")
Back to Journals » International Journal of Nanomedicine » Volume 17
A Novel Dextran-Based Dual Drug Conjugate Targeted Tumors with High Biodistribution Ratio of Tumors to Normal Tissues
Authors Liu J, Zhang N, Wu J, Dong P, Lv H, Wang Q, Wang S, Yang H, Wang S, Li X, Hu J, Wang A, Li DJ, Shi Y
Received 29 June 2022
Accepted for publication 21 September 2022
Published 17 October 2022 Volume 2022:17 Pages 4895—4910
DOI https://doi.org/10.2147/IJN.S379758
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yan Shen
Jiaojiao Liu,1 Naining Zhang,1 Jiaan Wu,1 Peng Dong,1 Hongshuai Lv,1 Qi Wang,1 Shenxu Wang,1 Haotong Yang,1 Si Wang,2 Xiaohai Li,2 Jinghua Hu,2 Anny Wang,2 Daisy J Li,2 Yikang Shi1
1National Glycoengineering Research Center, Shandong Key Laboratory of Carbohydrate Chemistry and Glycobiology, NMPA Key Laboratory for Quality Research and Evaluation of Carbohydrate Based Medicine, Shandong University, Qingdao, Shandong, 266237, People’s Republic of China; 2Santolecan Pharmaceuticals LLC, Jupiter, Florida, 33458, USA
Correspondence: Yikang Shi, Shandong University, 72 Binhai Road, Jimo, Qingdao, 266237, People’s Republic of China, Tel/Fax +86-532-5863-1418, Email [email protected]
Purpose: Most chemotherapeutic agents possess poor water solubility and show more significant accumulations in normal tissues than in tumor tissues, resulting in serious side effects. To this end, a novel dextran-based dual drug delivery system with high biodistribution ratio of tumors to normal tissues was developed.
Methods: A bi-functionalized dextran was developed, and several negatively charged dextran-based dual conjugates containing two different types of drugs, docetaxel and docosahexaenoic acid (DTX and DHA, respectively) were synthesized. The structures of these conjugates were characterized using nuclear magnetic resonance and liquid chromatography/mass spectrometry (1H-NMR and LC/MS, respectively) analysis. Cell growth inhibition, apoptosis, cell cycle distribution, and cellular uptake were measured in vitro. Drug biodistribution and pharmacokinetics were investigated in mice bearing 4T1 tumors using LC/MS analysis. Drug biodistribution was also explored by in vivo imaging. The effects of these conjugates on tumor growth were evaluated in three mice models.
Results: The dextran–docosahexaenoic acid (DHA)– docetaxel (DTX) conjugates caused a significant enhancement of DTX water solubility and improvement in pharmacokinetic characteristics. The optimized dextran–DHA–DTX conjugate A treatment produced a 2.1- to 15.5-fold increase in intra-tumoral DTX amounts for up to 96 h compared to parent DTX treatment. Meanwhile, the concentrations of DTX released from conjugate A in normal tissues were much lower than those of the parent DTX. This study demonstrated that DHA could lead to an improvement in the efficacy of the conjugates and that the conjugate with the shortest linker displayed more activity than conjugates with longer linkers. Moreover, conjugate A completely eradicated all MCF-7 xenograft tumors without causing any obvious side effects and totally outperformed both the conventional DTX formulation and Abraxane in mice.
Conclusion: These dextran-based dual drug conjugates may represent an innovative tumor targeting drug delivery system that can selectively deliver anticancer agents to tumors.
Keywords: docetaxel, dextran, docosahexaenoic acid, nanoparticle, tumor, chemotherapy
Introduction
Chemotherapy is one of the three traditional strategies for treating cancer. However, many chemotherapeutic agents have limited effectiveness due to high toxicity to normal healthy tissues.1 Many small molecule tyrosine kinase inhibitors have exhibited nonspecific biodistribution. Many studies have confirmed that epidermal growth factor receptor (EGFR) inhibitors, such as erlotinib, gefitinib, and others accumulate much less in tumors than in normal tissues.2–6 More than 30 monoclonal antibodies have been approved for targeting cancer therapy; however, a previous report indicated that four antibodies used clinically showed a similar biodistribution pattern with higher uptake in the livers, spleens, and kidneys than in the tumors.7 The immune checkpoint inhibitor, Pembrolizumab, which targets the programmed cell death 1 (PD-1) pathway shows impressive antitumor effects. However, serious immune-related toxicity has been reported due to higher concentration of pembrolizumab in livers and spleens than in tumors.8,9 Many nanoparticles, such as liposomes and micelles, which are conjugated to drugs, exert their anticancer efficacy through active and passive tumor targeting.10,11 Unfortunately, although most nano-drug formulations do deliver more drugs to tumor sites, they also accumulate in the reticuloendothelial systems, such as livers and spleens. For example, the cyclodextrin-based camptothecin conjugate, CRLX101, was tested in multiple Phase I and II studies, and more camptothecin was found in livers than in tumors.12 Therefore, an urgent need to develop drugs that preferentially enter tumors more than normal tissues is present.
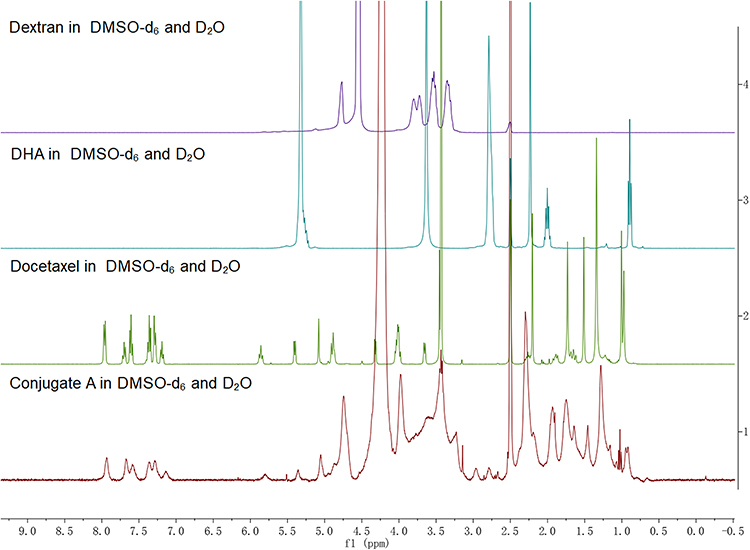 |
Figure 1 1H spectra of dextran, DHA, DTX and the conjugate A. |
Docetaxel (DTX) is one of the most important anticancer drugs that is widely used for cancer treatment. However, due to its poor water solubility, the commercial formulation of DTX in which Tween 80 and ethanol are used as solubility enhancers causes serious adverse effects. Moreover, DTX lacks tumor targeting and results in serious systemic toxicities.13,14 In recent years, several strategies have been utilized to improve the solubility and tumor targeting properties of DTX, including chemical conjugation to hydrophilic polymers in addition to physical encapsulation in liposomes, polymeric micelles, and dendrimers.15–17 Many of these new DTX-based formulations exhibit significantly enhanced antitumor efficacies when compared with conventional DTX in preclinical studies. A few of these formulations, such as BIND-014, CRXL301 and CPC634, have entered clinical trials; however, none of them have been approved for the clinical market yet.10,15,18,19
Dextran is a natural and excellent biomaterial for drug delivery application due to its water solubility, biocompatibility, biodegradability, and non-immunogenicity.20,21 As a drug delivery carrier, some dextran-based nanoparticles have shown potent therapeutic efficacy in cancer treatment.21–24 However, all of these physically loaded and chemically conjugated dextran-based nanoparticles were found to distribute less active drugs into tumors than into major normal tissues.23–28 The area under the curve (AUC) value of free paclitaxel in livers was 3.1-fold greater than in tumors after a paclitaxel-carboxymethyl dextran conjugate (AZ10992) was injected into mice carrying colon 26 cancer cells.24 Intra-tumoral active drug was less than that in livers at 12 h post-injection of a dextran-based conjugate Dex–g–(DOX+BTZ)/cRGD, which was coupled with doxorubicin (DOX), bortezomib (BTZ), and a targeted peptide (cRGD).25 In fact, there are many challenges, such as drug loading contents, stability, linkers, quality control, and the purity of polysaccharide drug conjugates, in the development of polysaccharide-based nano-drug formulations. All factors must be considered and optimized when developing polysaccharide-based nanoparticles.
Docosahexaenoic acid (DHA), an omega-3 fatty acid, has been reported by many studies to inhibit tumor growth either alone or in combination with chemotherapeutic agents.29,30 Existing evidence reveals that DHA can also sensitize tumor cells to chemotherapy, while not increasing their toxicity on normal tissues.31–34 DHA has been shown to improve the treatment efficacy of DTX.35,36 Furthermore, DHA–Paclitaxel (Taxoprexin) has exhibited enhanced anticancer activity with favorable efficacy and minimal toxicity profiles in clinical trials.37,38 DHA has also been loaded into or conjugated with polymers to construct nanoparticles for cancer treatment.39 DHA may be enriched in tumor cells because rapidly growing tumor cells need more energy and preferentially uptake unsaturated fatty acids.40,41 Moreover, G-protein-coupled receptors 40 and 120 (GPR40 and 120, respectively) are mainly overexpressed in many types of cancer cells in which they function as DHA receptors.42,43 Therefore, DHA may actively target tumor cells through recognition of GPR40 and 120 receptors.
In this study, the synthesis of four dextran-based dual conjugates containing two different types of drugs, DTX and DHA, in addition to different linkers is reported. After dextran–DHA–DTX conjugates were obtained, the pharmacokinetics, biodistribution, and antitumor activity were studied. Results indicate that the conjugate with the shortest linker selectively accumulates in tumors and completely eliminates all MCF-7 xenograft tumors in nude mice. This technology involving the construction of dextran–DHA–DTX conjugates exhibits a promising strategy for providing a new potential DTX formation for cancer treatment.
Materials and Methods
Materials
Anhydrous docetaxel was purchased from Wuxi Zishan Pharmaceutical Co., Ltd. Docetaxel injection Duopafei® was a commercial production produced by Qilu Pharmaceutical Co., LTD. in China. Dextran (Mr=100,000) was purchased from Sigma-Aldrich, DHA was purchased from Aladdin (Shanghai), Boc-Lys(N3)-OH, trichloromethyl chloroformate, 1-hydroxybenzotriazole (HOBt), o-benzotriazole-N,N,N’,N’-tetramethyluronium hexafluophosphate (HBTU), ultra-dry N,N-dimethylformamide (DMF), ultra-dry tetrahydrofuran (THF), ultra-dry dichloromethane (DCM), 1 - (3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDCI), 4-dimethylaminopyridine (DMAP), N-hydroxysuccinimide (NHS), sodium ascorbate, N6-BOC-L-lysine, glycine methyl ester hydrochloride, BOC-glycine, allyl chloroformate (Alloc-Cl), and tetrabutylammonium fluoride (TBAF) were purchased from Beijing Innokai Technology Co., Ltd; other chemical reagents were bought from Sinopharm Chemical Reagent Co., Ltd.
Characterization of Conjugates Dextran-DHA-DTX
The morphologies of the dextran–DHA–DTX conjugates were observed under the transmission electron microscopy ([TEM] JEM-1200EX II, Japan). A sample (2 mg) was dissolved in 1 mL distilled water, stained with a 2% aqueous solution of phosphotungstic acid, and allowed to dry before being observed under the TEM. Solutions of dextran–DHA–DTX (1 mg/mL) were prepared with deionized water. The size distribution and zeta potential of conjugates in the resulting solutions were determined using a Malvern particle size analyzer (ZEN3700, UK).
Determination of DTX Loading Contents Through Alkaline Hydrolysis of Conjugates
For quantifying the DTX loading content in these conjugates, measurements of the release of C-13 side chains of DTX by alkaline hydrolysis were obtained. The procedure is described briefly. Samples were incubated with 0.5 mol/L sodium hydroxide solution (methanol:water = 1:1) for 3 h at room temperature with shaking at 200 rpm. The sample was then adjusted to pH 6.0 by adding glacial acetic acid and then used for high-performance liquid chromatography/mass spectroscopy (HPLC/MS) analysis. The peak area of the C-13 side chains was recorded to calculate the DTX content. DTX at varying concentrations was hydrolyzed in 0.5 mol/L sodium hydroxide solution using a similar procedure after which a standard curve was prepared by the integrated peak area of the C-13 side chain corresponding to the amount of DTX. The amount of DTX in the conjugates was calculated depending on the standard curve. These dextran–DHA–DTX conjugates completely released the C-13 side chains under alkaline conditions, and the C-13 side chains were stable for at least 6 h. Samples was analyzed on an Agilent 7890A HPLC and mass spectrometer SCIEX TripleQuad 5500 at 254 nm wavelength using an Agilent C18 column (3.5 µm, 2.1×100 mm).
Cell Lines and Cell Culture
The lung cancer cell-line NCI-H460 (HTB-177), breast cancer cell-line MCF-7 (HTB-22), and mouse breast cancer cells 4T1 (CRL-2539) were purchased from the Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences. All cell lines were maintained in a humidified atmosphere containing 5% CO2 at 37 °C in RPMI-1640 medium supplemented with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin.
Sulforhodamine B (SRB) Assay
Cell growth inhibition was determined using the SRB assay. Briefly, the cells were incubated in 96-well microtiter plates for 24 h. Following the addition of dextran–DHA–DTX conjugates, the plates were incubated at 37 °C for an additional 72 h. The culture medium was then discarded, and the cells were fixed in situ by the gentle addition of 100 μL of cold 10% (w/v) trichloroacetic acid and incubated for 60 min at 4 °C. The supernatant was discarded, and the plates were washed five times with tap water and air dried. SRB solution (100 μL) at 0.4% (w/v) in 1% acetic acid was added and plates were incubated for 20 min at room temperature. After staining, unbound dye was removed by washing five times with 1% acetic acid after which the plates were air dried. Bound stain was dissolved with 10 mM Tris (pH 10.5), and the absorbance was read at 515 nm on a Bio-Rad 550 ELISA microplate reader. Cell viability was calculated as a percentage of the control group. The IC50 of each group was calculated using Graph Pad Prism 7.0 software.
Cell Cycle Analysis by Flow Cytometry
H460 and MCF-7 cells seeded on the 6-well plates were treated with dextran–DHA–DTX conjugates for the indicated time. At the end of the incubation, cells were detached with 0.05% trypsin, washed twice with cold phosphate-buffered saline (PBS), and then fixed with 70% cold ethanol at −20 °C. Prior to analysis, the samples were washed twice in PBS and resuspended in a solution of 50 mg/mL propidium iodide (PI) and RNase A (0.5 mg/mL) in PBS for 30 min in the dark. Data collected from each 10,000-cell sample was analyzed by Flow cytometry (Becton-Dickinson Co., USA).
Annexin V-FITC/Propidium Iodide Assay
An annexin V-FITC/PI binding assay was used to determine the percentage of apoptotic cells. Following the recommended protocols of the Annexin V-FITC kit (BD Pharmingen, USA), the cells were seeded at 4×105 cells/mL per well in 6-well plates. After treatment with dextran–DHA–DTX conjugates, the cells were harvested and washed twice with ice-cold PBS and resuspended in 100 μL of binding buffer. A total of 5 μL of Annexin V-FITC and 10 μL of PI were added, and the mixture was incubated for 30 min in the dark. Finally, 400 μL of binding buffer was added to the cells, and the mixture was analyzed with a flow cytometer.
In vivo and ex vivo Imaging to Test Tumor Targeting
To study the targeting properties, the fluorescent dye, Cy7.5, was covalently coupled to form a conjugated dextran–DHA–Cy7.5. The tumor model was established by subcutaneous injection of breast cancer cells 4T1 into the lower right flank of BALB/C mice. When the tumor volume reached approximately 200 mm3, mice were intravenously injected with Cy7.5 and dextran–DHA–Cy7.5 (30 μg/kg) via the tail vein. Mice were anesthetized and examined using the imaging system at different time points after administration. Fluorescence intensity was captured using the Xenogen IVIS Lumina system (Caliper Life Sciences, USA). For ex vivo imaging, the mice were sacrificed, and the major organs were obtained from mice after conjugate administration at different times. Results were analyzed using Living Image 4.1 software.
In vitro Release of DTX from Conjugates Dextran-DHA-DTX in Plasma
To investigate the profile of DTX release from conjugates, conjugates were incubated with rat plasma and the concentration of released DTX was measured. Briefly, 100 μL of each conjugate (10 mg/mL in PBS) was mixed with 900 μL rat plasma and then incubated at 37 °C with shaking. At predetermined time points, the incubation was stopped by adding protein precipitation solution (acetonitrile: methanol = 1:1) into samples. The mixture was rigorously vortexed and put on ice for 5 min after which it was centrifuged at 10,000 rpm for 15 min. The supernatant was filtered with a 0.22 μm membrane filter and analyzed by reverse phase (RP)-HPLC (Agilent 1220 Infinity II) with a C18 column.
Determination of Released DTX and Total DTX Contents in Plasma and Tissue
In blood and tissues, some of the DTX was released from dextran–DHA–DTX; however, some DTX remained conjugation with dextran. In this study, released DTX and total DTX (free and conjugated DTX) were examined.
For determination of DTX release, tissue samples or blood samples were mixed with PBS (pH 7.4) and homogenized on ice. The mixture was added with three volumes of the internal standard paclitaxel containing the solvent methanol:acetonitrile (1:1) after which the samples were centrifuged at 12,000 rpm for 15 min. The supernatant was filtered with a 0.22 μm membrane filter and subject to HPLC/MS quantification to determine free DTX (released DTX from conjugates).
For quantifying the total DTX content in blood and tissues, alkaline hydrolysis of these conjugates was used. Tissue or blood samples were mixed with PBS (pH 7.4) and homogenized on ice. The mixture was mixed with the internal standard, Boc-L-Phenylalanine, containing 3 times the volume of methanol:acetonitrile (1:1) for extraction, and samples were centrifuged at 12,000 rpm about 15 min. The supernatant was added to a sodium hydroxide aqueous solution at a final concentration of 50 mg/mL and was incubated at room temperature with shaking at 200 rpm. After incubation for 3 h, the sample was adjusted to pH 6.0 by adding glacial acetic acid and then centrifugated at 12,000 rpm. The supernatant was used for HPLC/MS analysis to test the total DTX including released DTX and dextran-conjugated DTX.
Released or total DTX was detected with HPLC/MS. HPLC separation of samples was carried out on Agilent 7890 system using an Agilent C18 column (3.5 µm, 2.1×100 mm). Detection was done using a mass spectrometer SCIEX TripleQuad5500 operated in the MRM mode under positive ion electrospray conditions for released DTX determination and under negative ion electrospray conditions for total DTX determination.
Determination of Released DTX and Total DTX Content in Cancer Cells
To quantify the intracellular amounts of released and dextran-coupled DTX after cancer cells were exposed to conjugates dextran–DHA–DTX, MCF-7 cells were plated in 10 cm dishes and cultured for 24 h after which parent DTX and the conjugate A were added to the medium at the final concentration of 200 ng/mL equivalent to DTX. After 0, 1, 3, 6, 12, 24, 48, and 72 h the cells were collected and washed with cold PBS.
For released DTX analysis, the cells were mixed with 1 mL internal standard (paclitaxel) containing a solution of acetonitrile:PBS (1:1). The mixture was sonicated for 2 min under probe sonication in an ice bath. Cell debris was removed by 1000 rpm centrifugation, and the supernatant was further filtered with a 0.22 μm membrane, and HPLC/MS analysis was done to determine the amount of released DTX.
For total DTX analysis, the cells were mixed with 1 mL of acetonitrile:PBS (1:1) containing the internal standard, Boc-L-Phenylalanine. The mixture was sonicated for 2 min under probe sonication in an ice bath. Cell debris was removed by 1000 rpm centrifugation, and the supernatant was further filtered using a 0.22 μm membrane. After adding sodium hydroxide aqueous solution at a final concentration of 50 mg/mL, the mixture was incubated at room temperature with shaking at 200 rpm for 5 h. The sample was then adjusted to pH 6.0 by adding glacial acetic acid after which it was centrifugated at 12,000 rpm. The supernatant was used for HPLC/MS analysis to evaluate the total amount of DTX.
Pharmacokinetic Analysis
All experiments of the animals in this study were approved by the Laboratory Animal Ethical and Welfare Committee of Shandong University and were performed following the guidelines and regulations of the Laboratory Animal Ethical and Welfare Committee of Shandong University. BALB/C mice were randomly divided into several groups (n = 5 in each group) and were intravenously injected with DTX and the dextran–DHA–DTX conjugates at a dose of 12 mg/kg (equivalent to DTX for conjugates). Blood (0.2 mL) was then collected from the inner canthus of mice at the predetermined time, immediately transferred to a heparinized, and centrifuges for 10 min at 3000 rpm. The time points were 30 min and 1, 2, 4, 6, 12, 24, and 48 h. The supernatants were tested for DTX amount following the procedure described in the section of determination of released DTX and total DTX content in plasma and tissue.
Biodistribution Analysis
BALB/C mice bearing breast cancer cells 4T1 were used to evaluate biodistribution of the dextran–DHA–DTX conjugate. When the tumor volume reached approximately 200 mm3, the mice were intravenously injected with parent DTX or conjugate A at 12 mg/kg via the tail vein. Mice were sacrificed, and major organs were excised and homogenized on ice for determination of released and total DTX according to the procedure in the section of determination of released DTX and total DTX content in plasma and tissues.
Inhibition of Tumor Growth in vivo
Three mice models were established in this study: (1) nude mice bearing human lung cancer cells H460, (2) nude mice bearing human breast cancer cells MCF-7, and (3) BALB/C mice bearing breast cancer cells 4T1. The animals were housed under pathogen-free conditions. Female BALB/c (nu/nu) mice (20 ± 2 g) were purchased from the Animal Center of the China Academy of Medical Sciences (Beijing, China). BALB/c mice (20 ± 2 g) were purchased from Shandong University. Cancer cells H460 (5.0 × 106), MCF-7 (5.0 × 106), and 4T1 (5.0 × 105) in 100 μL PBS were inoculated subcutaneously into the lower right flank of mice. When the tumors reached 100–150 mm3, the mice were divided randomly into different groups (n = 6 in each group). The control group received PBS only. The treated groups were intravenously injected once a week with parent DTX or dextran–DHA–DTX conjugate at indicated doses via tail vein. The diameter of the tumor and the body weights were measured twice a week. Tumor volume was calculated with the following formula: v = ab2/2 in which a and b are the long and perpendicular short diameter of the tumors, respectively. The major organs were isolated after mice were sacrificed at the end of the experiment.
Histological Examination of Tissues
Tissues obtained from mice were fixed with 10% formalin and then embedded in paraffin after which 5 μm sections were cut and stained with hematoxylin and eosin (H&E) staining.
Hematological Analysis
For hematological analysis, bloods were taken from different groups of 4T1-bearing mice for serum biochemistry experiments on day 14 after treatment had been administered twice.
Statistical Analysis
A Student’s t-test was used to analyze the statistical comparisons between two groups, and differences were considered statistically significant when P < 0.05. All results were reported as mean ± standard deviation (SD).
Results and Discussion
Chemical Synthesis of the Conjugates
Previous studies revealed that the negative charged carboxymethyl dextran with molecular weight more than 80 kDa and a degree of substitution within 0.2 to 0.6 resulted in a much better treatment outcome.24,44–46 Hence, 100 kDa dextran was selected, and a degree of substitution of 0.5 to 0.6 for the modified dextran was used in this study. Dextran was bi-functionalized for the first time with two different types of functional groups concurrently: (1) glutamic acid and (2) azido-lysine (Scheme 1 and Scheme S1, Supplementary Material). Glutamic acid with two carboxyl group endowed the dextran carrier with negative charges, which prolonged the circulation time in blood stream and caused a dramatic reduction in drug distribution in normal tissues.44,45 Azido-lysine provided the azide for conjugation with two drug molecules, DTX and DHA. Second, the 7-OH and 10-OH positions of DTX were protected by addition of Alloc-Cl (Scheme S2, Supplementary Material). It was previously reported that modification of PTX at the 7-OH position could cause a dramatic decrease in the antitumor efficacy of the conjugates.47 The modified DTX allowed linkers to specifically react with the 2´-OH of the DTX. Third, DHA was modified with a linker composed of lysine and glycine with different number and position in addition to two ends: (1) carboxylic group acid end and (2) alkyne end. The carboxylic acid end of DHA was further specifically attached to 2´- OH position of DTX to produce three different DHA-DTX-linkers (Scheme S3–Scheme S5, Supplementary Material). Meanwhile, non-DHA DTX-linker was also synthesized (Scheme S6, Supplementary Material). Finally, the alkyne end of the DHA–DTX and DTX linkers were coupled to bi-functionalized dextran under mild click chemistry conditions to yield the desired conjugates, respectively (Scheme S7–Scheme S10, Supplementary Material). In this study, we synthesized three dextran-DHA-DTX conjugates (A, B and C) and one non-DHA-coupled dextran-DTX conjugate D, respectively (Scheme 1). The linkers of the conjugates B and C have one more glycine than the conjugate A. The difference between conjugates B and C is the difference position of a glycine in the linker. Conjugate D is identical to conjugate A except for the lack of a DHA.
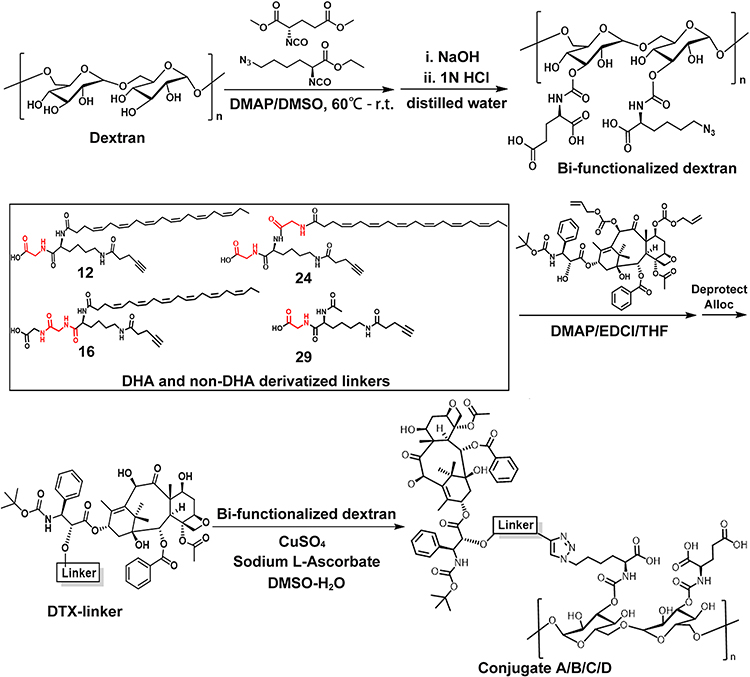 |
Scheme 1 Schematic diagram of the synthetic route of the conjugates dextran-DHA-DTX and dextran-DTX. |
Characterization of Conjugates
The structures of the dextran-based conjugates were confirmed by comparing the 1H spectra of dextran, DTX, and DHA (Figure 1 and Figures S1–S4, Supplementary Materials), and their purity was detected by gel permeation chromatography (GPC)–HPLC and confirmed to be ≥98% (Figure S5, Supplementary Material). The DTX and DHA contents of the dextran-based conjugates were quantified via the HPLC/MS quantitative method after alkaline hydrolysis of DTX of the conjugates. The drug contents of DTX in all four conjugates ranged from 17.0±1.8 to 19.7±2.1% (w/w) as shown in Table S1 (Supplementary Material). The nanoparticle shape of the dextran-based conjugates was examined by TEM, and each of the four conjugates had a spherical shape with the diameters ranging from 76.9 ± 0.4 nm to 93.0 ± 4.7 nm (Figure 2A and B, Table S1, Supplementary Material). No differences in the surface charge of all four conjugates were found as all then were around −25 mV (Figure 2C and Table S1, Supplementary Material). All four conjugates could easily be dissolved in water to yield a light-yellow solution (Figure 2D). The water solubility of the four conjugates ranged from 32.8 to 42.5 mg/mL, all of which were dramatically enhanced compared to parent DTX (Table S1, Supplementary Material). All four conjugates gradually released DTX upon incubation with plasma, while the parent DTX was released rapidly and fully at the first time point, indicating that dextran–DHA–DTX conjugates released DTX in a sustainable manner (Figure 2E).
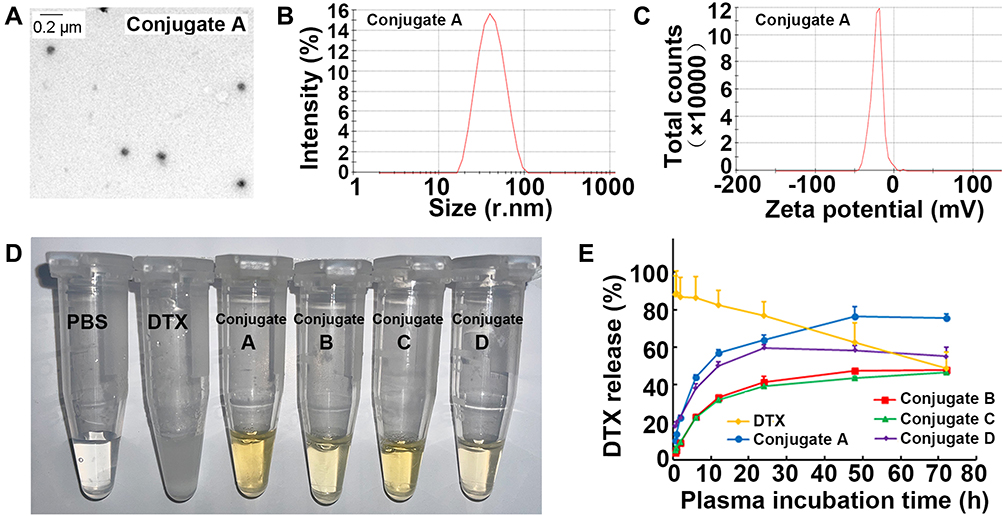 |
Figure 2 Characterization of conjugates dextran-DHA-DTX. (A) TEM images of the conjugate A (1 mg/mL). (B) Size distribution of the conjugate A. (C) Surface charge of the conjugate A. (D) Image of four conjugates dissolved in PBS. (E) DTX release from four conjugates in rat plasma. Data were presented as mean±SD (n = 3). |
In vitro Cytotoxicity Evaluation and Cellular Uptake of the Conjugate A
Lung cancer and breast cancer cells (H460 and MCF-7, respectively) were used to test the effects of all four conjugates on cell viability. No significant differences in cell viability and IC50 values were observed between parent DTX and conjugates A, B, and C in both types of cancer cells (Figures 3A and S6, Supplementary Material). When compared with parent DTX, exposure of the four conjugates for 72 h did not result in significant differences in apoptosis in either H460 or MCF-7 cells (Figures 3B and S7, Supplementary Material). As with parent DTX, all four conjugates also caused the arrest of cancer cells at the cell cycle G2/M phase (Figures 3C and S8, Supplementary Material). The above-described results indicated that all conjugates could inhibit cell growth by inducing apoptosis and cell cycle arrest.
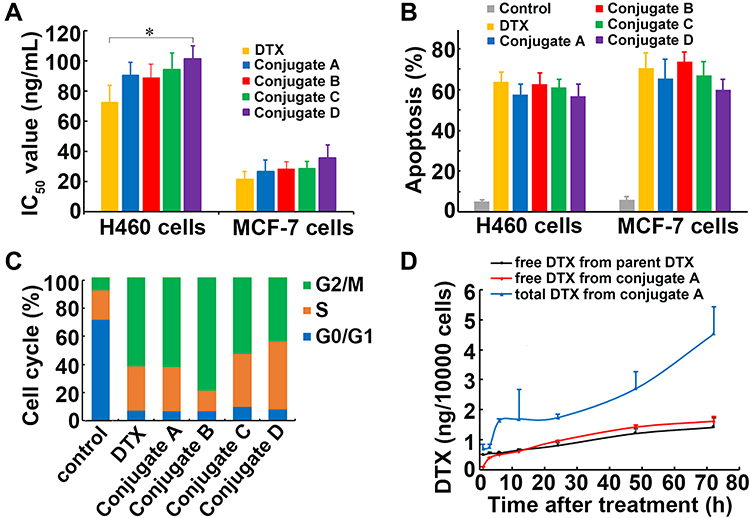 |
Figure 3 The effects of dextran-DHA-DTX conjugates on cell growth and cellular uptake. (A) IC50 values of four conjugates in cancer cells upon treatment for 72 h. (B) Conjugates induced apoptosis in cancer cells upon treatment for 72 h. (C) Conjugates induced cell cycle arrest in MCF-7 cells upon treatment for 72 h. (D) Amount of released DTX and total DTX in MCF-7 cells after incubation with parent DTX and the conjugate A, respectively. *P<0.05. Data were presented as mean±SD. |
Cellular uptake of dextran-DHA-DTX conjugate A was investigated in MCF-7 cells. As shown in Figure 3D, the amount of released DTX gradually increased and reached a maximum at 72 h after MCF-7 cells were exposed to either parent DTX or the conjugate A. The concentrations of released DTX from the conjugate A were significantly lower than that from parent DTX after 1 and 3 h of incubation (P < 0.001), indicating that conjugate A released DTX at a slower rate than did the parent DTX. Although free DTX released from the conjugate A was slightly higher than that from parent DTX, no significant difference was observed between the treatment of parent DTX and conjugate A from 24 to 96 h incubation. Total intracellular DTX contents were significantly higher than released DTX after MCF-7 cells were incubated with conjugate A for all indicated time points, indicating that conjugate A continuously released DTX in cancer cells (Figure 3D).
In vivo Imaging to Confirm Tumor Targeting
The conjugate dextran-DHA-Cy7.5 was synthesized (Scheme S11, Supplementary Material) and was intravenously injected into mice bearing 4T1 cells to confirm conjugates dextran–DHA–DTX can specifically target tumor. As shown in Figures 4A and S9 (Supplementary Material), the fluorescence intensity of dextran–DHA–Cy7.5 in the tumor gradually increased, reached a maximum at 72 h, and was still strong up to 96 h. The fluorescent signal of dextran–DHA–Cy7.5 in the tumors was dramatically higher than that of Cy7.5 throughout the 96-h period (P < 0.001). Individual Cy7.5 administration resulted in much more fluorescence accumulation in the liver than the administration of dextran–DHA–Cy7.5 (P < 0.001, Figures 4B and S9, Supplementary Material). These results suggested that the dextran-based dual conjugates could selectively accumulate in tumor tissues and cause a dramatic reduction of its distribution in normal tissues.
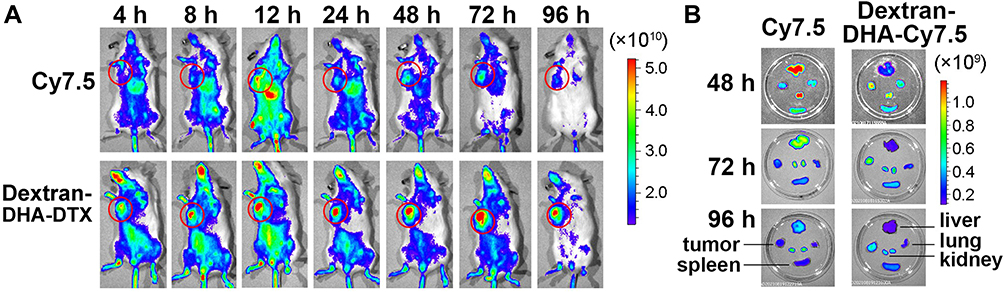 |
Figure 4 In vivo fluorescence imaging of mice and major organs. (A) In vivo fluorescence imaging of 4T1 tumor-bearing mice after intravenous administration of Cy7.5 and dextran-DHA-Cy7.5. The tumor foci were marked with red circles. (B) Fluorescence imaging of tumor and major organs from mice treated with Cy7.5 and dextran-DHA-Cy7.5, respectively. |
In vivo Antitumor Efficacy of Conjugates
The antitumor effects of these conjugates were assessed in three mice models. Based on our pilot experiments, 12 mg/kg DTX treatment caused mice to die, and 8 mg/kg DTX dosage was then selected as intravenous administration dosage in nude mice models. Parent DTX, Abraxane, and conjugates were intravenously injected via the tail on days 0 and 7 in nude mice bearing lung cancer cells H460. As shown in Figure 5A, all conjugates at both dosages of 8 and 12 mg/kg (equivalent to DTX) led to a significant inhibition of tumor growth compared to the control group (P < 0.001). When compared with the control group, conjugate A treatment at the dose of 12 mg/kg led to inhibition of tumor growth by 75.1%, which was greater than any of another three conjugates at 12 mg/kg (P < 0.05). As shown in Figure 5B, 8 mg/kg DTX treatment resulted in a 10.9% body weight loss, and all conjugate treatments at both dosages produced an increase in mice body weights, demonstrating that conjugates even at 12 mg/kg did not produce obvious side effects. Additionally, all treated groups had significantly longer survival times than the untreated control (P < 0.001), and 12 mg/kg treatment groups had significantly longer survival times than each 8 mg/kg treatment group of conjugates A, B and C (P < 0.05, Figure 5C). All six mice in the 12 mg/kg conjugate A group were alive on day 28, and 12 mg/kg conjugate A group yielded a significantly longer survival than any other group (P < 0.05). All results proved that 12 mg/kg of conjugate A produced the greatest tumor growth inhibition without causing any systemic side effects. Based on preliminary experiments, mice were also administrated intravenous albumin-bound paclitaxel (Abraxane) at 12 mg/kg twice a week for two weeks. Figure 5A also shows that conjugate A treatment once a week caused a significant decrease in tumor volume versus Abraxane treatment twice a week (P < 0.01), indicating that conjugate A had stronger efficacy than Abraxane.
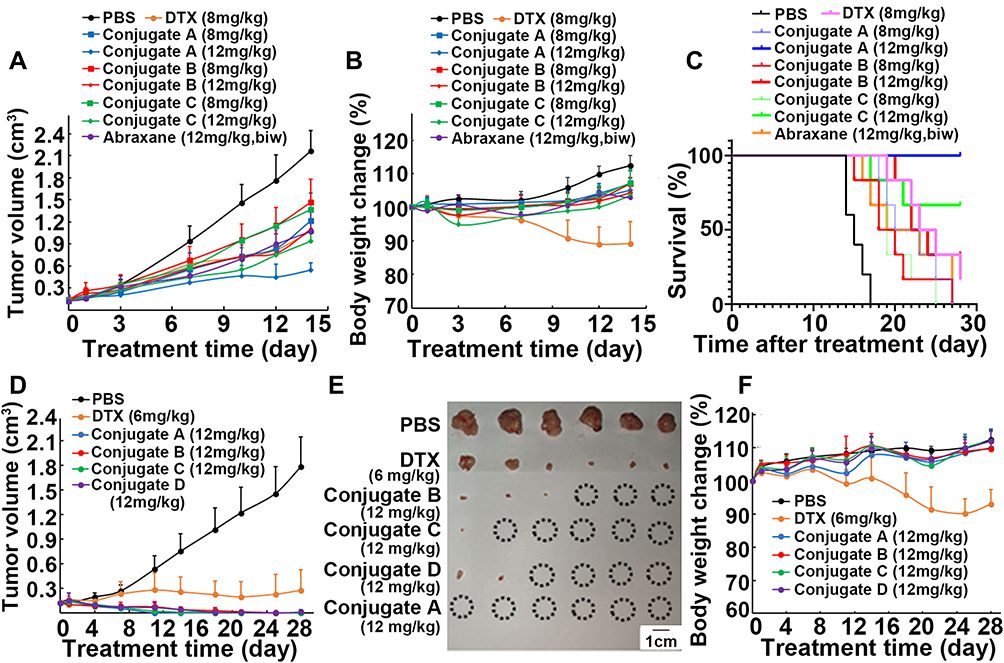 |
Figure 5 In vivo anticancer effects of four conjugates in mice. (A–C) Conjugates treatment influenced tumor volume (A), body weight (B) and mice survival (C) in nude mice bearing H460 cells. Mice were intravenously injected on day 0 and day 7, respectively. (D–F) Conjugates treatment influenced tumor volume (D), tumor image (E) and body weight (F) in nude mice bearing MCF-7 cells. Mice were intravenously injected on day 0, 7 and 14, respectively. Circle marked in tumor image represents complete eradication of xenograft tumor upon treatment. |
DTX (6 mg/kg) was chosen to use in the next mice experiment because 8 mg/kg DTX caused more than 10% mice body weight loss in nude mice bearing lung cancer H460 cells. DTX (6 mg/kg) and 12 mg/kg of all four conjugates (equivalent to DTX) were intravenously injected into nude mice bearing breast cancer cells MCF-7 on days 0, 7 and 14. Surprisingly, 12 mg/kg conjugate A caused a decrease in the tumor volume from 115.0 ± 19.6 mm3 on day 0 to 38.9 ± 15.5mm3 on day 7 and eliminated all the tumors in six mice on day 11 (Figure 5D and E). Conjugate C (12 mg/kg) treatment also led to eradication of all tumors in five of six mice by day 21. The tumors completely disappeared in three of the six mice in the group treated with 12 mg/kg conjugate B on day 28. In contrast to non-DHA-coupled conjugate D, DHA-coupled conjugate A exhibited more potent efficacy, suggesting that DHA enhanced the inhibitory effect of DTX. More interestingly, mice body weights in the groups treated with four conjugates progressively increased during the 28-day treatment period (Figure 5F). No significant differences in body weights between groups treated with four conjugates and control were found; however, the 6 mg/kg DTX group showed a decrease in body weights by 7.0% compared to the initial body weights on day 0. These results demonstrate that all four conjugates showed enhanced efficacy and did not cause any systemic side effects in nude mice bearing MCF-7 cells. Remarkably, conjugate A treatment led to eradication of all tumors after two administrations.
To confirm the excellent therapeutic efficacy in breast cancer-bearing mice, all four conjugates were evaluated in BALB/C mice with 4T1 breast cancer cells. When compared with the control group, treatment with the 12 mg/kg conjugates A, B, C, and D led to inhibition of tumor growth by 59.1%, 37.4%, 51.3%, and 15.4%, respectively (Figure 6A–C). In contrast, 6 mg/kg DTX treatment caused a reduction in tumor volume by 23.1%. Although similar in structure, conjugate A produced greater growth inhibition than non-DHA-coupled conjugate D, suggesting that DHA led to an improvement in the antitumor activity of DTX. The treatment with all four conjugates did not lead to a decrease in body weights; however, 6 mg/kg DTX treatment led to a reduction in body weights by 12.1% (Figure 6D). The side effects were further evaluated by histological examinations of the major tissues (Figure S10, Supplementary Material). H&E staining showed that all treatments produced no obvious pathological abnormalities in kidneys, spleens, and lungs. The tumor cells were arranged in a cord or vortex shape with high nucleocytoplasmic ratio, and a pathological mitotic phase can be seen in tumor tissues in all groups. However, tumor metastasis was found in the liver in control group and in all treatment groups. On the whole, DTX treatment caused degeneration and necrosis of liver cells, indicating that DTX alone had toxic effects on liver cells. In addition, serum biochemical detection also verified the biological safety of conjugates treatment. No significant differences in alanine transaminase (ALT), asparate aminotransferase (AST), blood urea nitrogen (BUN), and urine creatinine (UCr) among all groups in this study were found (Table S2, Supplementary Material). Taken together, although conjugates did not exhibit the same excellent therapeutic efficacy in 4T1-bearing mice as that in MCF-7-bearing nude mice, conjugate A also showed greater antitumor activity and less side effects than the parent DTX.
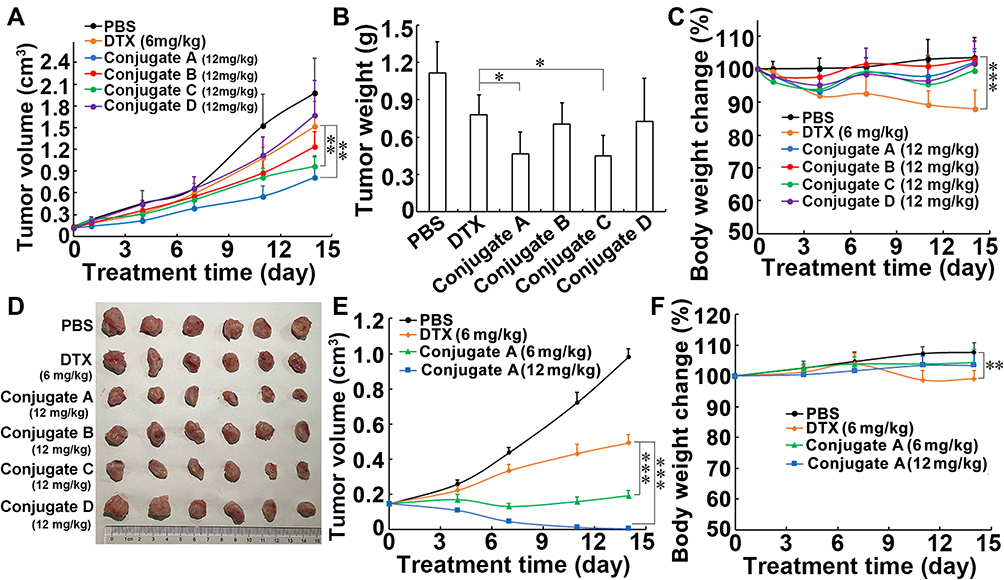 |
Figure 6 In vivo anticancer effects of four conjugates in mice. (A–D) Conjugates treatment influenced tumor volume (A), tumor weight (B), tumor image (C) and body weight (D) of mice bearing breast cancer cells 4T1. Mice were intravenously injected on day 0 and day 7, respectively. (E and F) The conjugate A treatment influenced tumor volume (E) and body weight (F) of mice bearing MCF-7 cells. Mice were intravenously injected on day 0 and day 7, respectively. Data were presented as mean±SD (n=6). *P<0.05, **P<0.01 and ***P<0.001. |
Because conjugate A exhibited the greatest antitumor effect among four conjugates in this study, only the antitumor efficacy of conjugate A against MCF-7 xenograft tumor in nude mice was verified again. As shown in Figure 6E, parent DTX and conjugate A at the same dosage of 6 mg/kg (equivalent to DTX) led to inhibition of tumor growth by 50.4% and 80.8%, respectively. Importantly, conjugate A treatment at 12 mg/kg after two administrations produced elimination of all xenograft tumors. Parent DTX caused significant body weight loss in the mice; however, conjugate A at both doses caused an increase in body weights (Figure 6F). The results further confirmed the great antitumor effect of conjugate A.
Plasma Pharmacokinetics and Biodistribution of Conjugates
For pharmacokinetic analysis, DTX and four conjugates at equivalent doses of 12 mg/kg DTX were individually administered to BALB/c mice bearing 4T1 cells via tail vein injection. The corresponding pharmacokinetic profiles are shown in Table 1 and Figure 7. No significant differences in pharmacokinetic parameters among the four conjugates in terms of released DTX or total DTX were found. Notably, the plasma concentration of both released DTX and total DTX from each conjugate was significantly higher than that of parent DTX (Figure 7A and B). Released DTX from conjugate A displayed 10.6-, 29.1- and 2.75-fold increases in AUC (0-∞), Cmax, and t1/2, respectively, then did those of the same parameters from the parent DTX group (Table 1). All dates indicate that conjugates retained a higher DTX concentration for extended periods in circulation than that of parent DTX, which suggests that the conjugate groups had long-term circulation of DTX.
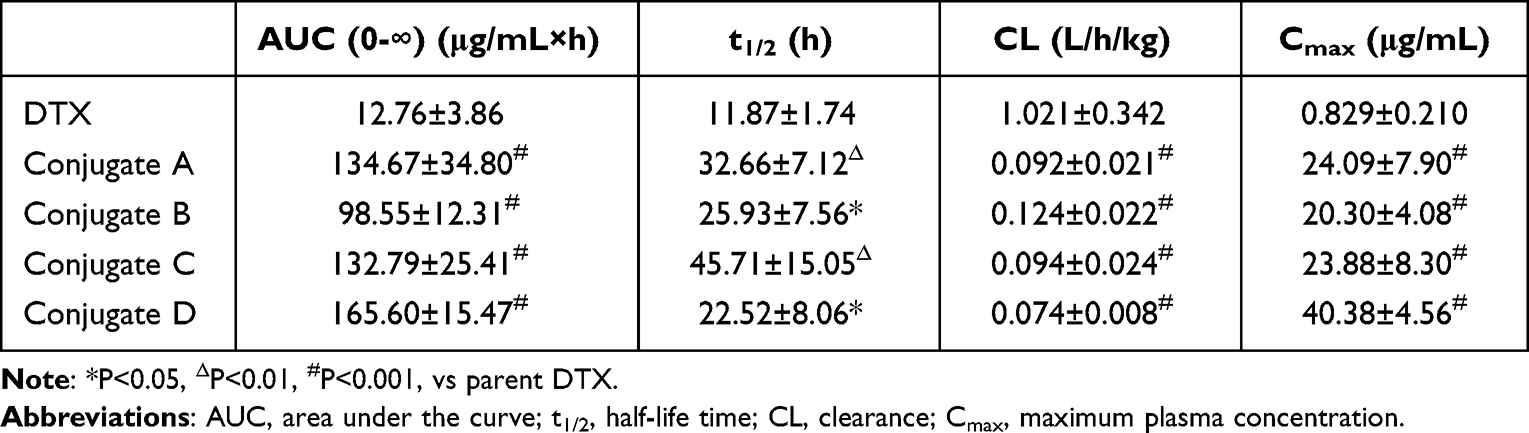 |
Table 1 Pharmacokinetic Parameters for Released DTX of Four Conjugates |
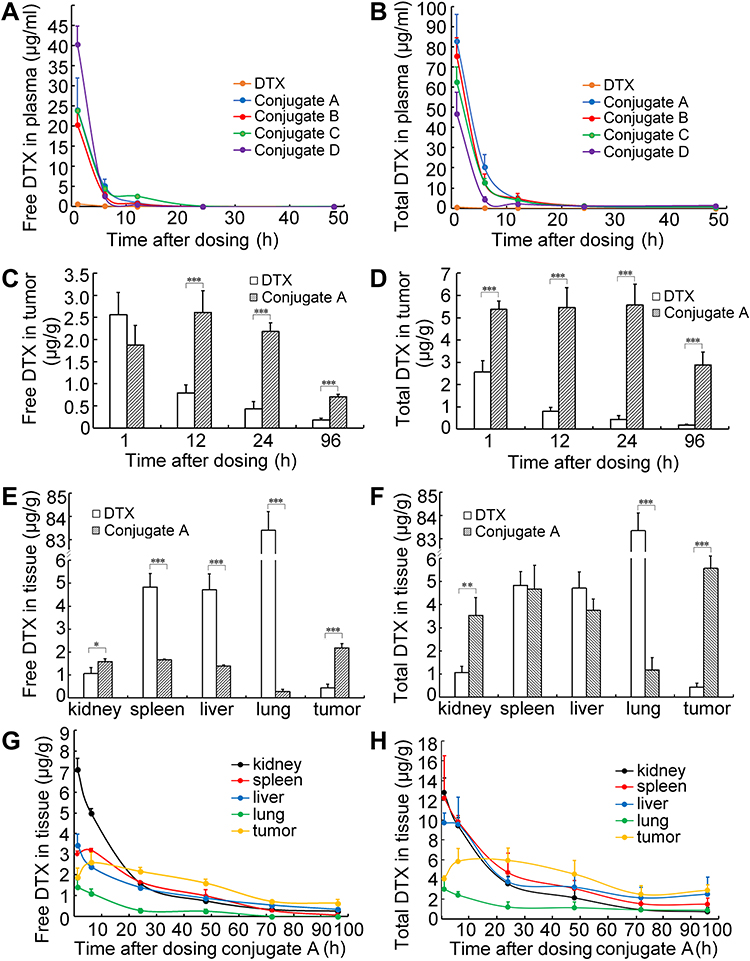 |
Figure 7 Pharmacokinetics and biodistribution of dextran-DHA-DTX conjugates in mice bearing breast cancer cells 4T1. (A and B) Free DTX and total DTX contents in plasma. (C) Free DTX contents in tumor after injection with parent DTX and the conjugate A. (D) Total DTX in tumor at indicated time points. (E) Free DTX contents in tissues after injection with parent DTX and the conjugate A for 24 h. (F) Total DTX in major organs after intravenous injection of parent DTX and the conjugate A for 24 h, respectively. (G and H) The curves of free DTX and total DTX contents in tissues after injection with the conjugate A. Data were presented as mean±SD (n = 5). *P<0.05, **P<0.01, ***P<0.001. |
Conjugate A was selected as the representative of the four conjugates for a study of its biodistribution properties because conjugate A exhibited the greatest antitumor efficacy in vivo than other three conjugates, B, C, and D. Results showed that the concentration of free DTX in the tumor rapidly decreased from 1 to 96 h in parent DTX-treated mice. In contrast, the released DTX in tumor peaked at 12 h and then began to decline gradually in the conjugate A-treated mice (Figure 7C). In terms of free DTX concentrations in tumor, the conjugate A group exhibited 0.73-, 3.29-, 5.02-, and 3.82-fold increases than those of the parent DTX at 1, 12, 24, and 96 h, respectively. The total DTX concentrations in tumors were 2.1-, 6.9-, 12.8-, and 15.5-fold higher after conjugate A treatment as compared to parent DTX treatment for 1, 12, 24, and 96 h, respectively (Figure 7D). Meanwhile, the DTX released from the conjugate A in spleens, livers, and lung tissues were only about 34%, 30%, and 2% of the parent DTX injection at 12 mg/kg dosage for 24 h (Figure 7E). Most importantly, both released and total DTX concentrations in tumors were higher than in any normal tissues of kidneys, spleens, livers, and lungs from 24 to 96 h, demonstrating that conjugate A largely accumulated in tumor versus normal tissues (Figure 7E–H). The total DTX was significantly higher than released DTX in tumors and normal tissues after conjugate A was administered to mice intravenously (Figure S11, Supplementary Material). The higher accumulation of DTX from conjugate A in the tumor and lower accumulation in the normal tissues could explain the general lack of systemic toxicity in mice receiving dextran–DHA–DTX conjugates.
Conjugate A showed superior biodistribution profiles versus taxane-based conjugates that were reported in previous studies. Albumin-bond paclitaxel is wildly used for treatment of many kinds of cancer. Unfortunately, albumin-bond paclitaxel did not show any significant differences in biodistribution as compared with parent paclitaxel at the same equivalent dose.48,49 Another formulation of paclitaxel, poly (L-glutamic acid)-paclitaxel conjugate (Xyotax, Poliglumex), was halted in Phase III clinical trials due to insignificant improvements over the current standard of care.50 A previous study showed that free paclitaxel released from Xyotax in tumors was less than in livers and that free paclitaxel in livers released from Xyotal was more than that from parent paclitaxel.51 Cellax, a polymer conjugate composed of acetylated carboxymethyl cellulose and DTX, released more free DTX in livers and spleens than in the tumors.52,53 In contrast, after retrieving literature, it was found that most clinically used cancer drugs exhibited higher concentrations in normal versus tumor tissues. Taken together, conjugate A exhibited remarkable pharmacokinetic profiles and selective tumor accumulation.
Conclusion
In summary, overcoming the reticuloendothelial system (RES) has long been a vital challenge for nanoparticle drug development; however, a novel strategy to synthesize polysaccharide-based dual drug conjugates dextran–DHA–DTX in which dextran was bi-functionalized with both glutamic acid and azide lysine concurrently was developed in this study. These dextran–DHA–DTX conjugates exhibited enhanced water solubility, prolonged plasma half-life, and selective tumor targeting. All conjugates produced greater tumor inhibition and less body weight loss than did the parent DTX treatment in three different tumor-xenografted mice models. Especially, conjugate A with the shortest linker among the conjugates fabricated in this study, led to elimination of all tumors in six mice after two sets of intravenous administrations. Importantly, the study showed that free DTX from conjugate A in tumors was greater than that in normal tissues and free DTX in normal tissues from the conjugate A group was dramatically less than that from parent DTX group. It was also proven that DHA could lead to an improvement in the therapeutic efficacy of the dextran–DHA–DTX conjugates. Conjugate A appears to be a promising candidate for clinical trials in cancer treatment and the strategy of synthesizing dextran–DHA–DTX conjugates provides a novel and very promising platform for drug-based nanoparticle delivery systems.
Acknowledgments
This study was supported by Shandong Province Major Science and Technology Innovation Project (No. 2018CXGC1402), the Fundamental Research Projects of Shandong University (No. 2017JC022), and the Natural Science Foundation of Shandong Province (No. ZR2019MH134).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Liu K, Jiang X, Hunziker P. Carbohydrate-based amphiphilic nano delivery systems for cancer therapy. Nanoscale. 2016;8(36):16091–16156. doi:10.1039/c6nr04489a
2. Memon AA, Jakobsen S, Dagnaes-Hansen F, Sorensen BS, Keiding S, Nexo E. Positron emission tomography (PET) imaging with [11C]-labeled erlotinib: a micro-PET study on mice with lung tumor xenografts. Cancer Res. 2009;69(3):873–878. doi:10.1158/0008-5472.CAN-08-3118
3. Jain A, Kameswaran M, Pandey U, Prabhash K, Sarma HD, Dash A. (68)Ga labeled Erlotinib: a novel PET probe for imaging EGFR over-expressing tumors. Bioorg Med Chem Lett. 2017;27(19):4552–4557. doi:10.1016/j.bmcl.2017.08.065
4. Su H, Seimbille Y, Ferl GZ, et al. Evaluation of [(18)F]gefitinib as a molecular imaging probe for the assessment of the epidermal growth factor receptor status in malignant tumors. Eur J Nucl Med Mol Imaging. 2008;35(6):1089–1099. doi:10.1007/s00259-007-0636-6
5. Gong J, Yang DJ, Kohanim S, Angelo LS, Kurzrock R. Molecular imaging of gefitinib activity in an epidermal growth factor receptor (EGFR)-bearing xenograft model. Cancer Biol Ther. 2009;8(23):2239–2247. doi:10.4161/cbt.8.23.9986
6. McKillop D, Partridge EA, Kemp JV, et al. Tumor penetration of gefitinib (Iressa), an epidermal growth factor receptor tyrosine kinase inhibitor. Mol Cancer Ther. 2005;4(4):641–649. doi:10.1158/1535-7163.MCT-04-0329
7. Bensch F, Smeenk MM, van Es SC, et al. Comparative biodistribution analysis across four different (89) Zr-monoclonal antibody tracers-The first step towards an imaging warehouse. Theranostics. 2018;8(16):4295–4304. doi:10.7150/thno.26370
8. van der Veen EL, Giesen D, Pot-de Jong L, Jorritsma-Smit A, De Vries EGE, Lub-de Hooge MN. 89 Zr-pembrolizumab biodistribution is influenced by PD-1-mediated uptake in lymphoid organs. J Immunother Cancer. 2020;8(2):e000938. doi:10.1136/jitc-2020-000938
9. Natarajan A, Mayer AT, Reeves RE, Nagamine CM, Gambhir SS. Development of novel ImmunoPET tracers to image human PD-1 checkpoint expression on tumor-infiltrating lymphocytes in a humanized mouse model. Mol Imaging Biol. 2017;19(6):903–914. doi:10.1007/s11307-017-1060-3
10. Ekladious I, Colson YL, Grinstaff MW. Polymer-drug conjugate therapeutics: advances, insights and prospects. Nat Rev Drug Discov. 2019;18(4):273–294. doi:10.1038/s41573-018-0005-0
11. Mitchell MJ, Billingsley MM, Haley RM, Wechsler ME, Peppas NA, Langer R. Engineering precision nanoparticles for drug delivery. Nat Rev Drug Discov. 2021;20(2):101–124. doi:10.1038/s41573-020-0090-8
12. Svenson S, Wolfgang M, Hwang J, Ryan J, Eliasof S. Preclinical to clinical development of the novel camptothecin nanopharmaceutical CRLX101. J Control Release. 2011;153(1):49–55. doi:10.1016/j.jconrel.2011.03.007
13. Engels FK, Mathot RA, Verweij J. Alternative drug formulations of docetaxel: a review. Anticancer Drugs. 2007;18(2):95–103. doi:10.1097/CAD.0b013e3280113338
14. A Razak A, Mohd Gazzali FA, Fisol,FA, et al. Advances in nanocarriers for effective delivery of docetaxel in the treatment of lung cancer: an overview. Cancers. 2021;13(3). doi:10.3390/cancers13030400
15. van Eerden RAG, Mathijssen RHJ, Koolen SLW. Recent clinical developments of nanomediated drug delivery systems of taxanes for the treatment of cancer. Int J Nanomedicine. 2020;15:8151–8166. doi:10.2147/IJN.S272529
16. He H, Liu L, Morin EE, Liu M, Schwendeman A. Survey of clinical translation of cancer nanomedicines-lessons learned from successes and failures. Acc Chem Res. 2019;52(9):2445–2461. doi:10.1021/acs.accounts.9b00228
17. Zhang E, Xing R, Liu S, Li P. Current advances in development of new docetaxel formulations. Expert Opin Drug Deliv. 2019;16(3):301–312. doi:10.1080/17425247.2019.1583644
18. Piha-Paul SA, Thein KZ, De Souza P, et al. First-in-human, phase I/IIa study of CRLX301, a nanoparticle drug conjugate containing docetaxel, in patients with advanced or metastatic solid malignancies. Invest New Drugs. 2021;39(4):1047–1056. doi:10.1007/s10637-021-01081-x
19. Atrafi F, Dumez H, Mathijssen RHJ, et al. A phase I dose-escalation and pharmacokinetic study of a micellar nanoparticle with entrapped docetaxel (CPC634) in patients with advanced solid tumours. J Control Release. 2020;325:191–197. doi:10.1016/j.jconrel.2020.06.020
20. Hu Q, Lu Y, Luo Y. Recent advances in dextran-based drug delivery systems: from fabrication strategies to applications. Carbohydr Polym. 2021;264:117999. doi:10.1016/j.carbpol.2021.117999
21. Huang G, Huang H. Application of dextran as nanoscale drug carriers. Nanomedicine. 2018;13(24):3149–3158. doi:10.2217/nnm-2018-0331
22. Raza K, Kumar N, Misra C, et al. Dextran-PLGA-loaded docetaxel micelles with enhanced cytotoxicity and better pharmacokinetic profile. Int J Biol Macromol. 2016;88:206–212. doi:10.1016/j.ijbiomac.2016.03.064
23. Han HS, Lee M, An JY, et al. A pH-responsive carboxymethyl dextran-based conjugate as a carrier of docetaxel for cancer therapy. J Biomed Mater Res B Appl Biomater. 2016;104(4):789–796. doi:10.1002/jbm.b.33581
24. Sugahara S, Kajiki M, Kuriyama H, Kobayashi TR. Complete regression of xenografted human carcinomas by a paclitaxel-carboxymethyl dextran conjugate (AZ10992). J Control Release. 2007;117(1):40–50. doi:10.1016/j.jconrel.2006.10.009
25. Li D, Su T, Ma L, et al. Dual-acidity-labile polysaccharide-di-drugs conjugate for targeted cancer chemotherapy. Eur J Med Chem. 2020;199:112367. doi:10.1016/j.ejmech.2020.112367
26. Zhang Q, He S, Kuang G, et al. Morphology tunable and acid-sensitive dextran-doxorubicin conjugate assemblies for targeted cancer therapy. J Mater Chem B. 2020;8(31):6898–6904. doi:10.1039/d0tb00746c
27. Huo M, Wang H, Zhang Y, et al. Co-delivery of silybin and paclitaxel by dextran-based nanoparticles for effective anti-tumor treatment through chemotherapy sensitization and microenvironment modulation. J Control Release. 2020;321:198–210. doi:10.1016/j.jconrel.2020.02.01
28. Li M, Tang Z, Zhang Y, Lv S, Li Q, Chen X. Targeted delivery of cisplatin by LHRH-peptide conjugated dextran nanoparticles suppresses breast cancer growth and metastasis. Acta Biomater. 2015;18:132–143. doi:10.1016/j.actbio.2015.02.022
29. Giordano C, Plastina P, Barone I, Catalano S, Bonofiglio D. n-3 Polyunsaturated fatty acid amides: new avenues in the prevention and treatment of breast cancer. Int J Mol Sci. 2020;21(7):2279. doi:10.3390/ijms21072279
30. VanderSluis L, Mazurak VC, Damaraju S, Field CJ. Determination of the relative efficacy of eicosapentaenoic acid and docosahexaenoic acid for anti-cancer effects in human breast cancer models. Int J Mol Sci. 2017;18(12):2607. doi:10.3390/ijms18122607
31. Bougnoux P, Hajjaji N, Maheo K, Couet C, Chevalier S. Fatty acids and breast cancer: sensitization to treatments and prevention of metastatic re-growth. Prog Lipid Res. 2010;49(1):76–86. doi:10.1016/j.plipres.2009.08.003
32. Das M, Das S. Identification of cytotoxic mediators and their putative role in the signaling pathways during docosahexaenoic acid (DHA)-induced apoptosis of cancer cells. Apoptosis. 2016;21(12):1408–1421. doi:10.1007/s10495-016-1298-2
33. D’Eliseo D, Di Renzo L, Santoni A, Velotti F. Docosahexaenoic acid (DHA) promotes immunogenic apoptosis in human multiple myeloma cells, induces autophagy and inhibits STAT3 in both tumor and dendritic cells. Genes Cancer. 2017;8(1–2):426–437. doi:10.18632/genesandcancer.131
34. Zhelev Z, Ivanova D, Lazarova D, Aoki I, Bakalova R, Saga T. Docosahexaenoic acid sensitizes leukemia lymphocytes to barasertib and everolimus by ROS-dependent mechanism without affecting the level of ROS and viability of normal lymphocytes. Anticancer Res. 2016;36(4):1673–1682.
35. Goupille C, Vibet S, Frank PG, Mahéo K. EPA and DHA fatty acids induce a remodeling of tumor vasculature and potentiate docetaxel activity. Int J Mol Sci. 2020;21(14):4965. doi:10.3390/ijms21144965
36. Newell M, Goruk S, Mazurak V, Postovit L, Field CJ. Role of docosahexaenoic acid in enhancement of docetaxel action in patient-derived breast cancer xenografts. Breast Cancer Res Treat. 2019;177(2):357–367. doi:10.1007/s10549-019-05331-8
37. Fracasso PM, Picus J, Wildi JD, et al. Phase 1 and pharmacokinetic study of weekly docosahexaenoic acid-paclitaxel, Taxoprexin, in resistant solid tumor malignancies. Cancer Chemother Pharmacol. 2009;63(3):451–458. doi:10.1007/s00280-008-0756-0
38. Homsi J, Bedikian AY, Papadopoulos NE, et al. Phase 2 open-label study of weekly docosahexaenoic acid-paclitaxel in patients with metastatic uveal melanoma. Melanoma Res. 2010;20(6):507–510. doi:10.1097/CMR.0b013e3283403ce9
39. Xie B, Wan J, Chen X, Han W, Wang H. Preclinical evaluation of a cabazitaxel prodrug using nanoparticle delivery for the treatment of taxane-resistant malignancies. Mol Cancer Ther. 2020;19(3):822–834. doi:10.1158/1535-7163.MCT-19-0625
40. Li S, Qin J, Tian C, et al. The targeting mechanism of DHA ligand and its conjugate with Gemcitabine for the enhanced tumor therapy. Oncotarget. 2014;5(11):3622–3635. doi:10.18632/oncotarget.1969
41. Mika A, Kobiela J, Pakiet A, et al. Preferential uptake of polyunsaturated fatty acids by colorectal cancer cells. Sci Rep. 2020;10(1):1954. doi:10.1038/s41598-020-58895-7
42. Oh DY, Talukdar S, Bae EJ, et al. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell. 2010;142(5):687–698. doi:10.1016/j.cell.2010.07.041
43. Nehra D, Pan AH, Le HD, et al. Docosahexaenoic acid, G protein-coupled receptors, and melanoma: is G protein-coupled receptor 40 a potential therapeutic target? J Surg Res. 2014;188(2):451–458. doi:10.1016/j.jss.2014.01.037
44. Harada M, Murata J, Sakamura Y, Sakakibara H, Okuno S, Suzuki T. Carrier and dose effects on the pharmacokinetics of T-0128, a camptothecin analogue-carboxymethyl dextran conjugate, in non-tumor- and tumor-bearing rats. J Control Release. 2001;71(1):71–86. doi:10.1016/s0168-3659(00)00372-2
45. Sugahara S, Kajiki M, Kuriyama H, Kobayashi TR. Carrier effects on antitumor activity and neurotoxicity of AZ10992, a paclitaxel-carboxymethyl dextran conjugate, in a mouse model. Biol Pharm Bull. 2008;31(2):223–230. doi:10.1248/bpb.31.223
46. Sugahara S, Kajiki M, Kuriyama H, Kobayashi TR. Paclitaxel delivery systems: the use of amino acid linkers in the conjugation of paclitaxel with carboxymethyldextran to create prodrugs. Biol Pharm Bull. 2002;25(5):632–641. doi:10.1248/bpb.25.632
47. Machulkin AE, Skvortsov DA, Ivanenkov YA, et al. Synthesis and biological evaluation of PSMA-targeting paclitaxel conjugates. Bioorg Med Chem Lett. 2019;29(16):2229–2235. doi:10.1016/j.bmcl.2019.06.035
48. Bhattacharyya J, Bellucci JJ, Weitzhandler I, et al. A paclitaxel-loaded recombinant polypeptide nanoparticle outperforms Abraxane in multiple murine cancer models. Nat Commun. 2015;6:7939. doi:10.1038/ncomms8939
49. Sparreboom A, Scripture CD, Trieu V, et al. Comparative preclinical and clinical pharmacokinetics of a cremophor-free, nanoparticle albumin-bound paclitaxel (ABI-007) and paclitaxel formulated in Cremophor (Taxol). Clin Cancer Res. 2005;11(11):4136–4143. doi:10.1158/1078-0432.CCR-04-2291
50. Zhao J, Koay EJ, Li T, Wen X, Li C. A hindsight reflection on the clinical studies of poly(l-glutamic acid)-paclitaxel. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2018;10(3):e1497. doi:10.1002/wnan.1497
51. Li C, Newman RA, Wu QP, et al. Biodistribution of paclitaxel and poly(L-glutamic acid)-paclitaxel conjugate in mice with ovarian OCa-1 tumor. Cancer Chemother Pharmacol. 2000;46(5):416–422. doi:10.1007/s002800000168
52. Ernsting MJ, Murakami M, Undzys E, Aman A, Press B, Li SD. A docetaxel-carboxymethylcellulose nanoparticle outperforms the approved taxane nanoformulation, Abraxane, in mouse tumor models with significant control of metastases. J Control Release. 2012;162(3):575–581. doi:10.1016/j.jconrel.2012.07.043
53. Ernsting MJ, Tang WL, MacCallum NW, Li SD. Preclinical pharmacokinetic, biodistribution, and anti-cancer efficacy studies of a docetaxel-carboxymethylcellulose nanoparticle in mouse models. Biomaterials. 2012;33(5):1445–1454. doi:10.1016/j.biomaterials.2011.10.061
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.