")
Back to Journals » The Application of Clinical Genetics » Volume 14
A Novel Intronic KMT2D Variant as a Cause of Kabuki Syndrome: A Case Report
Authors Aristizábal E, Diaz-Ordóñez L , Candelo E , Pachajoa H
Received 27 April 2021
Accepted for publication 22 August 2021
Published 5 October 2021 Volume 2021:14 Pages 409—416
DOI https://doi.org/10.2147/TACG.S317723
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Martin Maurer
Erica Aristizábal,1 Lorena Diaz-Ordóñez,1 Estephania Candelo,1,2 Harry Pachajoa1,2
1Center for Research on Congenital Anomalies and Rare Diseases (CIACER), Department of Basic Medical Sciences, Universidad Icesi, Cali, Valle del Cauca, Colombia; 2Fundación Valle del Lili, Cali, Valle del Cauca, Colombia
Correspondence: Lorena Diaz-Ordóñez
Center for Research on Congenital Anomalies and Rare Diseases (CIACER), Department of Basic Medical Sciences, L Building, Zip: 760031, Universidad Icesi, Cali, Colombia
Tel +57 2 5552334 Ext: 8542
Email [email protected]
Abstract: Kabuki syndrome (KS) is an autosomal dominant genetic disorder in which most cases are caused by de novo mutations. KS type 1 is caused by mutations in KMT2D (OMIM: #147920) and is more common. KS type 2 is caused by mutations in KDM6A (OMIM: #300867). Both genes encode proteins that modify histones and are involved in epigenetic regulation. The enzyme histone-lysine N-methyltransferase 2D, the product of KMT2D, is expressed in most adult tissues and is essential for early embryonic development. The main clinical manifestations of KS include dysmorphic facial features, such as elongated palpebral fissures, eversion of the lateral third of the lower eyelids, and short nasal columella with a broad and depressed nasal tip. Additionally, patients also present with skeletal abnormalities, dermatoglyphic features, mild-to-moderate intellectual disability, hearing loss, and postnatal growth deficiency. We describe an 11-year-old girl from Colombia, who presented with characteristic clinical signs of KS. Genetic studies showed a KMT2D intronic variant (KMT2D NM_003482.3: c.511‐2A> T) as a cause of KS.
Keywords: Kabuki syndrome, coloboma, rare disease, RNA splicing, sensorineural hearing loss
Introduction
Kabuki syndrome (KS) is a rare disease of genetic origin, first described in Japan by Niikawa & Kuroki in 1981. Its name is owed to the similarity of the facial dysmorphism characteristic of this condition with the makeup and kabuki masks used in traditional Japanese theater.1,2 KS prevalence is 1:32,000 live births in Japan and possibly similar in the rest of the world. In Colombia, the official number of diagnosed cases is unknown. However, 8 cases have been reported. KS does not have a predominance of ethnicity, age, or sex.2,3
This syndrome is characterized by dysmorphic facial features, such as elongated palpebral fissures with eversion of the lateral third of the lower eyelids, arched eyebrows, and short nasal columella with a wide and depressed nasal tip, skeletal conditions, such as cleft palate, spinal column abnormalities, brachydactyly, brachymesophalangia, clinodactyly, and craniofacial anomalies, dermatoglyphic anomalies, mild-to-moderate intellectual disability, and postnatal growth deficiency.2,3 In some cases, urogenital and cardiac abnormalities, visual deficiencies, conductive, sensorineural or mixed hearing loss due to recurrent otitis media in infancy, and endocrine disorders including precocious puberty, diabetes insipidus, thyroid dysfunction, and obesity may occur.3 Several cases with the characteristics described above have been reported in the literature (Table 1).
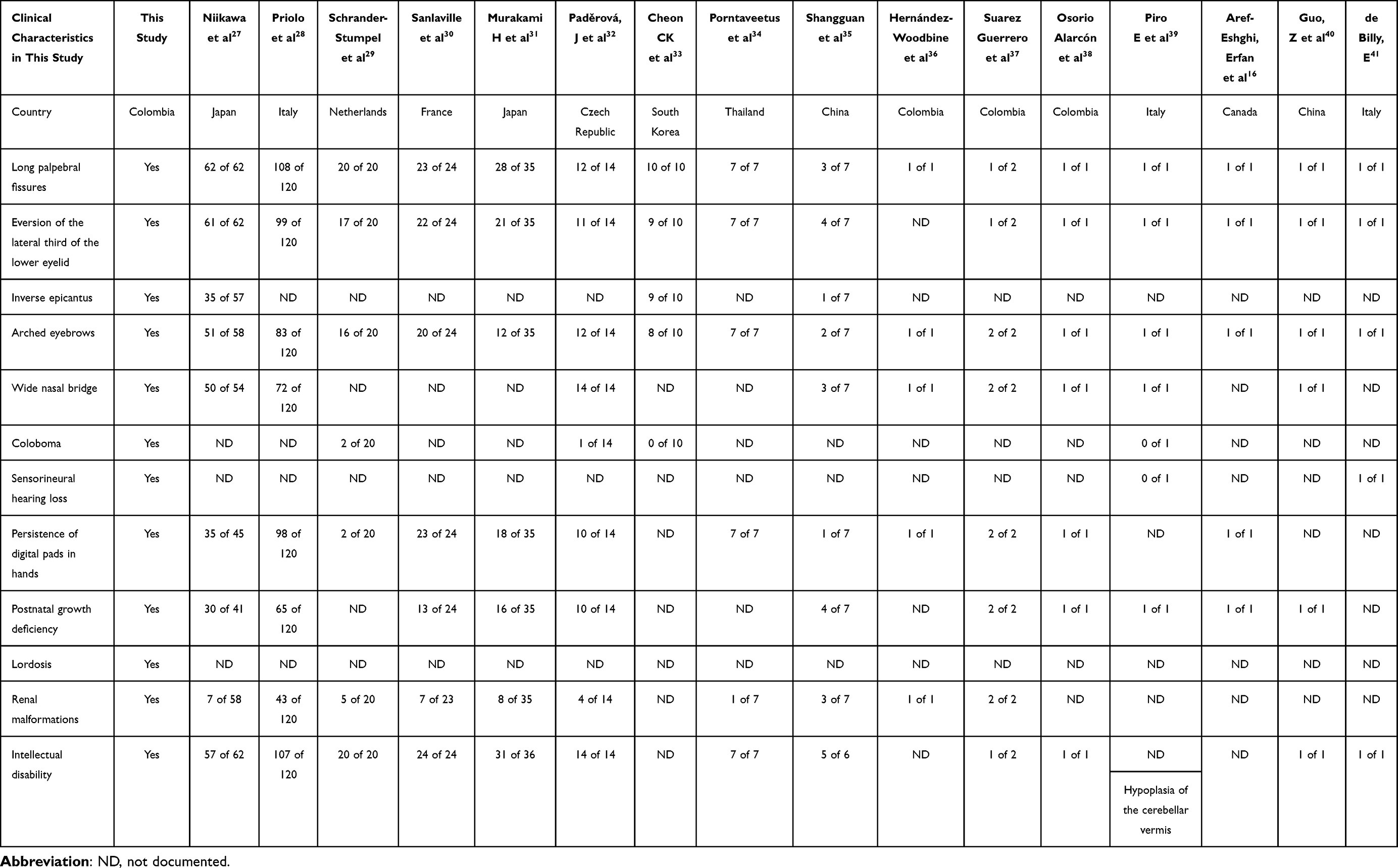 |
Table 1 Clinical Characteristics Found in Our Patient and Literature |
The genetic mutations associated with KS are mainly found in the KMT2D and KDM6A genes.4,5 KMT2D (also called MLL2 or MLL4) is located on the long arm of chromosome 12 (12q13.12). It encodes the histone-lysine methyltransferase 2D protein responsible for di- and tri-methylation of H3K4 in the enhancers and promoters necessary for transcriptional activation.6,7 This enzyme contains 5262 amino acid residues, 1 catalytic SET domain, 5 PHD domains, 2 ePHD domains, 1 HMG-I binding motif, and FY-rich motifs.8,9 Alterations in KMT2D are the leading cause of KS type 1.7 KDM6A (formerly known as UTX) is associated with KS type 2 and encodes a demethylase that eliminates the trimethylation of histone 3 lysine 27 and interacts with KMT2D in the regulation of gene expression.10,11 KS has variable expression, is inherited in an autosomal dominant fashion, and is associated with de novo mutations in most cases.1,12
More than 650 genetic variants associated with KS have been identified. These include nonsense and missense mutations, indels, duplications, frameshifts and splice site variants. Most lead to truncated protein synthesis.12–14 Several cases of splicing variants linked with this syndrome have been reported (Table 2), and here we describe the case of a patient with clinical characteristics compatible with KS and an intronic variant in KMT2D that explains her phenotype.
 |
Table 2 Splice-Site Variants |
Case Presentation
An 11-year-old Colombian girl was delivered at 38 weeks of pregnancy without complication. Her mother and father were non-consanguineous and aged 32 years and 44 years, respectively, at the time of birth. Birth weight was 2625 g (3rd−10th percentile), and height was 49 cm (50th and 75th percentile). No facial or phenotypic abnormalities were noticed at birth. However, at the age of 16 months and at 3 years, she underwent a pupilloplasty due to the reparation of the coloboma. Her family history was unremarkable.
At age 8 years, she underwent surgical correction of her right eyelid. One year later, ophthalmologic examination showed microphthalmia in the right eye with an irregular pupil, inferonasal coloboma, iridectomized iris, and soft-tissue protrusion at the superolateral pouch with dermoid fat herniation. The left eye had an irregular pupil with iris atrophy. Audiometric examination showed mild-to-moderate sensorineural hearing loss in the right ear (PTA 44dB); and normal hearing in the left (PTA 10dB). Estimated radiographic bone age was 6 years. Cardiac echocardiography showed normal ejection fraction (76%). Renal ultrasonography showed ectasia of the left renal sinus. Renal scintigraphy revealed bilateral postinfectious scarring with preserved parenchymal function, a dilated left renal sinus, renal sinus ectasia with interruption, and a probable duplex calyx system; renal function was 56% in the right kidney and 44% in the left. Spinal radiography showed a lumbar curvature to the right with slightly increased lumbar lordosis.
At age 11 years, the Intelligence Quotient (IQ) assessment Wechsler Intelligence Scale for Children IV (WISC-IV) was performed with a total intelligence quotient testing score of 67; verbal comprehension score was 81 (average), perceptual reasoning score was 75 (low), working memory score was 68 (deficient), and information processing speed score was 62 (deficient). Height was 133 cm (4th percentile), weight was 40 kg (66th percentile), and head circumference was 52 cm (22nd percentile). Physical examination showed bilateral iris coloboma, right microphthalmia, inverse epicanthus, elongated palpebral fissures that inclined upwards, eversion of the lateral portion of the lower eyelid, anteverted nostrils, short columella, long philtrum, and normal ear position (Figure 1A and B). Acanthosis nigricans was observed in the neck and armpits, and she had overdeveloped breast tissue. Cardiopulmonary auscultation was normal. Her abdomen had an enlarged panniculus adiposus and was soft and depressible without mass or organomegaly. Extremity examination showed persistent digital pads and a Beighton score of 4/6. Neurologically, she was alert and cooperative.
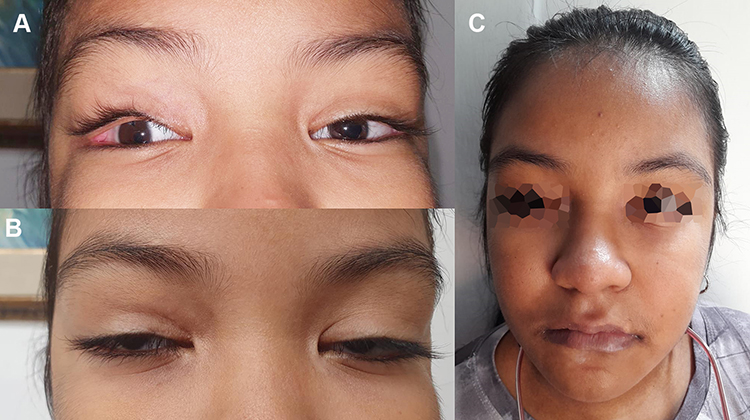 |
Figure 1 Facial characteristics. (A) Image of the patient where microphthalmia and eversion of the lateral third of the eyelid are observed. (B) Image of the patient with arched eyebrows, long palpebral fissures and wide nasal bridge, facial characteristics of kabuki syndrome. (C) Patient’s whole face image shows the dysmorphic characteristics previously described. |
Massive Parallel Sequencing of the Multigen Panel for KS was performed to evaluate KMT2D, KDM6A, RAP1A, and RAP1B. The mean vertical coverage was 325.14X, and the horizontal coverage was 99.46% (coding and splicing regions). This genetic panel detected an intronic KMT2D variant (KMT2D NM_003482.3:c.511–2A>T) in a heterozygous state. This variant was located in the fourth intron’s acceptor site, which would produce an alteration in the splice between the fourth and fifth exon. Sanger sequencing using an ABI 3500 sequencer (Applied Biosystems, Thermo Fisher Scientific, Waltham, MA, USA) was then performed and confirmed the results. This variant has not been annotated in databases, such as Clinvar, ExAC, 1000 genomes and HGMD. Furthermore, it presents a DANN score of 0.9917 and it is classified as a probably pathogenic variant according to the recommendations of the American College of Medical Genetics and Genomics (ACMG) due to experimental data being required to confirm the pathogenicity of the variant. Besides, a segregation study was performed and showed a negative result in the parents, demonstrating that the patient’s variant was de novo (Figure 2).
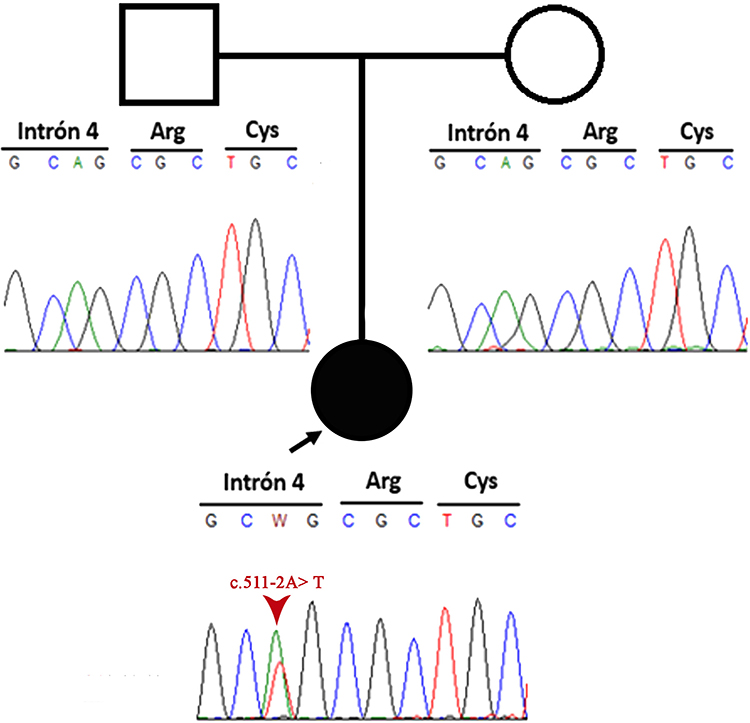 |
Figure 2 Pedigree information of the patient and Sanger sequencing electropherogram of both patient and her parents. The image shows a de novo mutation. |
Discussion
KS is diagnosed clinically by the presence of facial, skeletal, and dermatoglyphic abnormalities, growth deficiency, and mild-to-moderate intellectual disability. The central genes etiologically associated with this condition are KMT2D and KDM6A, which are involved in epigenetic regulation.15
The patient in this case report had developmental delay and intellectual disability and exhibited a probably pathogenic KMT2D splice site variant (NM_003482.3: c.511‐2A> T). This type of variant can produce various effects such as exon skipping, intron retention, transcribed non-coding sequences or non-transcribed coding sequences, resulting in partially or completely non-functional proteins.16 Therefore, according to the diagnostic criteria established by an international group of experts in 2018, it met the criteria for a definitive diagnosis of KS. Typical dysmorphic features, such as elongated palpebral fissures, arched eyebrows, a short columella with a sunken nasal tip (Figure 1A and B), and persistent finger pads, were also evident.4
In addition to the typical KS features, the patient presented with coloboma (Figura 1A), a less commonly reported feature (Table 1). However, the incidence of coloboma is considerably higher in KS patients (3.2%) than in the general population (0.011%).17,18 Coloboma can affect the iris, choroid, retina, and/or optic nerve.18,19 Hearing loss is also frequent in KS patients, but sensorineural hearing loss is rare.20,21 Some studies have reported dysplastic cochlea, enlarged vestibules, and even absence of the cochlea and semicircular canals in KS patients, demonstrating that a wide variety of abnormalities at the ear level can occur.20,21 These findings can be explained by KMT2D disease-causing variant effects on histone trimethylation, which is required for the open chromatin conformation needed for gene expression, especially in the control of genes essential for embryonic development, including HOX genes.22,23 The splice score was calculated by Human Splicing Finder HSF algorithm and the variation c.522‐2A>T was ‐35.16%. This finding indicates that the KMT2D variant results in alteration of the wild-type acceptor site and might affect the splicing process. The KMT2D variant in our patient was located at the acceptor site of the fourth intron. Bioinformatic analysis (HSF v 3.1) predicted splice alteration that leads to a modified splicing process between the fourth and fifth exons. The fifth exon encodes for a zinc finger plant homeodomain (PHD) that consists of 2 β sheets and 2 structure-stabilizing zinc atoms anchored by the Cys4-His-Cys3 motif.23 This homeodomain recognizes the methylation state of N-terminal lysine residues in histone H3, whether H3K4 di-, tri-, or non-methylated, thus acting as a histone reader.24,25 This interaction allows the recognition and retention of the entire protein complex (in this case, KMT2D) at a specific site in the chromatin and contributes to the regulation of enzymatic activity in response to modifications present in histones.26 Therefore, the variation c.522‐2A>T can result in skipping the succeeding exon or the use of a cryptic splicing site, within exon 5, or downstream of the wild-type splice-acceptor site (Figure 3). However, experimental data are required to confirm this hypothesis. Anyhow, this intronic variant would lead to the altered synthesis of one of the PHD domains, which plays an essential role in selection of the histone-lysine methyltransferase 2D enzyme-binding site.
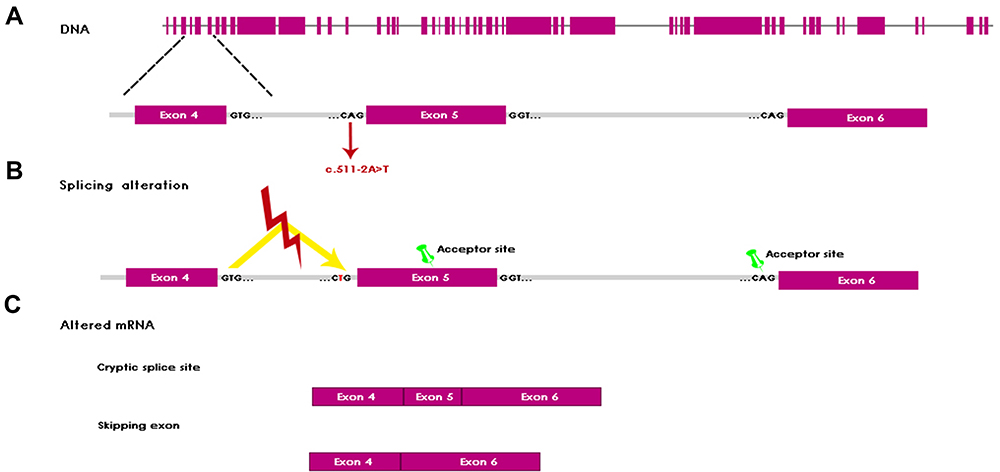 |
Figure 3 Alteration of splicing mechanism. (A) Schematic of the KMT2D genomic locus and variant c.511–2A>T. (B) Shows the splicing alteration, which represent a mis-splicing of KMT2D by mutation at the intron 4 resulting in the activation of alternative splice sites (green pin). (C) Shows the alteration of mRNA. The activation of cryptic splicing site in exon 5 results in an incomplete inclusion of this exon in the mRNA. Recognition of wild-type acceptor site of exon 6 results in a complete exclusion of exon 5 in the mRNA. |
Conclusion
We report a new intronic KMT2D variant as a probable cause of KS. Most characteristics described in our patient are similar to those previously reported. However, our patient also exhibited coloboma and sensorineural hearing loss, which are less common.
Abbreviations
ACMG, American College of Medical Genetics and Genomics; HSF, Human Splicing Finder; IQ, Intellectual Quotient; KS, Kabuki syndrome; KMT2D, Lysine methyltransferase 2D gene; KDM6A, Lysine Demethylase 6A gene; KMT2D, Lysine methyltransferase 2D; KDM6A, Lysine Demethylase 6A; WISC, Wechsler Intelligence Scale for Children.
Data Sharing Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics Approval and Informed Consent
This study was approved by the Ethics Committee of Fundacion Valle del Lili, Colombia (human study protocol #1504) and performed in accordance with the ethical standards found in the Declaration of Helsinki. Written informed consent was obtained from the parents of the subjects. Information revealing the subject’s identity was not included in the manuscript. The patient was identified by number and not by her real name.
Consent for Publication
Institutional approval was required to publish the case details. All the images and patient material presented in this study have been consented for publication and they are available to see upon request.
Acknowledgments
We thank the patient and her parents for agreeing to the pub HSF: Human Splicing Finder publication of this report. We also thank the people who have contributed to the development and execution of this study.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Funding
All forms of support and funding for study design, data collection, data analysis and manuscript writing were provided by the employers of the authors (Universidad Icesi and Fundación Valle del Lili).
Disclosure
The authors have no conflicts of interest to declare.
References
1. Liu S, Hong X, Shen C, et al. Kabuki syndrome: a Chinese case series and systematic review of the spectrum of mutations. BMC Med Genet. 2015;16(1):1–10. doi:10.1186/s12881-015-0171-4
2. Silva-andrade N, López-ortega K, Gallottini M. Orofacial features and medical profile of eight individuals with Kabuki syndrome. Med Oral Patol Oral Cir Bucal. 2019;24(5):e630.
3. Adam MP, Hudgins L, Hannibal M. Kabuki syndrome. Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews®. Seattle: University of Washington, Seattle. September 1, 2011:1993–2020.
4. Adam MP, Banka S, Bjornsson HT, et al. Kabuki syndrome: international consensus diagnostic criteria. J Med Genet. 2019;56(2):89–95. doi:10.1136/jmedgenet-2018-105625
5. Aref-eshghi E, Schenkel LC, Lin H, et al. The defining DNA methylation signature of Kabuki syndrome enables functional assessment of genetic variants of unknown clinical significance. Epigenetics. 2017;12(11):923–933. doi:10.1080/15592294.2017.1381807
6. Schwenty-lara J, Nehl D, Borchers A. The histone methyltransferase KMT2D, mutated in Kabuki syndrome patients, is required for neural crest cell formation and migration. Hum Mol Genet. 2020;29(2):305–319. doi:10.1093/hmg/ddz284
7. Weizmann Institute of Science GeneCards Human Gene Database. KMT2D gene; 2019. Available from: https://www.genecards.org/cgi-bin/carddisp.pl?gene=KMT2D&keywords=kmt2d.
8. Weksberg R, Tycko B. Epigenetics. In: En: Emery and Rimoin’s Principles and Practice of Medical Genetics.
9. Froimchuk E, Jang Y, Ge K. Histone H3 lysine 4 methyltransferase KMT2D. Gene. 2017;627:337–342. doi:10.1016/j.gene.2017.06.056
10. Boisgontier J, Tacchella JM, Lemaître H, et al. Anatomical and functional abnormalities on MRI in kabuki syndrome. Neuro Image Clin. 2018;21(101610):1–6.
11. Weizmann Institute of Science GeneCards Human Gene Database. KDM6A gene; 2020. Available from: https://www.genecards.org/cgi-bin/carddisp.pl?gene=KDM6A&keywords=kdm6a.
12. Cheon C, Ko JM. Kabuki syndrome: clinical and molecular characteristics. Korean J Pediatr. 2015;58(9):317–324. doi:10.3345/kjp.2015.58.9.317
13. Cuvertino S, Hartill V, Colyer A, et al. A restricted spectrum of missense KMT2D variants cause a multiple malformations disorder distinct from Kabuki syndrome. Genet Med. 2019;22(5):867–877. doi:10.1038/s41436-019-0743-3
14. Wang Y, Li N, Li X, et al. The phenotypic spectrum of Kabuki syndrome in patients of Chinese descent: a case series. Am J Med Genet. 2019;1981(7):1–12. doi:10.1002/ajmg.a.61467
15. Gatto LAM, Sousa LHA, Koppe GL, Junior ZD. Carotid artery occlusion in Kabuki syndrome: case report and literature review. Surg Neurol Int. 2017;8(1):8–11. doi:10.4103/sni.sni_427_16
16. Aref-Eshghi E, Bourque DK, Kerkhof J, et al. Genome-wide DNA methylation and RNA analyses enable reclassification of two variants of uncertain significance in a patient with clinical Kabuki syndrome. Hum Mutat. 2019;40(10):1684–1689. doi:10.1002/humu.23833
17. Imtiaz C, Farrukh S, Hisham A, Abdulrahman AS. Ocular manifestations in Kabuki syndrome: the first report from Saudi Arabia. Int Ophthalmol. 2008;28:131–134. doi:10.1007/s10792-007-9118-x
18. Ming JE, Russell KL, Bason L, Mcdonald-mcginn DM, Zackai EH. Coloboma and other ophthalmologic anomalies in kabuki syndrome: distinction from charge association. Am J Med Genet. 2003;123A:249–252. doi:10.1002/ajmg.a.20277
19. Chen Y, Sun M, Hsia S, Lai C, Wu W. Rare ocular features in a case of Kabuki syndrome (Niikawa-Kuroki syndrome). BMC Ophthalmol. 2014;14(1):2–4. doi:10.1186/1471-2415-14-143
20. Barozzi S, Di Berardino F, Atzeri F, et al. Audiological and Vestibular Findings in the Kabuki Syndrome. Am J Med Genet Part A. 2009;149A:171–176. doi:10.1002/ajmg.a.32610
21. Tekin M, Fitoz S, Arici S, Cetinkaya E, Incesulu A. Niikawa — Kuroki (Kabuki) syndrome with congenital sensorineural deafness: evidence for a wide spectrum of inner ear abnormalities. Int J Pediatr Otorhinolaryngol. 2006;70:885–889. doi:10.1016/j.ijporl.2005.09.025
22. Butcher DT, Cytrynbaum C, Turinsky AL, et al. CHARGE and kabuki syndromes: gene-specific DNA methylation signatures identify epigenetic mechanisms linking these clinically overlapping conditions. Am J Hum Genet. 2017;100(5):773–788. doi:10.1016/j.ajhg.2017.04.004
23. Van Laarhoven PM, Neitzel LR, Quintana AM, et al. Kabuki syndrome genes KMT2D and KDM6A: functional analyses demonstrate critical roles in craniofacial, heart and brain development. Hum Mol Genet. 2015;24(15):4443–4453. doi:10.1093/hmg/ddv180
24. Roberto Sanchez -M-MZ. The PHD finger: a versatile epigenome reader. Trends Biochem Sci. 2012;36(7):364–372.
25. Chignola F, Gaetani M, Rebane A, et al. The solution structure of the first PHD finger of autoimmune regulator in complex with non-modified histone H3 tail reveals the antagonistic role of H3R2 methylation. Nucleic Acids Res. 2009;37(9):2951–2961. doi:10.1093/nar/gkp166
26. Morrison EA, Musselman CA. The role of PHD fingers in chromatin signaling: mechanisms and functional consequences of the recognition of histone and non-histone targets. In: Binda O, Fernandez-Zapico ME, eds. En: Chromatin Signaling and Diseases. Iowa, USA: Elsevier Inc.; 2016:127–147.
27. Niikawa N, Kuroki Y, Kajii T, Matsuura N. Kabuki make-up (Niikawa-Kuroki) syndrome: a study of 62 patients. Am J Med Genet. 1988;31:565–589. doi:10.1002/ajmg.1320310312
28. Priolo M, Micale L, Augello B, et al. Absence of deletion and duplication of MLL2 and KDM6A genes in a large cohort of patients with Kabuki syndrome. Mol Genet Metab. 2012;107(3):627–629. doi:10.1016/j.ymgme.2012.06.019
29. Schrander-Stumpel CTRM, Spruyt L, Curfs LMG, Defloor T, Schrander JJP. Kabuki syndrome: clinical data in 20 patients, literature review, and further guidelines for preventive management. Am J Med Genet A. 2005;132A(3):234–243. doi:10.1002/ajmg.a.30331
30. Sanlaville D, Amiel J, Genevieve D, et al. Failure to detect an 8p22 – 8p23. 1 duplication in patients with Kabuki (Niikawa –Kuroki) syndrome. Eur J Hum Genet. 2005;13:690–693. doi:10.1038/sj.ejhg.5201383
31. Murakami H, Tsurusaki Y, Enomoto K, et al. Update of the genotype and phenotype of KMT2D and KDM6A by genetic screening of 100 patients with clinically suspected Kabuki syndrome. Am J Med Genet A. 2020;182(10):2333–2344. doi:10.1002/ajmg.a.61793
32. Paděrová J, Holubová A, Simandlová M, et al. Molecular genetic analysis in 14 Czech Kabuki syndrome patients is confirming the utility of phenotypic scoring. Clin Genet. 2016;90(3):230–237. doi:10.1111/cge.12754
33. Cheon CK, Choi HY, Park SH, Jung JH, Kim SJ. Ocular manifestations in kabuki syndrome: a report of 10 cases and literature review. Ophthalmic Genet. 2021;42(2):101–104. doi:10.1080/13816810.2020.1861308
34. Porntaveetus T, Abid MF, Theerapanon T. Expanding the oro-dental and mutational spectra of kabuki syndrome and expression of KMT2D and KDM6A in human tooth germs. Int J Biol Sci. 2018;14(4):381–389. doi:10.7150/ijbs.23517
35. Shangguan H, Su C, Ouyang Q, et al. Kabuki syndrome: novel pathogenic variants, new phenotypes and review of literature. Orphanet J Rare Dis. 2019;14(1):1–7. doi:10.1186/s13023-019-1219-x
36. Hernández-Woodbine MJ, Del Castillo-Rix DS, Baquero-Mejía IC. Reporte de una nueva mutación en Colombia: un paciente con síndrome de Kabuki. [Report of a new mutation in Colombia: a patient with Kabuki syndrome]. Iatreia. 2019;33(1):78–83. doi:10.17533/udea.iatreia.38
37. Suarez Guerrero JL, Ordónez Suarez AA, Contreras García GA. Síndrome de Kabuki. [Syndrome Kabuki]. An Pediatr. 2012;77(1):51–56. doi:10.1016/j.anpedi.2012.01.016
38. Osorio Alarcón C, Mantilla DO, Redondo CS, Galofre PG. Kabuki syndrome: a case report and literature review. Rev Salud Uninorte. 2016;32(3):565–575. doi:10.14482/sun.32.2.9754
39. Piro E, Schierz IAM, Antona V, et al. Neonatal hyperinsulinemic hypoglycemia: case report of kabuki syndrome due to a novel KMT2D splicing-site mutation. Ital J Pediatr. 2020;46(1):1–7. doi:10.1186/s13052-020-00902-8
40. Guo Z, Liu F, Li HJ. Novel KDM6A splice-site mutation in kabuki syndrome with congenital hydrocephalus: a case report. BMC Med Genet. 2018;19(1):206. doi:10.1186/s12881-018-0724-4
41. de Billy E, Strocchio L, Cacchione A, et al. Burkitt lymphoma in a patient with Kabuki syndrome carrying a novel KMT2D mutation. Am J Med Genet A. 2019;179(1):113–117. doi:10.1002/ajmg.a.60674
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.