")
Back to Journals » International Medical Case Reports Journal » Volume 12
A novel case report of spinal muscular atrophy with progressive myoclonic epilepsy from Iran
Authors Shervin Badv R, Nilipour Y, Rahimi-Dehgolan S , Rashidi-Nezhad A , Ghahvechi Akbari M
Received 18 January 2019
Accepted for publication 9 April 2019
Published 30 May 2019 Volume 2019:12 Pages 155—159
DOI https://doi.org/10.2147/IMCRJ.S202046
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ronald Prineas
Reza Shervin Badv,1 Yalda Nilipour,2 Shahram Rahimi-Dehgolan,3 Ali Rashidi-Nezhad,4 Masood Ghahvechi Akbari5
1Children’s Medical Center, Pediatrics Center of Excellence, Tehran University of Medical Sciences (TUMS), Tehran, Iran; 2Pediatric Pathology Research center, Research Institute for Children Health, Mofid Hospital, Shahid Beheshti University of Medical Sciences, Tehran, Iran; 3Physical Medicine and Rehabilitation Department, Imam Khomeini Hospital Complex (IKHC), Tehran University of Medical Sciences (TUMS), Tehran, Iran; 4Maternal, Fetal and Neonatal Research Center, Imam khomeini Hospital Complex, Tehran University of Medical Sciences, Tehran, Iran; 5Physical Medicine and Rehabilitation Department, Children’s Medical Center, Tehran University of Medical Sciences (TUMS), Tehran, Iran
Abstract: Spinal muscular atrophy (SMA) is a disorder characterized by decreased motor function due to the muscle atrophy in the background of degenerated anterior horn cells and motor cells of lower cranial nerves nuclei. The most frequent form is inherited as an autosomal recessive trait resulting from mutations in the survival motor neuron gene (SMN-1). On the other hand, a rare variant of this condition, named progressive myoclonic epilepsy subtype (SMA-PME) occurs in the result of a mutation in N-acylsphingosine amidohydrolase-1 gene (ASAH-1). The latter gene is responsible for lysosomal acid-ceramidase production. SMA-PME has been characterized by a progressive muscle weakness from ages 3–7 years, accompanied by epilepsy, an intractable seizure, and sometimes sensorineural hearing loss. In this report, we have presented a 15-year old female patient with SMA-PME that was attended to neurology clinic for a new onset tremor, seizure and proximal weakness in all limbs. We identified a homozygous mutation in exon II on her ASAH-1 gene [c.173C>T (p. Thr58Met)]. Also, a modest reduction was found in ceramidase-activity. As was expected patient`s seizures did not respond to conventional therapies.
Keywords: muscular atrophy, case report, myoclonic epilepsies, seizures
A corrigendum for this paper has been published
Introduction
Spinal muscular atrophy (SMA) is a heterogeneous disease characterized by degeneration of the lower motor neurons and is usually linked to mutations of SMN-1 gene.1 The condition often presents by decreased motor function and muscle atrophy due to degeneration of anterior horn cell in the spinal cord and motor cells of lower cranial nerves nuclei.2 SMA with progressive myoclonic epilepsy (PME; OMIM #159950) is a very rare autosomal-recessive (AR) condition, that is not the result of mutation in SMN-1 gene, but is caused by mutation in the N-acylsphingosine amidohydrolase-1 (ASAH-1) gene, which is responsible for lysosomal acid-ceramidase production. Ceramides and their metabolites, as the lipid mediators, contribute to many cellular functions including cell survival.3 The catabolism of ceramides occurs through the activity of ceramidase; this enzyme has three different subtypes including alkaline-, neutral- and acid-ceramidase.4 Deficient activity of the latter one occurs in two rare inherited disorders: Farber disease and SMA-PME.2 SMA-PME has been characterized by a progressive muscle weakness from ages 3–7 years, plus to epilepsy, an intractable seizure, and sometimes tremor.3,5 The SMA-PME diagnosis could be confirmed using a biallelic mutation in ASAH-1 gene, or a modest (6–32%) deficiency of acid-ceramidase activity in white blood cells or fibroblasts of skin tissue.1,6 Here, we have reported the first case of SMA-PME from Iran.
Case presentation
A 15-year old female patient was attended to neurology clinic for recent onset tremor, seizure, and weakness in limbs. She was the first offspring of a family with relative parents; she was born from normal spontaneous delivery. The psychomotor developmental course was normal. The symptoms initiated with a resting tremor in both hands since the fifth year of age. When she was seven years old, new presentations such as staring episodes and myoclonus were added. The latter feature occurred several times a day lasting for a few seconds. Myoclonus was stimulus-sensitive, sometimes activated by volitional movement. Five years ago, head drop attacks and multiple episodes of generalized tonic-clonic seizure (GTCS), were initiated. Also, since the 10th year of age, a progressive limb weakness was gradually started, especially in attempting for stair-up, standing, and hand washing.
In physical examination, the patient was cooperative and fully oriented. She had waddling gait pattern in walking, and her Gower`s sign was positive indicating of proximal weakness. Muscle forces were evaluated according to the medical research council muscle scale. Neck extensor and flexor muscles obtained 3/5, and strength of proximal muscles in upper limbs (shoulder abductors) were 2/5. This pattern was less severe in distal muscles, where elbow and hand muscles obtained 3/5 and 4/5, respectively. In her lower extremities, the strength was 3/5 for both hip and knee muscles; and it was again higher in distal muscles of legs (4/5). Also, a mild weakness in facial muscles was found; while the extra-ocular muscles were spared. Deep tendon reflexes in Biceps brachii, Triceps brachii, Brachioradialis, and Achilles were 1+ and the patellar knee jerk was absent. Sensory exam for small and large fibers was not remarkable. Resting tremor was present in both hands. Patient`s tongue was obviously atrophic with fasciculation movements. Several staring attacks and myoclonus were observed during the physical examination.
Brain MRI was done, but it did not reveal any pathologic features. Laboratory blood tests including complete blood count (CBC), creatine phosphokinase (CPK), lactate dehydrogenase (LDH), and ammonia, plus thyroid, renal, and liver function tests were all within normal ranges. In order to further confirming disease, the ceramidase-activity level was obtained that revealed about 30% reduction. Moreover, a muscle biopsy of left Biceps brachii was obtained using an open technique (Figures 1 and 2); Histo-pathological examination confirmed a neuropathic pattern with muscle atrophy of mainly type-II fibers, with concomitant denervation and reinnervation processes. The video EEG monitoring was performed using a standard 10–20 system with additional electrodes and bipolar settings; no drug was administrated to induce sedation. Both sleep and awake phases were recorded that has been presented in Figure 3. The patient had a total of frequent epileptic events during recording, the main features were epileptic poly-myoclonus with generalized 2.5–3 Hz irregular sharp and slow wave complexes that was accentuated during intermittent photic stimulation (IPS).
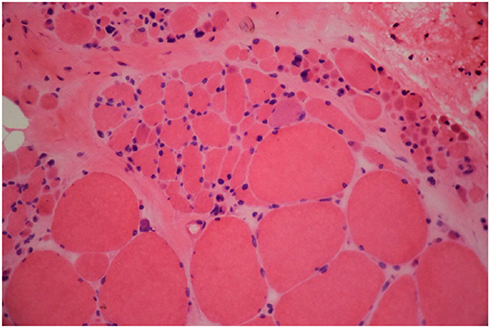 | Figure 1 H&E stain revealed atrophic fibers of round or angular shapes that arranged in small and large groups with hypertrophied fibers. Fascicular atrophy was seen. Nuclear clumps were noted. |
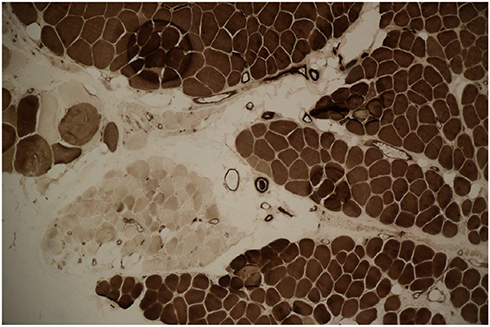 | Figure 2 ATPase reaction PH 4.35 revealed fiber type grouping with type-I fibers predominance. Atrophic fibers are mostly type-II. |
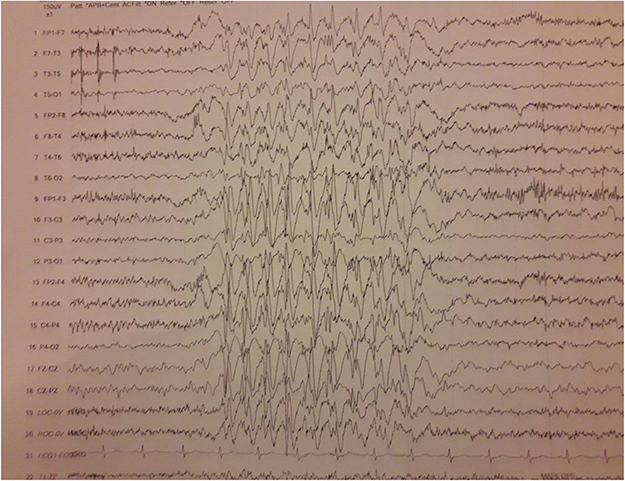 | Figure 3 Recorded EEG in the patient, It was recorded by bipolar setting, with sensitivity of 150 microvolt, high frequency filter of 70 Hz and low frequency filter of 1 Hz.There are generalized epileptiform 2.5–3 Hz irregular sharp and slow wave complexes. |
In needle electromyography (EMG) and nerve conduction study (NCS), three limbs and two cranial nerves (V, XII) were investigated. The left upper limb was preserved for performing a muscle biopsy. After ensuring the standard temperature in tested limbs (T=37.0), compound muscle action potential (CMAP) [for Median, Ulnar, Tibial, and Peroneal nerves], sensory nerve action potential (SNAP) [for Median and Sural nerves], and F-waves [from Tibial, Median and Ulnar nerves] were recorded. All NCS responses were normal. Needle-EMG demonstrated a neuropathic pattern with high amplitude and long duration motor unit action potentials (MUAPs), associated with reduced recruitment and spontaneous potentials such as fibrillation (Fib), positive sharp waves (PSW), and fasciculation (Fasc). These findings confirmed a chronic denervation process with ongoing regeneration that was compatible with the category of anterior horn cell diseases. Afterward, the genetic study revealed a homozygous mutation on ASAH-1 gene [c.173C>T (p. Thr58Met)]; According to Gene Review® database of Washington University, this mutation was a known one; also the parents` genetic study revealed a heterozygous mutation.6 Then, treatment was started; at the first line, Depakin 500 mg/TDS, Ethosuximide 250 mg/BID, and Amantadine 100 m/BID were prescribed for the patient. But she had not a good response on the follow-up visits. We added Clonazepam 1 mg/BID and Sabril 500 mg/QD to her previous medications; however, the patient did not achieve any improvement and the symptoms were progressive. Fortunately, she is alive now and still under therapeutic regimen of the recombinant enzyme.
Discussion
In the current report, we presented an adolescent female patient with SMA-PME. Jancovic and Rivera reported this rare condition for the first time with three main features of myoclonus, epilepsy, and signs of SMA; all of them had the onset in adulthood and a benign course.7 Lance and Marjanovic reported next similar cases.8,9 Lance et al reported a male patient of SMA-PME associated with sensorineural deafness;8 while Marjanovic described a family with two boys suffered from SMA-PME.9 Haliloglu et al in 2002 reported another four cases of SMA-PME in two families, with myoclonic epilepsy, progressive weakness, and tremor in common.10 More recent similar Asian cases were reported by Liyanage.2 Also, Rubboli in Italy reported the clinical manifestations of three SMA-PME patients.11 The main clinical presentations in their patients were earlier onset in childhood with progressive proximal muscle weakness and uncontrolled generalized epilepsy. Another Italian researcher, Filosto reported two patients with SMA-PME with a very slowly progressive SMA disease since their childhood.12 There was no history of myoclonus or seizures and EEG was unremarkable. The genetic study of ASAH-1 gene discovered a homozygote nucleotide variation. The biochemical laboratory test of cultured fibroblasts confirmed a concomitant reduction in ceramidase-activity and subsequent accumulation of ceramide. In another case report, Dyment et al in Canada introduced an adolescent female presenting with atonic and absence seizures, in addition to myoclonic jerks.13 Although the genetic and metabolic workup were unable to diagnose the disease, an increased level of ceramide was found in the patient’s fibroblasts. Progressive muscle weakness could be perfectly explained by the lower motor neurons involvement. However, the pathogenic cause of central nervous system (CNS) among SMA-PME patients would be probably unrelated to the accumulation of ceramide.1
As was mentioned, our patient presented with a resting tremor from the fifth year of age as the first manifestation. This has not been a typical presenting feature for SMA-PME. PME typically initiates after the onset of weakness.6 The course of SMA-PME is generally progressive and sometimes might lead to the respiratory muscle involvement and subsequent death before the third decade of life.5 Fortunately, none of these catastrophic endpoints occurred in our reported patient yet.
Literature review
Among all SMA-PME reported cases, the most comprehensive ones have been discussed here. The majority of reports have described the lower motor neuron involvement as the first presenting symptom with the mean onset age of 5 years;6 however, there are some exceptions.5,12 The initial clumsiness/frequent falls might progress to a more advanced stage such as waddling gait pattern. Respiratory muscles are also at risk of LMN involvement; which may present with the frequent episodes of aspiration.6
In respect to myoclonus and epileptic episodes, it is noteworthy that a wide spectrum of seizures including GTCS, PME, atonic head drop, and even absence epilepsy have been reported up to now; Myoclonic epilepsy usually presents in the upper extremity, as well as in the palpebra.6 Regarding the EEG findings, earlier studies have revealed a generalized spike-and-wave pattern accompanied with myoclonic seizures,1,11 similar to our EEG results in Figure 3. Although brain MRI is almost always normal; the epilepsy episodes gradually become resistant and usually do not respond to conventional therapies.5 A clinically-significant relationship has been evidently observed between the frequency of myoclonus and proximal weakness.11,13
As was said, tremor and cognition impairment following the aforementioned symptoms have been reported.6 In about one-half of individuals, a generalized tremulousness has been recorded; while sensorineural hearing loss (SNHL) and scoliosis have been detected in few subjects.3,13 Although cognition was overall intact; one child with profound impairment was reported,11 and in another patient an intellectual disability was the first feature.14 Most subjects die in their late teens, however, the estimated time from onset to death was recorded between 5–15 years.6
Standard management of seizures, tremor or deafness are the only treatments could be considered for the condition. Utilizing a powered wheelchair or other orthotics and assistive devices would probably improve gait balance. Epilepsy episodes, nutritional status, muscle strength, respiratory function, and mobility capacity should be intermittently evaluated. Eventually, SMA-PME should be suspected in a previously well child with the normal milestones who has had a recent-onset proximal weakness at an average age of five years, along with several episodes of myoclonic or atonic epilepsy that do not respond to the routine medications. The mentioned clinical features should be confirmed by a combination of para-clinic tools including evidence of chronic denervation in EMG; generalized poly spike-and-wave complexes in EEG; neurogenic pattern in muscle biopsy; and simultaneously absence of homozygous mutations in SMN1 gene.
Ethical approval and consent
Since our case was under age of 18 years, a written informed consent was fulfilled by her father (as the patient's legal guardian) to agree about publishing the case details, without any accompanying image. Also, the institutional (Tehran University of Medical Sciences Ethics Committee) approval was required to publish the case details. In accordance with our ethics committee rules, the patient herself was provided with all necessary information about this publication and another informed consent was obtained.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Zhou J, Tawk M, Tiziano FD, et al. Spinal muscular atrophy associated with progressive myoclonic epilepsy is caused by mutations in ASAH1. Am J Hum Genet. 2012;91(1):5–14. doi:10.1016/j.ajhg.2012.05.001
2. Liyanage DS, Pathberiya LS, Gooneratne IK, Vithanage KK, Gamage R. Association of type IV spinal muscular atrophy (SMA) with myoclonic epilepsy within a single family. Int Arch Med. 2014;7(1):42. doi:10.1186/1755-7682-7-42
3. Gan JJ, Garcia V, Tian J, et al. Acid ceramidase deficiency associated with spinal muscular atrophy with progressive myoclonic epilepsy. Neuromuscul Disord. 2015;25(12):959–963. doi:10.1016/j.nmd.2015.09.007
4. Yildiz EP, Yesil G, Bektas G, et al. Spinal muscular atrophy with progressive myoclonic epilepsy linked to mutations in ASAH1. Clin Neurol Neurosurg. 2017;164:47–49. doi:10.1016/j.clineuro.2017.11.008
5. Topaloglu H, Melki J. Spinal muscular atrophy associated with progressive myoclonus epilepsy. Epileptic Disord. 2016;18(S2):128–134. doi:10.1684/epd.2016.0858
6. Dyment DA, Bennett SAL, Medin JA, et al. ASAH1-related disorders. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle: University of Washington, Seattle; March 29, 2018:1993–2019. Available from:
7. Jankovic J, Rivera VM. Hereditary myoclonus and progressive distal muscular atrophy. Ann Neurol. 1979;6:227–231. doi:10.1002/ana.410060309
8. Lance WJ, Evans WA. Progressive myoclonic epilepsy. Clin Exp Neurol. 1984;20:141–151.
9. Marjanovic B, Todorovic S, Dozic S. Association of progressive myoclonic epilepsy and spinal muscular atrophy. Paediatr Neurol. 1993;9:147–150. doi:10.1016/0887-8994(93)90053-F
10. Haliloglu G, Chattopadhyay A, Skorodis L, et al. Spinal muscular atrophy with progressive myoclonic epilepsy: report of new cases and review of the literature. Neuropediatrics. 2002;33(6):314–319. doi:10.1055/s-2002-37087
11. Rubboli G, Veggiotti P, Pini A, et al. Spinal muscular atrophy associated with progressive myoclonic epilepsy: a rare condition caused by mutations in ASAH1. Epilepsia. 2015;56(5):692–698. doi:10.1111/epi.12977
12. Filosto M, Aureli M, Castellotti B, et al. ASAH1 variant causing a mild SMA phenotype with no myoclonic epilepsy: a clinical, biochemical and molecular study. Eur J Hum Genet. 2016;24(11):1578–1583. doi:10.1038/ejhg.2016.28
13. Dyment DA, Sell E, Vanstone MR, et al. Evidence for clinical, genetic and biochemical variability in spinal muscular atrophy with progressive myoclonic epilepsy. Clin Genet. 2014;86(6):558–563. doi:10.1111/cge.12307
14. Sathe S, Pearson T. Phenotypic characterization of the spinal muscular atrophy with progressive myoclonus epilepsy syndrome caused by ASAH1 mutations. Mol Genet Metab. 2013;111:S93. doi:10.1016/j.ymgme.2013.12.226
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.