")
Back to Journals » International Journal of Nanomedicine » Volume 11
A novel double-targeted nondrug delivery system for targeting cancer stem cells
Authors Qiao S, Zhao Y, Geng S, Li Y, Hou X, Liu Y, Lin FH, Yao L, Tian W
Received 1 July 2016
Accepted for publication 30 October 2016
Published 8 December 2016 Volume 2016:11 Pages 6667—6678
DOI https://doi.org/10.2147/IJN.S116230
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lei Yang
Shupei Qiao,1,* Yufang Zhao,1,* Shuai Geng,2,* Yong Li,1,* Xiaolu Hou,1,3 Yi Liu,1 Feng-Huei Lin,4,5 Lifen Yao,6 Weiming Tian1
1School of Life Science and Technology, Harbin Institute of Technology, 2Department of Pharmacology, Harbin Medical University, 3Department of Cardiology, The Fourth Affiliated Hospital of Harbin Medical University, Harbin, People’s Republic of China; 4Institute of Biomedical Engineering, National Taiwan University, Taipei, Taiwan; 5Institute of Biomedical Engineering and Nanomedicine, National Health Research Institutes, Miaoli, Taiwan; 6Department of Neurology, The First Affiliated Hospital of Harbin Medical University, Harbin, People’s Republic of China
*These authors contributed equally to this work
Abstract: Instead of killing cancer stem cells (CSCs), the conventional chemotherapy used for cancer treatment promotes the enrichment of CSCs, which are responsible for tumor growth, metastasis, and recurrence. However, most therapeutic agents are only able to kill a small proportion of CSCs by targeting one or two cell surface markers or dysregulated CSC pathways, which are usually shared with normal stem cells (NSCs). In this study, we developed a novel nondrug delivery system for the dual targeting of CSCs by conjugating hyaluronic acid (HA) and grafting the doublecortin-like kinase 1 (DCLK1) monoclonal antibody to the surface of poly(ethylene glycol) (PEG)–poly(d,l-lactide-co-glycolide) (PLGA) nanoparticles (NPs), which can specifically target CD44 receptors and the DCLK1 surface marker – the latter was shown to possess the capacity to distinguish between CSCSs and NSCs. The size and morphology of these NPs were characterized by dynamic light scattering (DLS), transmission electron microscopy (TEM), and scanning electron microscopy (SEM). This was followed by studies of NP encapsulation efficiency and in vitro drug release properties. Then, the cytotoxicity of the NPs was tested via Cell Counting Kit-8 assay. Finally, the 4T1 CSCs were obtained from the alginate-based platform, which we developed as an in vitro tumor model. Tumor-bearing nude mice were used as in vivo models to systematically detect the ability of NPs to target CSCs. Our results showed that the DCLK1–HA–PEG–PLGA NPs exhibited a targeting effect toward CSCs both in vitro and in vivo. These findings have important implications for the rational design of drug delivery systems that target CSCs with high efficacy.
Keywords: alginate, CD44, PEG–PLGA, DCLK1, hyaluronic acid
Introduction
Cancer stem cells (CSCs) are a rare cell population localized within solid tumors that overexpress CD44 receptors; they are usually regarded as a determinant of cancer resurrection and metastasis.1–5 Several studies have suggested that the elimination of CSCs serves as an opportunity to cure various tumors.6,7 Conventional treatments for cancer, such as radiation therapy and chemotherapy, tend to kill the majority of differentiated cancer cells, and they are often associated with severe side effects toward normal tissues due to the incomplete responses of tumor cells; however, CSCs possess the properties of multidrug resistance and cannot be killed. In addition, treatments always promote the enrichment of CSCs.8,9 The surviving CSCs result in tumor recurrence via self-renewal and differentiation into multiple tumor cell types. Over the course of the past decade, the development of CSC-targeted therapies has offered hope to improve the survival of cancer patients. However, most of the CSC therapeutic agents can only kill a small proportion of CSCs by targeting one or two cell surface markers or dysregulated CSC pathways, which are usually shared with normal stem cells (NSCs).10–14
Recently, nanoparticles (NPs) have attracted much attention in the field of cancer therapeutics, as they can easily reach their targets through physicochemical modification. Moreover, considerable amounts of drugs can be introduced into NPs through simple loading and chemical conjugates.15,16 One study has shown that long-circulating NPs accumulated within tumor cells, while only a few were present in normal cells due to both the small size of the NPs and the leaky tumor vasculature, which is referred to as the enhanced permeability and retention (EPR) effect.17 Furthermore, several recent studies showed that NPs can enhance the accumulation of anticancer drugs in three-dimensional (3D) breast, glioblastoma, and gastric spheroids enriched with CSCs; all of these results demonstrated that the use of NPs holds considerable potential in cancer therapy.14,18,19 In this study, poly(ethylene glycol) (PEG)–poly(D,L-lactide-co-glycolide) (PLGA) NPs were synthesized through the single-emulsion solvent evaporation method. PEG–PLGA NPs have been largely explored for their ability to deliver a range of therapeutics to specific cells or tissues. Both NP compositions have been approved by the US Food and Drug Administration (FDA) due to their biocompatibility and resorbability.
Furthermore, hyaluronic acid (HA) has been extensively investigated and has typically been used to target CD44, which is always highly expressed in multiple tumor tissues or CSCs, in the diagnosis and treatment of various cancers.3 Previous research has shown that HA coatings can selectively and specifically target and deliver drugs to CSCs.20,21 Therefore, HA was conjugated to the PEG–PLGA NPs to improve their targeting property to the CSCs in this study.
Other studies have suggested that the targeting of therapeutic NPs via antibodies can further enhance their accumulation in target cells, as well as improve their retention, ultimately allowing for decreased effective dosages.22–24 Cetuximab, a 152 kDa chimeric monoclonal antibody that specifically binds to the human epidermal growth factor receptor (EGFR) – a potential marker for glioblastoma stem-like cells – was conjugated to iron oxide NPs as a targeted therapy for glioblastoma.13 Given the number of profound studies that have been published on CSCs, an increasing number of markers have been found and identified. These findings have frequently been used to develop cancer-targeting treatments. Swaminathan et al25 grafted the CD133 monoclonal antibody to a maleimide-modified PLGA NP for targeting CSCs in chemotherapy. However, CSC markers are often shared by NSCs; thus, therapies that target these markers may cause severe injury to normal tissues.26 Recently, a marker specific to CSCs (known as doublecortin-like kinase 1 [DCLK1]) was found, which was shown to have the capacity to distinguish between tumor tissues and NSCs.27 It was demonstrated that DCLK1 could be a more specific marker for CSCs, and it was further suggested that targeting DCLK1-positive tumor cells could serve as a promising treatment for cancer.27,28
Here, we developed a novel nondrug delivery system for the dual-targeting of CSCs by conjugating HA and grafting the DCLK1 monoclonal antibody to the surface of the PEG–PLGA NPs. As was shown in our previous study,29 CD44 was highly expressed and CD24 had low expression in the 4T1 tumor spheroids, that were screened by our alginate-based platform when compared with the 3D control (this involved only culturing the 4T1 breast cancer cells in hydrogel without screening) and the 4T1 breast cancer cells that were cultured in two-dimensional (2D) rigid dishes. Furthermore, we noticed that DCLK1, which is known as a putative marker for intestinal and pancreatic stem cells, including the CSCs of those organs, was highly expressed in 4T1 tumor spheroids. Therefore, in this investigation, the 4T1 breast cancer stem-like cells, which overexpress CD44 and DCLK1, were used as an in vitro model. The targeting activity of DCLK1–HA–PEG–PLGA NPs to CSCs in vitro and in vivo was investigated. Surprisingly, we found that the dual-targeting NPs we synthesized were able to effectively target CSCs. These findings have important implications for the rational design of drug delivery systems that effectively target CSCs.
Materials and methods
Materials
Alginate, HA, and poly(vinyl alcohol) (PVA) (MW =16 kDa; 98% hydrolyzed) were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). PLGA (molar ratio of D,L-lactic to glycolic acid, 50:50; MW: 7 kDa) was purchased from Shandong Freda Biopharmaceutical Co., Ltd. (Shandong, People’s Republic of China). N-Hydroxysuccinimide (NHS) and 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) were purchased from Alfa Aesar (Ward Hill, MA, USA). PEG diamine (NH2-PEG-NH2) was purchased from Beijing Kaizheng Bioengineering Development Co., Ltd. (Beijing, People’s Republic of China).
Preparation of the NPs
Synthesis of the PEG–PLGA polymer
The PEG–PLGA polymer was synthesized by EDC and NHS chemistry according to previous studies.30,31 Briefly, PLGA was preactivated prior to PEGylation with PEG diamine. First, PLGA was dissolved in methylene chloride (10 mg/mL). Then, EDC and NHS were added to the PLGA solution with a molar ratio of 1:5:5 with respect to PLGA under magnetic stirring; the activation reaction of the carboxyl of PLGA continued for 24 hours within a nitrogen gas chamber at room temperature. Vacuum rotary evaporation was used to remove a part of the methylene chloride solvent, and the mixed solution was then added in a dropwise manner to precooled ether. The solution was centrifuged (15,000× g; 10 minutes) at 4°C. The precipitate was collected, and the PLGA–NHS that was obtained was placed in the vacuum-drying oven (24 hours). For the synthesis of PEG–PLGA polymer, PLGA–NHS was dissolved in methylene chloride (80 mg/mL). PEG was added to the PLGA solution with a PLGA to PEG molar ratio of 1:1, and this solution was magnetically stirred for 10 minutes. The mixed solution was added to a three-necked flask and magnetically stirred for 12 hours within a nitrogen gas chamber at room temperature. Rotary evaporation was used to remove a part of the methylene chloride solvent in a vacuum, and then the mixed solution was added in a dropwise manner to precooled methanol. The solution was centrifuged (15,000× g; 10 minutes) at 4°C. The precipitate was collected, and the light yellow precipitate was placed in the vacuum-drying oven (24 hours). The PEG–PLGA polymer was obtained by freeze drying.
Preparation of the PEG–PLGA NPs
The NPs were prepared using the single-emulsion solvent evaporation method.32 PEG–PLGA (150 mg) polymer was dissolved in methylene chloride (4 mL) and mixed well. Then, the mixture was added to a precooled PVA solution (2%, wt/vol) and sonicated at 4°C for 2 minutes, followed by magnetic stirring for 2.5 hours at room temperature in the fume hood. The PEG–PLGA NPs were further isolated using the centrifugation method, as described earlier. Subsequently, the PEG–PLGA NPs were obtained by centrifugation at 15,000× g for 15 minutes (4°C), washed thrice with water, and lyophilized. The dried NPs were stored in the refrigerator at 4°C.
Preparation of the HA–PEG–PLGA NPs
The HA was conjugated to the PEG–PLGA via chemical conjugation; the HA enabled the carboxyl to react with the amino acid from PEG–PLGA using the EDC/NHS coupling reaction. First, HA (0.423 g) was dissolved in 10 mL of 2-ethanesulfonic acid (MES) buffer (pH 4.7) and mixed well. Subsequently, EDC and NHS were added to the HA solution with an HA to EDC to NHS molar ratio of 1:10:10; magnetic stirring was continued for 2 hours. The PEG–PLGA NPs (50 mg) were dissolved in 10 mL of deionized water followed by ultrasonic dispersion for 5 minutes. Then, the mixture was added to the HA solution and magnetically stirred for 12 hours. Subsequently, the HA–PEG–PLGA NPs were obtained by centrifugation at 15,000× g for 15 minutes (4°C); they were washed thrice with water and lyophilized. The dried NPs were stored in the refrigerator at 4°C.
Preparation of the DCLK1–HA–PEG–PLGA NPs
The DCLK1 antibody was grafted to the HA–PEG–PLGA NPs by the Schiff base reaction. First, a DCLK1 antibody solution (10 μL) was added to 5 mL of sodium periodate solution (10 mg/mL) at room temperature, and it was reacted under vibration for 30 minutes. Then, 600 μL of HA–PEG–PLGA NP solution (50 mg/mL) was added to the above solution at room temperature, and it was reacted via shocking for 4 hours in the dark. Moreover, 1 mL of NaBH4 solution (1%, wt/vol) was added dropwise to the system, and it was reacted for 20 minutes. The DCLK1–HA–PEG–PLGA NPs were further isolated by the centrifugation method described earlier.
Preparation of the doxorubicin (DOX)-loaded NPs
First, DOX HCl, which is soluble in methylene, was collected through chemical modification. Briefly, DOX HCL (5.3 mg) was stirred with excess trimethylamine (3.1 mg) at room temperature under nitrogen to obtain the DOX base. DOX was added to the methylene chloride in addition to the PEG–PLGA polymer to prepare the DOX-loaded NPs, as described earlier.
Preparation of the R6G-loaded fluorescein isothiocyanate (FITC)–HA–PEG–PLGA NPs
R6G was added to methylene chloride, along with the PEG–PLGA polymer, prior to preparing the PEG–PLGA NPs; DCLK1 was substituted by FITC to obtain the R6G-loaded FITC–HA–PEG–PLGA NPs with the same method described earlier.
NP morphology, size distribution, and zeta potential analysis
A morphological examination of the NPs was performed via transmission electron microscopy (TEM) (Hitachi, Tokyo, Japan) and scanning electron microscopy (SEM) (Hitachi Company). The average size and zeta potentials of the NPs were determined by dynamic light scattering (DLS) using a ZetaSizer Nano series Nano-ZS (Brookhaven Companies, Brookhaven, GA, USA). Each batch was analyzed in triplicate.
In vitro drug release test
The establishment of a standard curve for DOX hydrochloride
DOX (1 mg) was resuspended in 1 mL of phosphate-buffered saline (PBS), and it was then diluted to concentrations of 0.02, 0.04, 0.08, 0.16, 0.24, and 0.32 μg/mL. The standard curve for adriamycin was established by measuring the fluorescence intensity with different concentrations of DOX solutions at an excitation wavelength of 480 nm and an emission wavelength of 590 nm.
Encapsulation efficiencies of NPs
The entrapment efficiency of the NPs was evaluated using an ultraviolet (UV) method.33 To determine the encapsulation efficiency of DOX in NPs, 5 mg of DOX–DCLK1–HA–PEG–PLGA NPs were mixed with 2 mL of dimethyl sulfoxide (DMSO) in order to break the structure of the NPs. The fluorescence intensity of the solution was measured at an excitation wavelength of 480 nm and an emission wavelength of 590 nm using a 752 UV–visible recording spectrophotometer. The drug-loading content (LC%) and encapsulation efficiency (EE%) of the PEG–PLGA NPs loaded with DOX were calculated using the following equations:
|
|
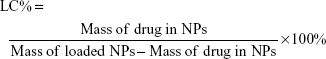
|
|
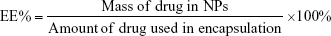
Each experiment was conducted in triplicate.
Drug-release profile in vitro
To investigate the release kinetics of DOX from NPs, the NPs were suspended in 1 mL of deionized water and they were dialyzed by a semipermeable membrane (40 kDa). The dialysis tubes were then immersed in 25 mL of PBS dialysis solution (10 mM; pH =7.4) at 37°C while being moderately shaken using a constant temperature water bath vibrator. The released DOX in the dialysis buffer was monitored at indicated time intervals over the course of 24 hours (for a total of 7 days). The concentration of the released DOX was determined by measuring the fluorescent density of DOX at an excitation wavelength of 480 nm and an emission wavelength of 590 nm using a UV spectrophotometer (Tecan, Durham, NC, USA).
Alginate-based platform prepared
Alginate-based platform was prepared as previously reported.19,34 Briefly, the oxidized alginate was obtained by mixing sodium peroxide and sodium alginate (in distilled water) with a mass ratio of 1:2. The reaction was conducted at 4°C for 2 hours and was terminated by the addition of ethylene glycol; then, the oxidized alginate was lyophilized at −20°C and dissolved in distilled water to achieve a 1.4% concentration. Epidermal growth factor (EGF) and basic fibroblast growth factor (bFGF) immobilized oxidized alginate was prepared by dissolving EGF and bFGF (50 ng/mL) in an oxidized alginate solution while stirring and was collected and lyophilized at −20°C. The alginate-based platform was prepared by using the EGF and bFGF immobilized oxidized alginate, and the addition of low molecular weight HA with 1.5% concentration, cross-linking with calcium ions.
Cell culture and cell seeding on the alginate-based platform
Human umbilical vein endothelial cell (HUVEC) and mouse 4T1 breast cancer cell lines were purchased from the Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences (Shanghai, People’s Republic of China). Mouse neural stem cells were purchased from the Cyagen Biosciences, Inc. (Guangzhou, People’s Republic of China). Mouse breast cancer 4T1 cells were cultured in Roswell Park Memorial Institute (RPMI)-1640 medium supplemented with 10% fetal bovine serum (FBS) (Thermo Fisher Scientific, Waltham, MA, USA) and incubated at 37°C in a humidified atmosphere with 5% CO2. Also, HUVECs were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% FBS and incubated at 37°C in a humidified atmosphere with 5% CO2. Mouse neural stem cells were cultured in neural stem cell growth medium (Cyagen Biosciences, Inc.) and incubated at 37°C in a humidified atmosphere with 5% CO2. The mouse 4T1 breast cancer cells were digested from monolayer cultures and seeded onto the alginate-based platform; they were then incubated in a humidified atmosphere of 5% CO2 at 37°C and grown in DMEM/F-12 media supplemented with 1% B-27; the media were changed every other day. The mouse neural stem cells were digested and seeded onto the alginate-based platform; they were then incubated in a humidified atmosphere of 5% CO2 at 37°C and grown in a neural stem cell growth medium.
Cell cytotoxicity assay
Cell cytotoxicity was determined by Cell Counting Kit-8 (CCK-8) (Dojindo, Kumamoto, Japan) according to the manufacturer’s instructions. A total of 5,000 HUVECs were seeded in 96-well plates after 24 hours of culture; the DCLK1–HA–PEG–PLGA NPs, HA–PEG–PLGA NPs, and PEG–PLGA NPs with different concentrations were added to the 96-well plates to incubate with HUVECs for 24 hours. The CCK-8 solution was added to the wells (20 μL/well); after 4 hours of incubation, absorbance at 450 nm was measured using a standard enzyme-linked immunosorbent assay (ELISA)-format spectrophotometer. Each experiment was repeated three times, and the data are represented as the mean of all measurements.
Immunostaining
4T1 tumor spheroids were released from the alginate-based platform and were fixed with 4% paraformaldehyde for 15 minutes and permeabilized with 1% Triton X-100 in PBS for 5 minutes at room temperature with gentle rocking. Then, the samples were blocked with 2% bovine serum albumin (BSA) (Sigma-Aldrich Co.) in PBS for 1 hour; they were then incubated with the mouse monoclonal DCLK1 primary antibody. Samples were washed three times with PBS prior to incubation with the tetramethyl rhodamin isothiocyanate (TRITC)-conjugated secondary antibody for 1 hour at room temperature. Cell nuclei were counterstained with DAPI (300 nM in Dulbecco’s-PBS; Thermo Fisher Scientific) for 10 minutes. Images were obtained using an inverted fluorescent microscope (Nikon Instruments, Melville, NY, USA).
Cell uptake
Cell uptake was examined using confocal laser scanning microscopy (UltraVIEW VoX System; PerkinElmer, Waltham, MA, USA). Briefly, the appropriate 2D and 3D mouse 4T1 breast cancer cells and 3D mouse 4T1 tumor spheroids were incubated with R6G–PEG–PLGA NP, R6G–HA–PEG–PLGA NP, and R6G–DCLK1–PEG–PLGA NP suspensions (200 μg/mL). After 2 hours, cells were washed three times; they were then incubated with the DAPI solution (1:1,000) and washed three times, once more. Finally, the cells were analyzed by confocal laser scanning microscopy.
Animal studies
Immune-deficient mice provided by the Shanghai Experimental Animal Resource Center served as the animal model and were housed in a specified chamber with regular temperature (22°C±2°C) and relative humidity (40%–60%). All animal care and experiments were approved by the Ethics Committee of the School of Life Science and Technology, Harbin Institute of Technology. Ethical and legal approval was obtained prior to the commencement of this study. All animal care and experiments were guided in line with the standards of the Animal Research Committee of the School of Life Science and Technology, Harbin Institute of Technology, and conducted in accordance with Chinese National Guidelines for Animal Welfare which were issued in 1988 and revised in 2013 (http://www.gov.cn/gongbao/content/2014/content_2692743.htm). Mouse 4T1 tumor spheroids were released from the alginate-based platform and were digested to single cells and counted; 1×103 4T1 cancer stem-like cells were subcutaneously injected to establish the tumor model. Tumor dimensions were measured with calipers to estimate the volume according to V = a2b/2, where a is the longest diameter and b is the shortest diameter. Only mice with a tumor volume of 1 cm3 were selected for the experiment to ensure consistency throughout the study. When the tumor grew to the proper size, the mice were anesthetized (n=5 for each group). R6G–PEG–PLGA NPs and R6G–DCLK1–HA-PEG–PLGA NPs within PBS solution were respectively injected into mice via the tail vein; each mouse was injected with 50 μL (50 mg/mL) of the solution. After 2 hours and then after 12 hours, the mice were anesthetized with ether, and they were observed using small animal imaging technology.
Results
Characterization of PEG–PLGA, HA–PEG–PLGA, and DCLK1–HA–PEG–PLGA NPs
Figure 1 shows the synthesis procedures of PEG–PLGA, HA–PEG–PLGA, and DCLK1–HA–PEG–PLGA NPs. First, the PEG–PLGA polymers were synthesized; the PEG–PLGA NPs were then prepared using the single-emulsion solvent evaporation method. Subsequently, HA was conjugated to the PEG–PLGA NPs via a carboxyl reaction with the amino acid of PEG–PLGA using the EDC/NHS coupling reaction. Finally, the DCLK1 antibody was grafted to the HA–PEG–PLGA NPs by the Schiff base reaction. As shown in Figure 2, the hydrodynamic diameters and morphologies of the PEG–PLGA and HA–PEG–PLGA NPs were examined by DLS, TEM, and SEM, respectively. According to the SEM and TEM images, the NPs were round in shape with a core–shell structure and their size was ~210–250 nm in diameter. This size was consistent with DLS measurements of the NPs in aqueous solutions, as shown in Figure 2A and D. The hydrodynamic size of the PEG–PLGA NPs was ~220.6 nm, and that of the HA–PEG–PLGA NPs was 230.4 nm. With respect to the DCLK1–HA–PEG–PLGA NPs that were prepared, an FITC-conjugated secondary antibody was used to validate whether this adopted method was effective, particularly since the monoclonal antibody was exorbitantly priced. First, red fluorescence-labeled NPs were obtained by adding R6G to the PEG–PLGA polymer during the preparation of PEG–PLGA NPs. Then, the FITC-conjugated secondary antibody was oxidized by sodium periodate before being grafted to the R6G–HA–PEG–PLGA NPs. As shown in Figure 3, the green fluorescence of FITC can overlap with the red fluorescence, resulting in yellow dots. This result showed that the method we used was able to immobilize antibodies on the HA–PEG–PLGA NPs. Finally, the DCLK1–HA–PEG–PLGA NPs were obtained with the same method.
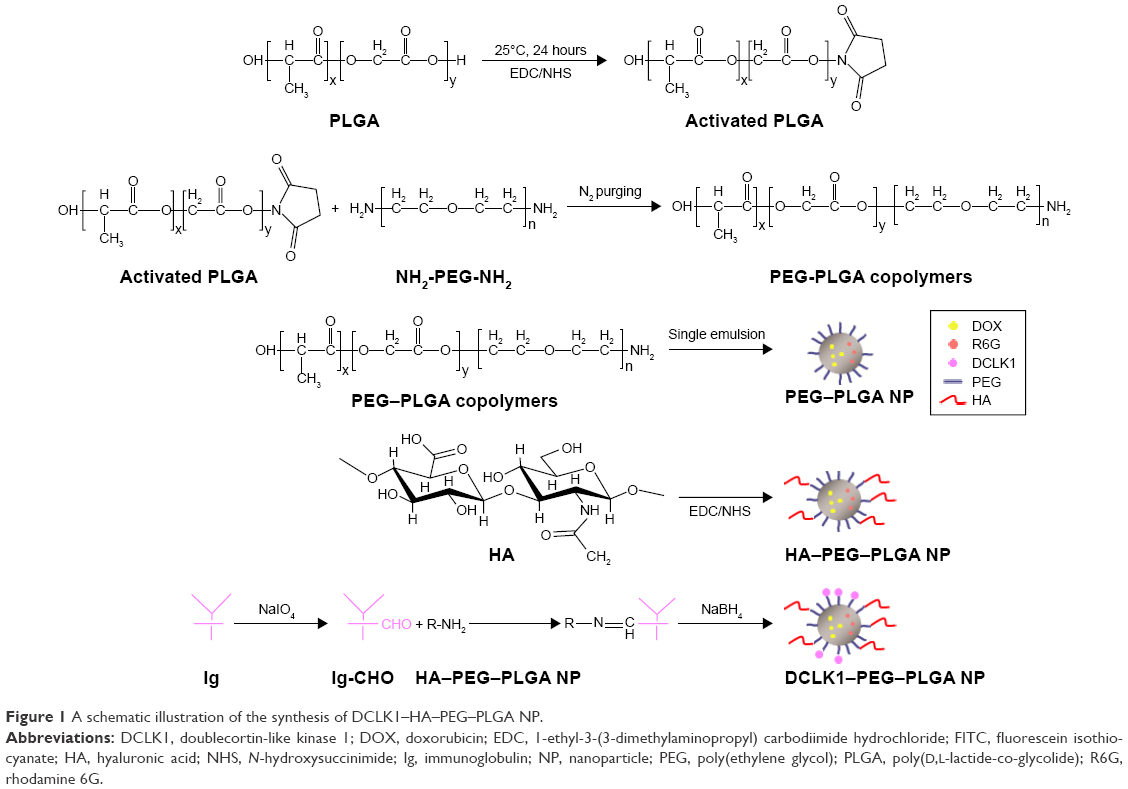 | Figure 1 A schematic illustration of the synthesis of DCLK1–HA–PEG–PLGA NP. |
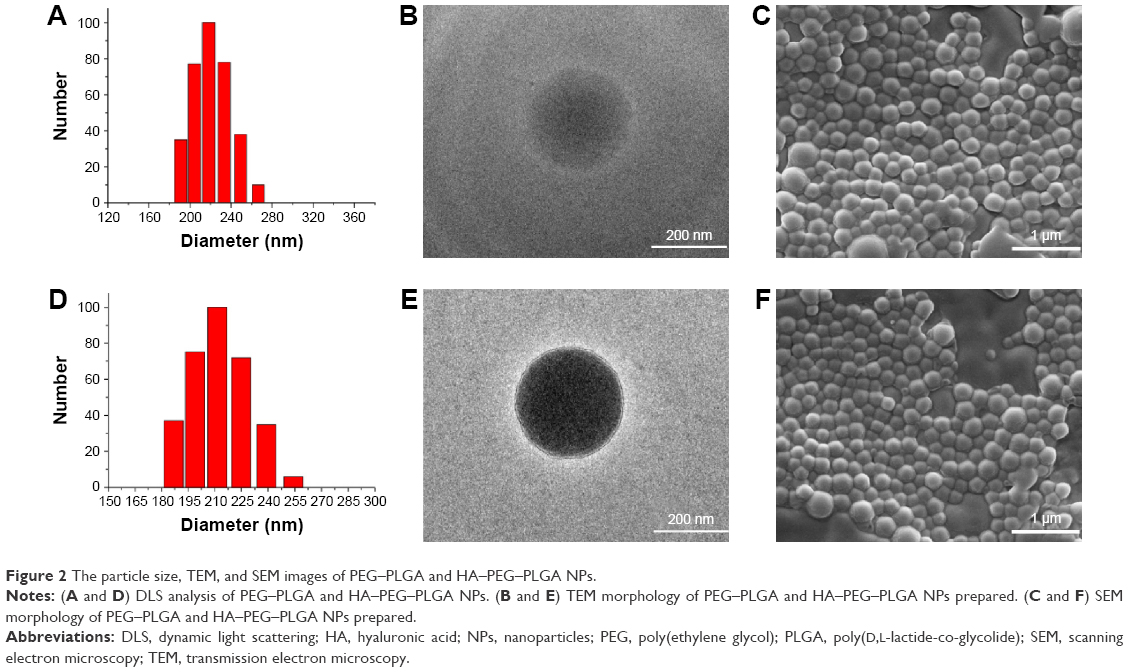 | Figure 2 The particle size, TEM, and SEM images of PEG–PLGA and HA–PEG–PLGA NPs. |
 | Figure 3 FITC grafted on the R6G-loaded HA–PEG–PLGA NPs. |
Research on the drug release properties of DCLK1–HA–PEG–PLGA NPs in vitro
The traditional anticancer drug DOX was used as the model drug to explore the drug release properties of NPs in vitro. In order to better determine the concentration of DOX, the solution was analyzed by fluorescence measurements at an excitation wavelength of 480 nm and an emission wavelength of 590 nm using a 752 UV–visible recording spectrophotometer to establish a standard curve for DOX. The results showed that from a concentration range of 20–320 μg/L, the DOX fluorescence intensity (Y) and concentration (X) showed a good linear relationship with the following linear regression equation (Figure 4A):
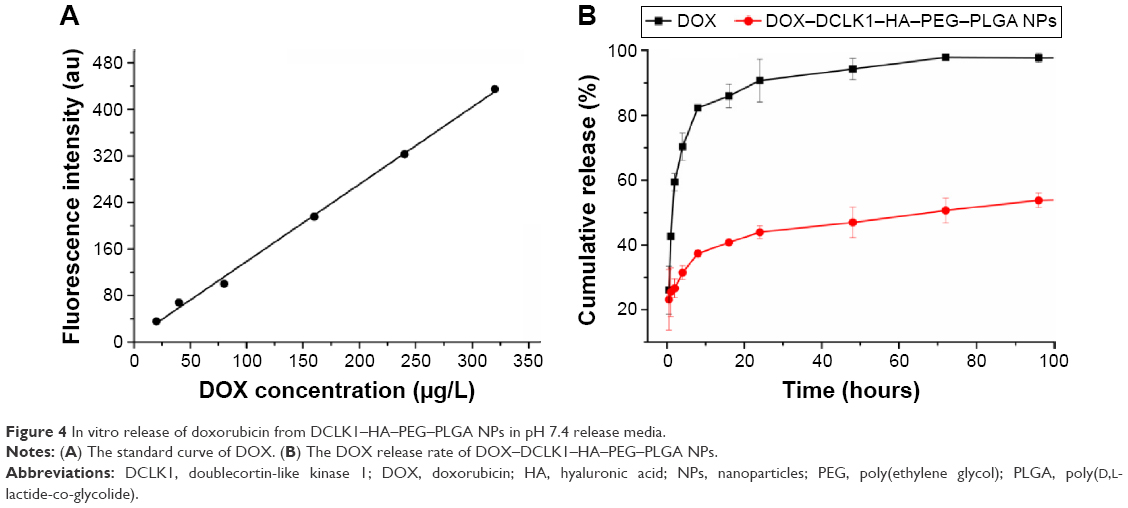 | Figure 4 In vitro release of doxorubicin from DCLK1–HA–PEG–PLGA NPs in pH 7.4 release media. |
Y = 1.32613X + 5.47726, R2=0.99737; | (3) |
When the molar ratio of DOX and the PEG–PLGA polymer was 1:20, the DOX encapsulation efficiency was 58.12% and its drug loading was 5.43%. Fluorescence spectroscopy was used to detect changes in the concentration of DOX in order to test the performance of the nondrug delivery carrier. The results showed that at 37°C, pH 7.4 in PBS solution, the free DOX release rate was faster than the release rate of DOX–DCLK1–HA–PEG–PLGA NPs. At 72 hours, almost all of the free DOX was released, while the NPs carrying DOX released only 46.98% (Figure 4B). The in vitro release results showed that once the drug was loaded, DCLK1–HA–PEG–PLGA NPs could prolong the drug release. Before the NPs targeted the tumor site, not much of the drug was released. Not only can this enhance the targeted killing of tumor tissues, but it can also reduce the side effects caused by the free drug.
In vitro cytotoxicity and targeting test
HUVECs have been shown to be more sensitive than other cell types to the same nanomaterials.35 In this study, the cytotoxicity of the PLGA–PEG, HA–PEG–PLGA, and DCLK1–HA–PEG–PLGA NPs was evaluated using this cell model. Figure 5 depicts the cell viability at different concentrations of PLGA–PEG, HA–PEG–PLGA, and DCLK1–HA–PEG–PLGA NPs. With increases in the concentrations of these three NPs, no prominent change in HUVEC viability was observed. The results indicated that even when the concentration of NPs reached 200 μg/mL, the cell viability continued >80%. In other words, these three types of NPs possess acceptable biocompatibility and they are ideal biological carriers for drug delivery in vivo.
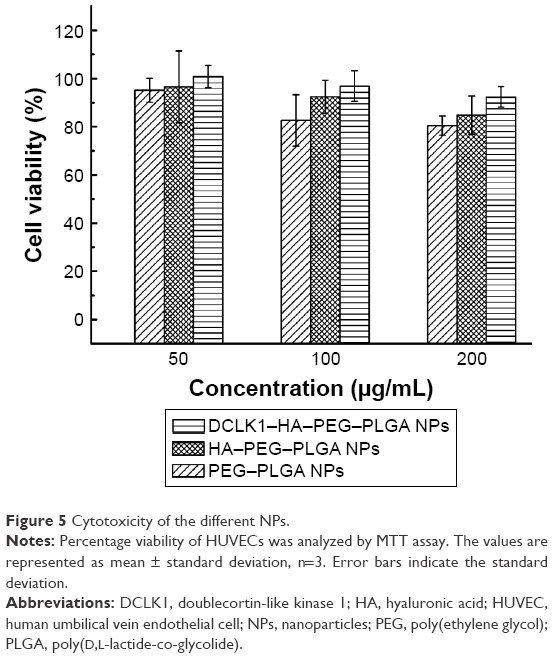 | Figure 5 Cytotoxicity of the different NPs. |
To verify whether DCLK1–HA–PEG–PLGA can target cancer stem-like cells, R6G–PEG–PLGA, R6G–HA–PEG–PLGA, and R6G–DCLK1–HA–PEG–PLGA NPs, which were stained with a red fluorescence molecule, were obtained. The mouse 4T1 breast cancer cells that were cultured in conventional 2D rigid dishes, 3D alginate hydrogel, and the 4T1 tumor spheroids that possessed CSC properties were sifted through the alginate-based platform we developed and used to compare the targeting behaviors of these three types of NPs. Moreover, the NSCs (neurospheres) were prepared and used as a negative control. Figure 6 presents typical images that illustrate the uptake of NPs together with the stained nuclei in 2D- and 3D-cultured 4T1 breast cancer cells and 4T1 tumor spheroids that were incubated for 2 hours with the three NPs. As expected, the red fluorescence of the NPs was not observable in the cells treated with empty NPs (R6G–PEG–PLGA). R6G–HA–PEG–PLGA and R6G–DCLK1–HA–PEG–PLGA, which were taken up by the 4T1 tumor spheroids after 3 hours of incubation at 37°C, were significantly higher than the 2D- and 3D-cultured 4T1 breast cancer cells due to the high expression of CD44 and DCLK1 of the 4T1 tumor spheroids, suggesting that HA and DCLK1 modification can greatly improve PEG–PLGA NPs’ ability to target cancer stem-like cells. However, there was no significant difference between the uptake of R6G–HA–PEG–PLGA and R6G–DCLK1–HA–PEG–PLGA NPs by the 4T1 tumor spheroids and the uptake of HA–PEG–PLGA and DCLK1–HA–PEG–PLGA NPs by neurospheres (data not shown).
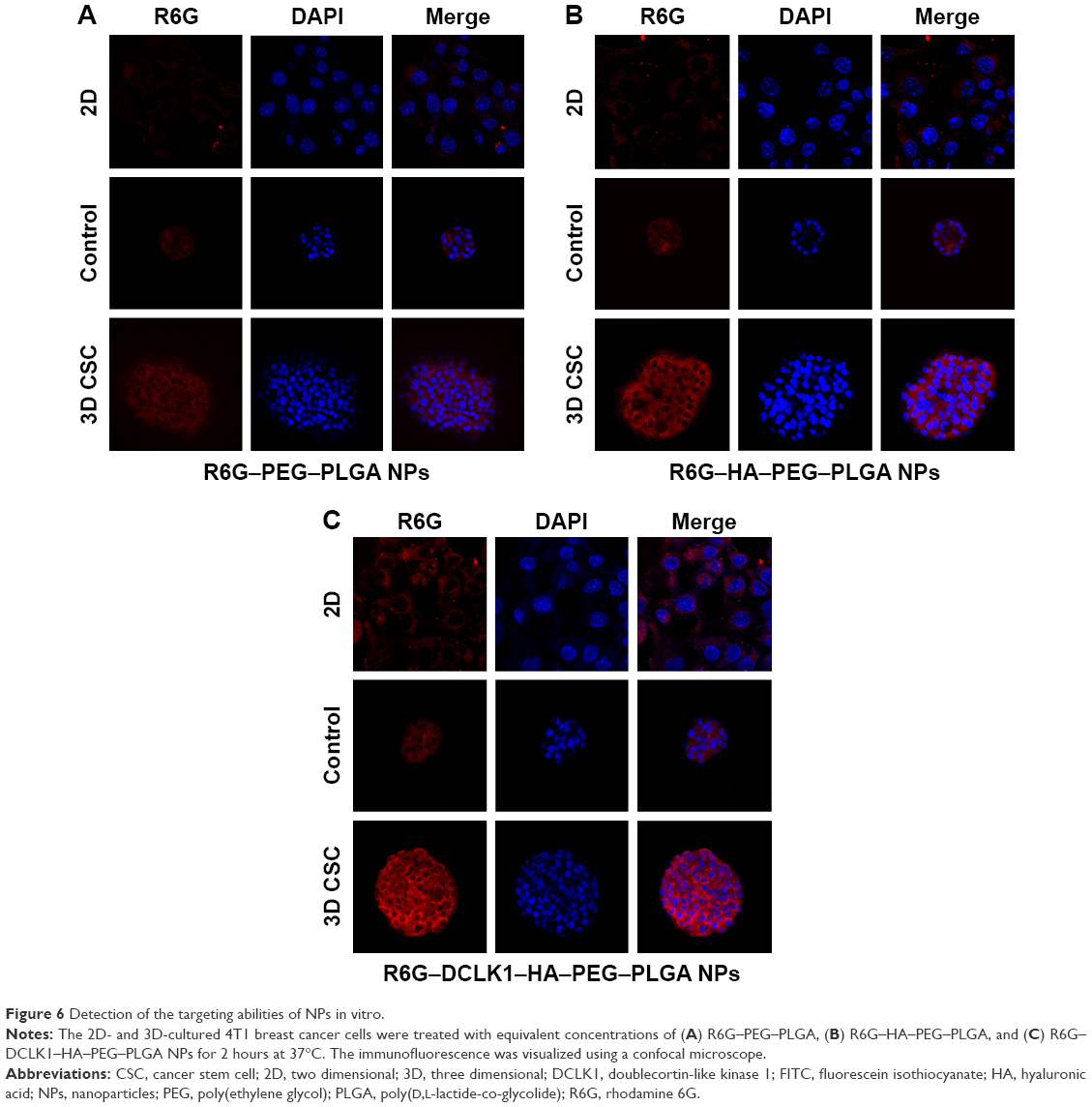 | Figure 6 Detection of the targeting abilities of NPs in vitro. |
In vivo tumor-targeting capability of DCLK1–HA–PEG–PLGA NPs
The ability of DCLK1–HA–PEG–PLGA NPs to target tumor tissues was evaluated in nude mice bearing 4T1 tumors. First, the 4T1 tumor spheroids were screened by culturing the 4T1 breast cancer cells on the alginate-based platform for 7 days without serum. The 4T1 tumor spheroids were released from the alginate–HA hydrogel and digested and counted. Four-week-old male nude mice with similar physiological characteristics were prepared, followed by 1,000 4T1 breast cancer stem-like cells, which were subcutaneously inoculated to the back of the nude mice. After 4 weeks, the tumor formed with normal physiological indexes. To evaluate the NPs’ tumor-targeting capability in vivo, NPs encapsulated with R6G were prepared once again. R6G–PEG–PLGA and R6G–DCLK1–HA–PEG–PLGA NPs were intravenously injected into the nude mice. Twelve hours after the injection, the fluorescence of these two groups was examined and recorded by an in vivo imaging system, as shown in Figure 7. R6G–PEG–PLGA NP-injected mice only showed a small amount of red fluorescence staining. However, a large amount of red fluorescence was observed at the tumor site after R6G–DCLK1–HA–PEG–PLGA NPs were injected into nude mice with formed tumors. These results indicate that grafting of the HA and DCLK1 antibody substantially improved NPs’ tumor-targeting abilities.
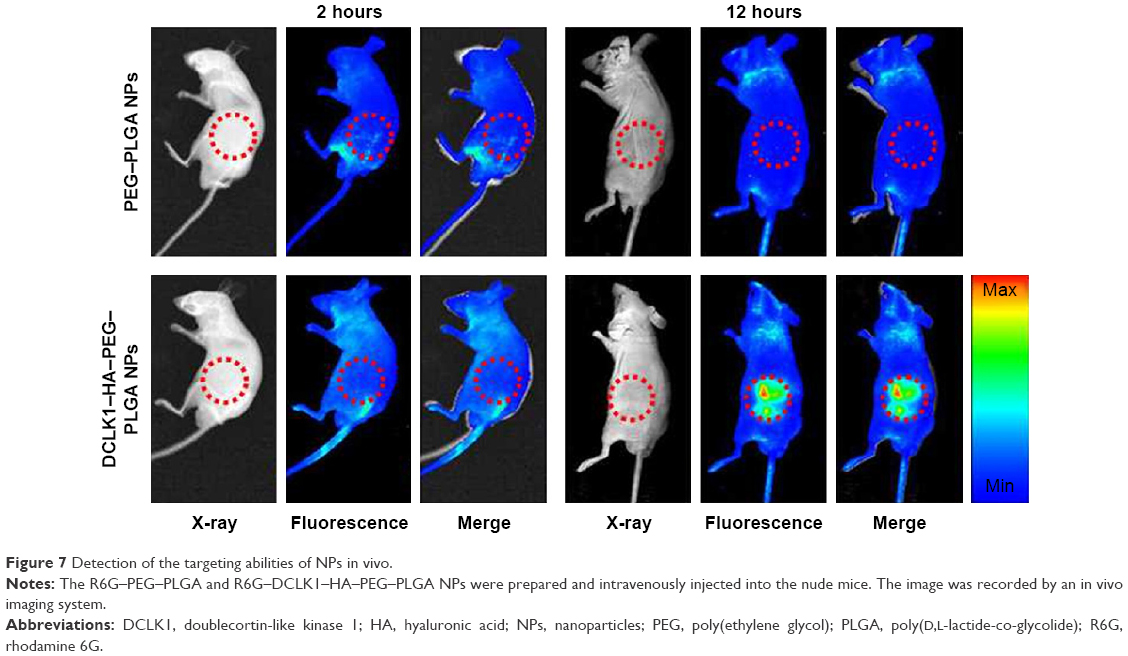 | Figure 7 Detection of the targeting abilities of NPs in vivo. |
Discussion
Previous research had revealed that CSCs account for the development of cancer resistance, rapid tumor growth, tumor recurrence, and migration/invasion.36,37 These findings suggest that CSCs can serve as therapeutic targets for the treatment of cancer progression and recurrence.37 Due to the EPR effect, nanocarriers have recently been used to deliver anti-CSC therapeutics to the tumor site to reduce the population of CSCs.38,39 However, the drugs delivered by NPs often fail to transport to the CSCs due to the heterogeneity of the tumor and the high drug resistance of CSCs, which would result in the development of many common cancer cells, while only a small amount of CSCs would be killed.28,39 Thus, developing CSC-targeted NPs may offer enhanced efficiency and specificity for drug delivery, ultimately eliminating CSCs in tumors. In this study, mouse 4T1 breast cancer cells were used as a model for NP-targeted research. Emerging evidence has supported the idea that 3D culture can promote cell reprogramming and tumor malignancy and that in the tumor microenvironment where CSCs and non-CSCs are balanced, 3D culture can promote the reprogramming of non-CSCs to CSCs.40,41 An alginate-based platform was developed in our previous study; this platform can efficiently select and enrich 4T1 breast CSCs by culturing 4T1 breast cancer cells in a 3D model without serum.29 The 4T1 breast CSCs exhibited tumor formation in a small number of Balb/c mice. Moreover, the 4T1 breast CSCs exhibited a high level of expression of CD44 and a low expression of CD24, which was consistent with other studies.29,42,43 Furthermore, it is worth noting that the 4T1 breast CSCs highly express DCLK1, which is a marker of a subpopulation of cancer cells exhibiting stem cell properties; however, these studies often aim at intestinal and pancreatic cancers.27,29,44,45
To develop a new therapeutic approach to target CSCs screened by our alginate–HA hydrogel system, a PLGA polymer was used to fabricate the NPs in our study due to its good biocompatibility, longstanding track record in biomedical applications, and well-documented utility for sustained drug release. As such, PLGA has been the center of focus in the development of drug-loaded NPs for cancer therapy.21,46,47 PEG was modified to the PLGA to obtain long-circulating NPs; this will be beneficial for the controlled release of drugs and site-specific drug delivery.46 From the aforementioned description, we know that the CSCs highly express CD44. It is generally accepted that HA is a recognized receptor for CD44 on the tumor’s cell surface; it mediates endocytosis. NPs that were synthesized in recent studies, which were used to selectively target and deliver drugs specifically to CSCs, tend to be coated with HA, and they are often accompanied by good success rates.20,21,48 Furthermore, previous studies showed that CD44 interacts with HA to reduce tumorigenicity and cancer cells’ migration ability, which enhances the sensitivity of cancer cells to the drug.49 Therefore, in our study, HA was conjugated to the PEG–PLGA NPs to improve CSC targeting, and our results demonstrated that HA–PEG–PLGA NPs are more likely to be taken up by the 4T1 breast CSCs than the other control groups. However, CD44 is widely expressed in various tissues; it has a short update cycle and it is easily saturated, which can reduce the specificity of drug delivery. During the study of CSC identification, abundant cell surface markers were discovered and their antibodies were often conjugated to the NPs to target CSCs. Recently, Ni et al50 developed PEG–PLGA NPs conjugated with the CD133 monoclonal antibody to target salinomycin delivery to CD133+ osteosarcoma CSCs; they obtained a positive result. Mesoporous silica NPs were modified with aptamer (Ap) against the epithelial cell adhesion molecule (EpCAM) to target drug delivery in the treatment of colon cancer.51 CD133 and EpCAM are also the most frequently documented CSC markers; however, growing evidence has demonstrated that CSCs share many similar markers with NSCs. The NPs conjugated with the CSCs markers’ antibody can target the delivery of anticancer drugs to eliminate CSCs, while they can also cause severe injury to normal tissues. Therefore, it is necessary to discover a marker that can distinguish CSCs from non-CSCs. DCLK1, which was recently identified as a CSC marker, was predicted to possess this ability. Therefore, in this study, the DCLK1 monoclonal antibody was conjugated to the HA–PEG–PLGA NPs to improve NP targeting. In summary, we reported DCLK1–HA–PEG–PLGA NPs as a new drug release system. HA combined with the double targets of the DCLK1 antibody allowed the NPs to attach to, and be consumed by, the CSCs. Our in vitro drug release tests showed that the particles are able to slowly release the drug, they highly target CSCs, and they also reduce the side effects of certain drugs. The in vivo results showed that DCLK1–HA–PEG–PLGA NPs can target the tumor more efficiently, thus representing a novel therapeutic approach for targeting CSCs.
Conclusion
In this study, DCLK1–HA–PEG–PLGA NPs were prepared to target CSCs. Based on the specific interaction between the DCLK1 monoclonal antibodies, the HA molecules coated on the PEG–PLGA NPs, the marker protein DCLK1, and the CD44 receptors that were overexpressed on the CSCs, this proposed method has been successfully utilized to target those breast CSCs that were selected by our alginate-based platform, which can be used to screen for various types of CSCs (in vitro) and tumors (in vivo). This unveils the great potential associated with this simple, convenient, and economical method that can be used to specifically target various types of CSCs, while also developing various cancer treatments in clinical applications.
Acknowledgments
This study is supported by the National Natural Science Foundation of China (grant 81361128005 and grant 50903024), the Fundamental Research Funds for the Central Universities (grant no HIT.MKSTISP.2016 37). We would like to acknowledge Mr Kai Guo and Mrs Chunfeng Li for the assistance with animal experiments. English-language editing of this article was provided by Journal Prep. All the authors have agreed on the article.
Disclosure
The authors report no conflicts of interest in this work.
References
Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8(10):755–768. | ||
Yan Y, Zuo X, Wei D. Concise review: emerging role of CD44 in cancer stem cells: a promising biomarker and therapeutic target. Stem Cells Transl Med. 2015;4(9):1033–1043. | ||
Chanmee T, Ontong P, Kimata K, Itano N. Key roles of hyaluronan and its CD44 receptor in the stemness and survival of cancer stem cells. Front Oncol. 2015;5:180. | ||
Jaggupilli A, Elkord E. Significance of CD44 and CD24 as cancer stem cell markers: an enduring ambiguity. Clin Dev Immunol. 2012;2012:708036. | ||
Plaks V, Kong N, Werb Z. The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell. 2015;16(3):225–238. | ||
Mukherjee N, Schwan JV, Fujita M, Norris DA, Shellman YG. Alternative treatments for melanoma: targeting BCL-2 family members to de-bulk and kill cancer stem cells. J Invest Dermatol. 2015;135(9):2155–2161. | ||
Cioffi M, Trabulo S, Hidalgo M, et al. Inhibition of CD47 effectively targets pancreatic cancer stem cells via dual mechanisms. Clin Cancer Res. 2015;21(10):2325–2337. | ||
Conley SJ, Gheordunescu E, Kakarala P, et al. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc Natl Acad Sci U S A. 2012;109(8):2784–2789. | ||
Hirsch HA, Iliopoulos D, Struhl K. Metformin inhibits the inflammatory response associated with cellular transformation and cancer stem cell growth. Proc Natl Acad Sci U S A. 2013;110(3):972–977. | ||
Uibo R, Laidmae I, Sawyer ES, et al. Soft materials to treat central nervous system injuries: evaluation of the suitability of non-mammalian fibrin gels. Biochim Biophys Acta. 2009;1793(5):924–930. | ||
Zuo ZQ, Chen KG, Yu XY, et al. Promoting tumor penetration of nanoparticles for cancer stem cell therapy by TGF-beta signaling pathway inhibition. Biomaterials. 2015;82:48–59. | ||
Haraguchi N, Ishii H, Mimori K, et al. CD13 is a therapeutic target in human liver cancer stem cells. J Clin Invest. 2010;120(9):3326–3339. | ||
Kaluzova M, Bouras A, Machaidze R, Hadjipanayis CG. Targeted therapy of glioblastoma stem-like cells and tumor non-stem cells using cetuximab-conjugated iron-oxide nanoparticles. Oncotarget. 2015;6(11):8788–8806. | ||
Wang CH, Chiou SH, Chou CP, Chen YC, Huang YJ, Peng CA. Photothermolysis of glioblastoma stem-like cells targeted by carbon nanotubes conjugated with CD133 monoclonal antibody. Nanomedicine. 2011;7(1):69–79. | ||
Tan GR, Feng SS, Leong DT. The reduction of anti-cancer drug antagonism by the spatial protection of drugs with PLA-TPGS nanoparticles. Biomaterials. 2014;35(9):3044–3051. | ||
Prabhu RH, Patravale VB, Joshi MD. Polymeric nanoparticles for targeted treatment in oncology: current insights. Int J Nanomedicine. 2015;10:1001–1018. | ||
Fang J, Nakamura H, Maeda H. The EPR effect: unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev. 2011;63(3):136–151. | ||
Yao HJ, Zhang YG, Sun L, Liu Y. The effect of hyaluronic acid functionalized carbon nanotubes loaded with salinomycin on gastric cancer stem cells. Biomaterials. 2014;35(33):9208–9223. | ||
Wang D, Huang J, Wang X, et al. The eradication of breast cancer cells and stem cells by 8-hydroxyquinoline-loaded hyaluronan modified mesoporous silica nanoparticle-supported lipid bilayers containing docetaxel. Biomaterials. 2013;34(31):7662–7673. | ||
Hu K, Zhou H, Liu Y, et al. Hyaluronic acid functional amphipathic and redox-responsive polymer particles for the co-delivery of doxorubicin and cyclopamine to eradicate breast cancer cells and cancer stem cells. Nanoscale. 2015;7(18):8607–8618. | ||
Wang H, Agarwal P, Zhao S, et al. Hyaluronic acid-decorated dual responsive nanoparticles of Pluronic F127, PLGA, and chitosan for targeted co-delivery of doxorubicin and irinotecan to eliminate cancer stem-like cells. Biomaterials. 2015;72:74–89. | ||
Flygare JA, Pillow TH, Aristoff P. Antibody-drug conjugates for the treatment of cancer. Chem Biol Drug Des. 2013;81(1):113–121. | ||
Shargh VH, Hondermarck H, Liang M. Antibody-targeted biodegradable nanoparticles for cancer therapy. Nanomedicine (Lond). 2016;11(1):63–79. | ||
Wang C, Bao C, Liang S, et al. HAI-178 antibody-conjugated fluorescent magnetic nanoparticles for targeted imaging and simultaneous therapy of gastric cancer. Nanoscale Res Lett. 2014;9(1):274. | ||
Swaminathan SK, Roger E, Toti U, Niu L, Ohlfest JR, Panyam J. CD133-targeted paclitaxel delivery inhibits local tumor recurrence in a mouse model of breast cancer. J Control Release. 2013;171(3):280–287. | ||
Li L, Neaves WB. Normal stem cells and cancer stem cells: the niche matters. Cancer Res. 2006;66(9):4553–4557. | ||
Nakanishi Y, Seno H, Fukuoka A, et al. Dclk1 distinguishes between tumor and normal stem cells in the intestine. Nat Genet. 2013;45(1):98–103. | ||
Metcalfe C, de Sauvage FJ. A tumor-specific stem cell. Nat Genet. 2013;45(1):7–9. | ||
Qiao SP, Zhao YF, Li CF, et al. An alginate-based platform for cancer stem cell research. Acta Biomater. 2016;37:83–92. | ||
Liu CW, Lin WJ. Polymeric nanoparticles conjugate a novel heptapeptide as an epidermal growth factor receptor-active targeting ligand for doxorubicin. Int J Nanomedicine. 2012;7:4749–4767. | ||
Yoon JJ, Nam YS, Kim JH, Park TG. Surface immobilization of galactose onto aliphatic biodegradable polymers for hepatocyte culture. Biotechnol Bioeng. 2002;78(1):1–10. | ||
Senthilkumar M, Mishra P, Jain NK. Long circulating PEGylated poly(D,L-lactide-co-glycolide) nanoparticulate delivery of Docetaxel to solid tumors. J Drug Target. 2008;16(5):424–435. | ||
Abdel-Latif A, Zuba-Surma EK, Case J, et al. TGF-beta1 enhances cardiomyogenic differentiation of skeletal muscle-derived adult primitive cells. Basic Res Cardiol. 2008;103(6):514–524. | ||
Fang R, Qiao S, Liu Y, et al. Sustained co-delivery of BIO and IGF-1 by a novel hybrid hydrogel system to stimulate endogenous cardiac repair in myocardial infarcted rat hearts. Int J Nanomedicine. 2015;10:4691–4703. | ||
Gutiérrez-Praena D, Pichardo S, Sánchez E, Grilo A, Cameán AM, Jos A. Influence of carboxylic acid functionalization on the cytotoxic effects induced by single wall carbon nanotubes on human endothelial cells (HUVEC). Toxicol In Vitro. 2011;25(8):1883–1888. | ||
Alison MR, Lin WR, Lim SM, Nicholson LJ. Cancer stem cells: in the line of fire. Cancer Treat Rev. 2012;38(6):589–598. | ||
Alison MR, Lim SM, Nicholson LJ. Cancer stem cells: problems for therapy? J Pathol. 2011;223(2):147–161. | ||
Malhi S, Gu X. Nanocarrier-mediated drugs targeting cancer stem cells: an emerging delivery approach. Expert Opin Drug Deliv. 2015;12(7):1177–1201. | ||
Shen S, Xia JX, Wang J. Nanomedicine-mediated cancer stem cell therapy. Biomaterials. 2016;74:1–18. | ||
Chaudhuri O, Koshy ST, Branco DCC, et al. Extracellular matrix stiffness and composition jointly regulate the induction of malignant phenotypes in mammary epithelium. Nat Mater. 2014;13(10):970–978. | ||
Han J, Chen L, Luo G, Dai B, Wang X, Dai J. Three-dimensional culture may promote cell reprogramming. Organogenesis. 2013;9(2):118–120. | ||
Hiraga T, Ito S, Nakamura H. EpCAM expression in breast cancer cells is associated with enhanced bone metastasis formation. Int J Cancer. 2016;138(7):1698–1708. | ||
Yang X, Sarvestani SK, Moeinzadeh S, He X, Jabbari E. Three-dimensional-engineered matrix to study cancer stem cells and tumorsphere formation: effect of matrix modulus. Tissue Eng Part A. 2013;19(5–6):669–684. | ||
Weygant N, Qu D, May R, et al. DCLK1 is a broadly dysregulated target against epithelial-mesenchymal transition, focal adhesion, and stemness in clear cell renal carcinoma. Oncotarget. 2015;6(4):2193–2205. | ||
Sureban SM, Qu D, Houchen CW. Regulation of miRNAs by agents targeting the tumor stem cell markers DCLK1, MSI1, LGR5, and BMI1. Curr Pharmacol Rep. 2015;1(4):217–222. | ||
Li Y, Pei Y, Zhang X, et al. PEGylated PLGA nanoparticles as protein carriers: synthesis, preparation and biodistribution in rats. J Control Release. 2001;71(2):203–211. | ||
Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Préat V. PLGA-based nanoparticles: an overview of biomedical applications. J Control Release. 2012;161(2):505–522. | ||
Zhong Y, Zhang J, Cheng R, et al. Reversibly crosslinked hyaluronic acid nanoparticles for active targeting and intelligent delivery of doxorubicin to drug resistant CD44+ human breast tumor xenografts. J Control Release. 2015;205:144–154. | ||
Zoller M. CD44: can a cancer-initiating cell profit from an abundantly expressed molecule? Nat Rev Cancer. 2011;11(4):254–267. | ||
Ni M, Xiong M, Zhang X, et al. Poly(lactic-co-glycolic acid) nanoparticles conjugated with CD133 aptamers for targeted salinomycin delivery to CD133+ osteosarcoma cancer stem cells. Int J Nanomedicine. 2015;10:2537–2554. | ||
Xie X, Li F, Zhang H, et al. EpCAM aptamer-functionalized mesoporous silica nanoparticles for efficient colon cancer cell-targeted drug delivery. Eur J Pharm Sci. 2015;83:28–35. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.