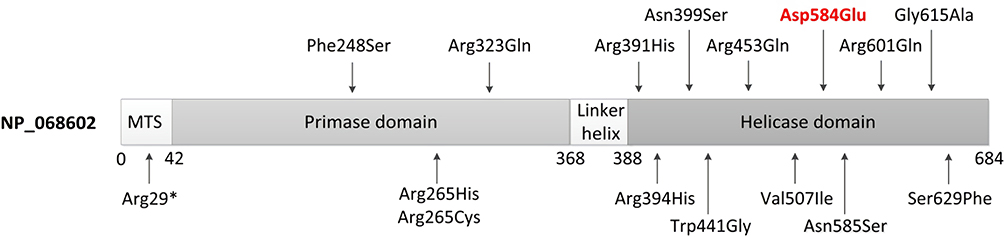 |
Figure 2 Scheme of Twinkle structure (NP_068602) and distribution of pathogenic variants associated with Perrault syndrome. The N-terminal contains the mitochondrial targeting sequence (MTS); then, there is an N-terminal primase domain, a C-terminal helicase domain, and a linker region connecting the two domains. The novel missense variant identified in the proband in this study is in red. |
Clinical Features and TWNK Gene Variants
Ten literature reports about PRLTS5 were retrieved, and 20 affected patients, including our patient (female: male = 18:2), from 12 different families have been reported so far.
The clinical features of patients with TWNK mutations of PRLTS5 were summarized in Table 1 (for more detailed information, see Table S1). The diagnosed age was 6–58 years. Karyotypes were 46XX in all tested female patients. All 20 patients (100%) had sensorineural hearing impairment (SNHL). The onset age was less than three years to ten years, except for one Japanese patient with hearing loss at age 48.12 Ovarian dysfunction developed in 85% of patients (17/20), 70% of patients (14/20) had primary amenorrhea, while 10% of patients (2/20) suffered from primary ovarian insufficiency (POI). These patients ceased menses at age 20 and 32, respectively. One male patient aged 29 had dysfunction of the gonadal axis.4 After ovarian dysfunction, thyroid dysfunction was the next most common endocrine abnormality (4/20). All 20 patients (100%) with TWNK mutations showed a significant degree of neurological involvement. There was peripheral neuropathy in 90% of patients (18/20). These patients had sensory or sensory-motor neuropathy with axonal or demyelinating or mixed lesions (predominantly axonal). Ataxia was found in 100% of patients (20/20). Abnormal neuroimaging, like mild to severe cerebellar atrophy and signal changes in the cerebellar white matter, were found in 65% of patients (13/20). Nystagmus was found in 70% of patients (14/20), and 30% of patients (6/20) had ophthalmoplegia. In the musculoskeletal system, muscle weakness, muscle atrophy, motor dysfunction, and different skeletal deformities (pes cavus, high-arched palate, scoliosis, clawed toes, clubbed-finger, hammertoes, cubitus valgus, etc.) were observed. Mental retardation was not common or just slightly below average (3/20).5,13 One patient had tonic-clonic seizures at seven years.2 One patient had increased deep tendon reflexes and bilateral pyramidal signs on both upper and lower limbs, which may have been due to the damage of upper motor neurons. This patient also had moderate depression, anxiety, querulous behavior, and paranoid personality traits,14 which was rarely seen in previous reports.
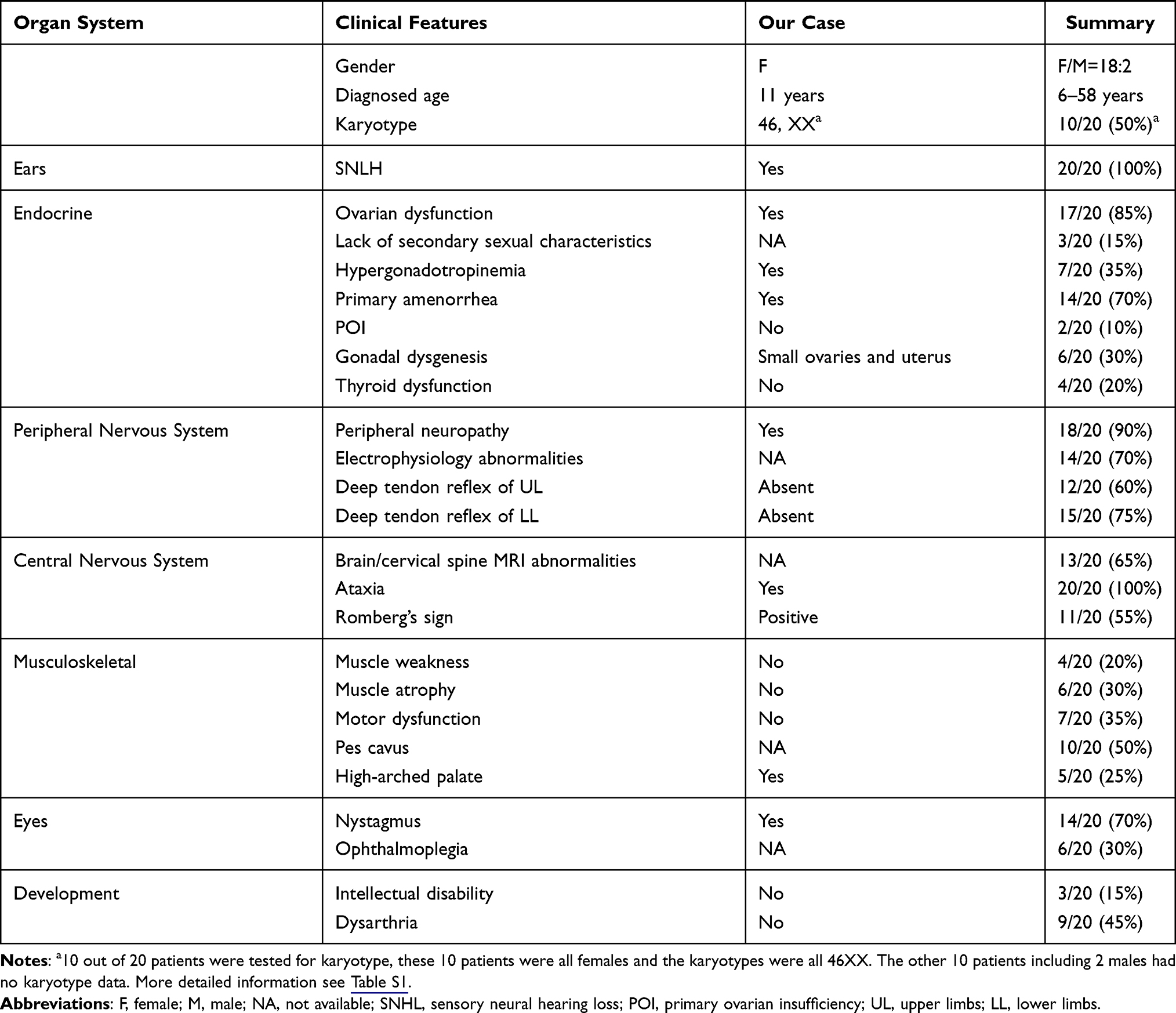 |
Table 1 Common Clinical Features of Patients with TWNK Mutations in PRLTS5 |
The TWNK variants in PRLTS5 are listed in Table 2. Two families (four patients) had homozygous mutations with variants c.793C>T and c.1358G>A. The other ten families (16 patients) had compound heterozygous mutations. A total of 40 biallelic mutations were identified with 16 different pathogenic variants (Figure 2). The most common pathogenic variant was c.1172G>A (5/40). The patients with c.1172G>A mutations were all from Asian countries (Japan and China), The second most common pathogenic variant was c.1196A>G (4/40), with all patients from Europe (Poland, Hungary, and Norway, respectively). This indicates that c.1172G>A and c.1196A>G may be the hot spots of PRLTS5 in Asian and European patients, respectively.
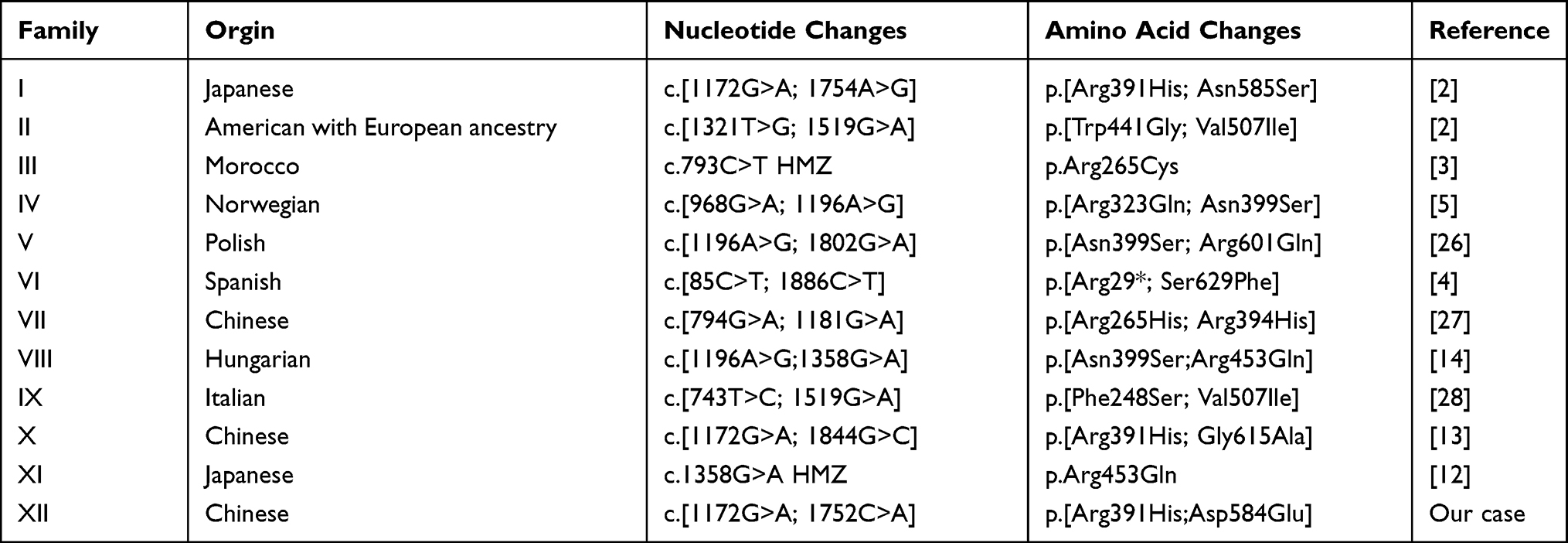 |
Table 2 Reported TWNK Mutations in PRLTS5 |
Discussion
Perrault syndrome is a very rare disease (<1/1,000,000), first described by Perrault in 1951.15 Morino first described compound heterozygous mutations in the TWNK gene (also known as C10orf2) in two families with Perrault syndrome in 2014.2
Although male patients have been reported in some literature, Perrault syndrome is more common in female patients. Women are more likely to have delayed sexual development. The onset age of SNHL is pre-lingual or post-lingual and appears in early childhood. Cochlear implants can be used to improve hearing. Ovarian dysfunction is one of the main characteristics of Perrault syndrome in female patients, and it is difficult to diagnose the disease before puberty. After puberty, the affected women may show a lack of secondary sexual characteristics, gonadal dysplasia, primary amenorrhea, POI, high gonadotropin, and low estrogen. Most women lack fertility. Women with POI have menarche, but their menstruation is terminated early. These patients retain fertility before menstruation stops. Patients with primary amenorrhea in adolescence can use hormone replacement therapy to induce puberty. Oocyte cryopreservation should be considered in patients at risk of POI. Patients with gonadal dysgenesis can only choose assisted reproduction through in vitro fertilization using donor eggs. Other endocrine abnormalities include Hashimoto’s thyroiditis or autoimmune hypothyroidism. All patients have abnormalities in the nervous system. Peripheral neuropathy is most common with sensory or sensorimotor abnormalities. Axonal abnormalities are more common than demyelination, suggesting a role of this gene in peripheral neurological pathways, especially damage to axons. Tendon reflexes are usually weakened or absent. Some patients have complications with central nervous system structural abnormalities, manifested as mild to severe cerebellar atrophy, cerebellar white matter signal changes, cervical spinal cord atrophy, medulla oblongata atrophy, and ventricular enlargement. We believe that the main criteria for clinical diagnosis of PRLTS5 include 1. TWNK gene mutation; 2. male or female SNHL; 3. female ovarian dysfunction with normal karyotype; 4. peripheral neuropathy; 5. ataxia. Other auxiliary diagnostic features include 1. nystagmus/ophthalmoplegia; 2. cerebellar atrophy; 3. high foot arch/high palatal arch; 4. dysarthria; 5. muscular atrophy/muscle weakness; 6. thyroiditis; 7. normal or mild mental impairment. We should note that different clinical manifestations have different onset times. SNHL often appears in early childhood, ovarian dysfunction is often discovered in adolescence or after puberty, and neurological signs can appear in early childhood to adulthood.
TWNK encodes the Twinkle (NP_068602) protein, which comprises 684 amino acids and forms hexamer complexes. Twinkle is the mitochondrial DNA replicative helicase and localizes to mitochondrial nucleosomes. Twinkle contains three functional domains (Figure 2): a 3-prime helicase region required for mtDNA replication, a linker region involved in hexamer formation, and a 5-primase domain.16 Twinkle may serve a dual role in mitochondria as both the replicative DNA helicase and a key regulator of mtDNA maintenance.17–19 The Twinkle helicase is the main helicase in mitochondria and is the sole replication helicase required for mtDNA maintenance. Most pathogenic variants (11/16, 68.8%) of the TWNK gene associated with Perrault syndrome are located in the helicase domain of Twinkle. These mutant variants show impaired helicase activity, which is thought to stall mtDNA replication and result in the slow accumulation of large mtDNA deletions. Mutations in the N-terminal domain demonstrated a dramatic decrease in ATPase activity and impaired ssDNA binding.20 Mutations in the linker region were shown to disrupt oligomerization and abolish DNA helicase activity.21,22
TWNK mutations result in three different phenotypes: dominant progressive external ophthalmoplegia type 3 (PEOA3; MIM 609286), recessive Perrault syndrome type 5 (PRLTS5; MIM 606075), and mitochondrial DNA depletion syndrome 7 (MTDPS7; MIM 271245). There is a substantial phenotypic overlap across the conditions resulting from TWNK mutations, particularly in regard to the neurological features. Hearing loss, ataxia, myopathy, neuropathy, and ophthalmoplegia have been reported in patients with each of these diseases. PEOA3 is characterized by ptosis and external ophthalmoplegia, exercise intolerance, peripheral neuropathy, deafness, and ataxia.23 MTDPS7 begins very early in life, in children under three years. The course of the disease is severe and includes infantile-onset spinocerebellar ataxia (IOSCA) and hepatocerebral syndrome. IOSCA is characterized by infantile-onset ataxia, athetosis, reduced tendon reflexes, ophthalmoplegia, sensorineural hearing loss, and sensory neuropathy.24 Hepatocerebral syndrome is characterized by neonatal or early infantile-onset hepatopathy, associated with renal tubulopathy and progressive neurodegenerative manifestations.25
In conclusion, we report a patient with early onset of sensorineural deafness, ovarian dysfunction with gonadal dysgenesis and hypergonadotropinemia, nystagmus, peripheral neuropathy, and ataxia. Unfortunately, we had not assessed the imaging and neuroelectrophysiology of the nervous system. By WES and Sanger sequencing, we identified one novel and one previously described missense mutation in TWNK, expanding the mutation spectrum of TWNK pathogenicity in PRLTS5. We find the hot spots c.1172G>A and c.1196A>G of PRLTS5 in Asian and European patients, respectively. The diagnosis of Perrault syndrome is further complicated by the variable expressivity of its clinical features, which are related to the age and sex of the patient and the diversity of genes involved. This study summarizes and reinforces that the neurological features of patients of PRLTS5—peripheral neuropathy and ataxia— are the two most common neurological disorders. When combined with SNHL in both sexes and ovarian dysfunction in females, TWNK gene testing should be considered. Because of a large number of overlapping phenotypes and differences in onset age and severity, we may refer to TWNK-related spectrum disease to cover PEOA3, PRLTS5, and MTDPS7. With more patients diagnosed in the future, we can get more information to study the correlation between genotype and phenotype.
Ethics Approval and Consent to Participate
This study was conducted with approval from the Ethics Committee of Tongji Hospital, Tongji Medical College, Huazhong University of Science & Technology (TJ-IRB20210871). This study was conducted in accordance with the declaration of Helsinki. Written informed consent for publication of the case details has been provided by the legal guardians of the patient.
Funding
National Key R&D Program of China (No. 2018YFC1002400). Special Science and Technology Major Project of Hubei Province (ZDZX2020000020).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Newman WG, Friedman TB, Conway GS, Demain LAM. Perrault syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews((R)). Seattle (WA); 1993.
2. Morino H, Pierce SB, Matsuda Y, et al. Mutations in Twinkle primase-helicase cause Perrault syndrome with neurologic features. Neurology. 2014;83(22):2054–2061. doi:10.1212/WNL.0000000000001036
3. Lerat J, Jonard L, Loundon N, et al. An application of NGS for molecular investigations in Perrault syndrome: study of 14 families and review of the literature. Hum Mutat. 2016;37(12):1354–1362. doi:10.1002/humu.23120
4. Dominguez-Ruiz M, Garcia-Martinez A, Corral-Juan M, et al. Perrault syndrome with neurological features in a compound heterozygote for two TWNK mutations: overlap of TWNK-related recessive disorders. J Transl Med. 2019;17(1):290. doi:10.1186/s12967-019-2041-x
5. Demain LA, Urquhart JE, O’Sullivan J, et al. Expanding the genotypic spectrum of Perrault syndrome. Clin Genet. 2017;91(2):302–312. doi:10.1111/cge.12776
6. Jenkinson EM, Clayton-Smith J, Mehta S, et al. Perrault syndrome: further evidence for genetic heterogeneity. J Neurol. 2012;259(5):974–976. doi:10.1007/s00415-011-6285-5
7. Pierce SB, Walsh T, Chisholm KM, et al. Mutations in the DBP-deficiency protein HSD17B4 cause ovarian dysgenesis, hearing loss, and ataxia of Perrault syndrome. Am J Hum Genet. 2010;87(2):282–288. doi:10.1016/j.ajhg.2010.07.007
8. Pierce SB, Chisholm KM, Lynch ED, et al. Mutations in mitochondrial histidyl tRNA synthetase HARS2 cause ovarian dysgenesis and sensorineural hearing loss of Perrault syndrome. Proc Natl Acad Sci U S A. 2011;108(16):6543–6548. doi:10.1073/pnas.1103471108
9. Jenkinson EM, Rehman AU, Walsh T, et al. Perrault syndrome is caused by recessive mutations in CLPP, encoding a mitochondrial ATP-dependent chambered protease. Am J Hum Genet. 2013;92(4):605–613. doi:10.1016/j.ajhg.2013.02.013
10. Chatzispyrou IA, Alders M, Guerrero-Castillo S, et al. A homozygous missense mutation in ERAL1, encoding a mitochondrial rRNA chaperone, causes Perrault syndrome. Hum Mol Genet. 2017;26(13):2541–2550. doi:10.1093/hmg/ddx152
11. Pierce SB, Gersak K, Michaelson-Cohen R, et al. Mutations in LARS2, encoding mitochondrial leucyl-tRNA synthetase, lead to premature ovarian failure and hearing loss in Perrault syndrome. Am J Hum Genet. 2013;92(4):614–620. doi:10.1016/j.ajhg.2013.03.007
12. Kume K, Morino H, Miyamoto R, et al. Middle-age-onset cerebellar ataxia caused by a homozygous TWNK variant: a case report. BMC Med Genet. 2020;21(1):68. doi:10.1186/s12881-020-01002-4
13. Chen Z, Tang S, Li H, Xu X, Lyu J. [Analysis of TWNK variant in a family affected with Perrault syndrome]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2020;37(7):739–742. Chinese. doi:10.3760/cma.j.issn.1003-9406.2020.07.009
14. Fekete B, Pentelenyi K, Rudas G, et al. Broadening the phenotype of the TWNK gene associated Perrault syndrome. BMC Med Genet. 2019;20(1):198. doi:10.1186/s12881-019-0934-4
15. Perrault M, Klotz B, Housset E. [Two cases of Turner syndrome with deaf-mutism in two sisters]. Bull Mem Soc Med Hop Paris. 1951;67(3–4):79–84. French.
16. Shutt TE, Gray MW. Twinkle, the mitochondrial replicative DNA helicase, is widespread in the eukaryotic radiation and may also be the mitochondrial DNA primase in most eukaryotes. J Mol Evol. 2006;62(5):588–599. doi:10.1007/s00239-005-0162-8
17. Tyynismaa H, Sembongi H, Bokori-Brown M, et al. Twinkle helicase is essential for mtDNA maintenance and regulates mtDNA copy number. Hum Mol Genet. 2004;13(24):3219–3227. doi:10.1093/hmg/ddh342
18. Korhonen JA, Gaspari M, Falkenberg M. TWINKLE Has 5ʹ -> 3ʹ DNA helicase activity and is specifically stimulated by mitochondrial single-stranded DNA-binding protein. J Biol Chem. 2003;278(49):48627–48632. doi:10.1074/jbc.M306981200
19. Milenkovic D, Matic S, Kuhl I, et al. TWINKLE is an essential mitochondrial helicase required for synthesis of nascent D-loop strands and complete mtDNA replication. Hum Mol Genet. 2013;22(10):1983–1993. doi:10.1093/hmg/ddt051
20. Holmlund T, Farge G, Pande V, Korhonen J, Nilsson L, Falkenberg M. Structure-function defects of the twinkle amino-terminal region in progressive external ophthalmoplegia. Biochim Biophys Acta. 2009;1792(2):132–139. doi:10.1016/j.bbadis.2008.11.009
21. Lee SJ, Richardson CC. The linker region between the helicase and primase domains of the gene 4 protein of bacteriophage T7. Role in helicase conformation and activity. J Biol Chem. 2004;279(22):23384–23393. doi:10.1074/jbc.M400857200
22. Peter B, Farge G, Pardo-Hernandez C, Tångefjord S, Falkenberg M. Structural basis for adPEO-causing mutations in the mitochondrial TWINKLE helicase. Hum Mol Genet. 2019;28(7):1090–1099. doi:10.1093/hmg/ddy415
23. Tafakhori A, Yu Jin Ng A, Tohari S, et al. Mutation in TWINKLE in a large Iranian family with progressive external ophthalmoplegia, myopathy, dysphagia and dysphonia, and behavior change. Arch Iran Med. 2016;19(2):87–91.
24. Lönnqvist T. Infantile-onset spinocerebellar ataxia. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews(®). Seattle (WA): University of Washington, Seattle Copyright © 1993–2021, University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved; 1993.
25. Sukhudyan B, Gevorgyan A, Sarkissian A, Boltshauser E. Expanding phenotype of mitochondrial depletion syndrome in association with TWNK mutations. Eur J Paediatr Neurol. 2019;23(3):537–540. doi:10.1016/j.ejpn.2019.02.002
26. Oldak M, Ozieblo D, Pollak A, et al. Novel neuro-audiological findings and further evidence for TWNK involvement in Perrault syndrome. J Transl Med. 2017;15(1):25. doi:10.1186/s12967-017-1129-4
27. Duan X, Wang W, Dong M, et al. [Clinical and genetic analysis of a patient with Perrault syndrome and additional neurological features]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2019;36(6):577–580. Chinese. doi:10.3760/cma.j.issn.1003-9406.2019.06.011
28. Gotta F, Lamp M, Geroldi A, et al. A novel mutation of Twinkle in Perrault syndrome: a not rare diagnosis? Ann Hum Genet. 2020;84(5):417–422. doi:10.1111/ahg.12384
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.