")
Back to Journals » Drug Design, Development and Therapy » Volume 9
A novel bispecific immunotoxin delivered by human bone marrow-derived mesenchymal stem cells to target blood vessels and vasculogenic mimicry of malignant gliomas
Authors Zhang Y, Sun X, Huang M, Ke Y, Wang J, Liu X
Received 17 December 2014
Accepted for publication 8 January 2015
Published 11 June 2015 Volume 2015:9 Pages 2947—2959
DOI https://doi.org/10.2147/DDDT.S79475
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Shu-Feng Zhou
This paper has been retracted.
Yonghong Zhang, 1,2 Xinlin Sun, 1 Min Huang, 1 Yiquan Ke, 1 Jihui Wang, 1 Xiao Liu 1
1National Key Clinic Specialty, Neurosurgery Institute of Guangdong Province, Guangdong Provincial Key Laboratory on Brain Function Repair and Regeneration, Department of Neurosurgery, Zhujiang Hospital, Southern Medical University, Guangzhou, 2Department of Neurosurgery, First Hospital of Lanzhou University, Lanzhou, People’s Republic of China
Background: In previous years, immunotoxins have been shown to be a greatly promising therapeutic tool for brain malignancies, such as gliomas. Human mesenchymal stem cells (hMSCs) exhibit tropism to tumor tissue. However, the effect of bispecific immunotoxins in malignant gliomas is still unknown. The aim of this study was to investigate the function of bispecific immunotoxins in human malignant gliomas.
Materials and methods: In the present study, the bispecific immunotoxin VEGF 165-ephrin A1-PE38KDEL was established using deoxyribonucleic acid shuffling and cloning techniques. The VEGF 165-ephrin A1-PE38KDEL was delivered by hMSCs to mouse malignant gliomas. The effects of the bispecific immunotoxins on glioma-derived blood vessels and vasculogenic mimicry to elucidate the molecular mechanisms underlying the antitumorigenic effects of immunotoxins were examined in vivo.
Results: In vitro, transfected hMSCs significantly inhibited the cell viability of gliomas cell lines U87 and U251 in a dose-dependent manner compared with untransfected hMSCs (P< 0.01). In vivo, the intratumoral injection of engineered hMSCs was effective at inhibiting tumor growth in a malignant glioma tumor model.
Conclusion: The bispecific immunotoxin secreted from hMSCs acts as a novel strategy for improving treatment options for malignant gliomas in the clinic.
Keywords: bispecific immunotoxin, human mesenchymal stem cells, ephrin A1, VEGF 165, malignant glioma
Introduction
Glioblastoma multiforme (GBM) is the most common and most malignant of glial tumors in adults, which presents one of the most significant treatment challenges in oncology. Despite considerable surgical and medical advancements, the 5-year survival rate for GBM has remained extremely low at 3.4% for the past 3 decades.1 In recent years, novel discovered strategies for GBM therapy have been associated with regard to increased specificity and efficacy. Among the treatment approaches for GBM, antiangiogenesis therapy has emerged as a potent tool, due to the abnormally rich vascular network in gliomas. Inhibition or blockage of angiogenesis can prevent tumor growth. Besides the vascular endothelium, the latest research has shown that vasculogenic mimicry (VM; a type of endothelial cell-independent microcirculation) also exists in malignant tumors, such as GBM.2,3 The phenomenon of VM not only accounts for the limitations of antiendothelial angiogenic therapy but also provides a new target for tumor therapy. In this study, we hypothesized that immunotoxin targeting of specific molecules of tumor vascular endothelial cells that simultaneously destroy the tumor vascular system and formation of VM may facilitate the blockage of blood supply to the tumor and effectively inhibit tumor growth.
Immunotoxins, hybrid molecules consisting of targeting and toxic moieties, have been developed as a novel treatment strategy for tumors in recent years.4 The toxins injure cells by damaging the plasma membrane or inactivating cytosolic protein synthesis.5 The specificity of immunotoxin therapy relies on the presence of a molecular marker that is highly overexpressed in tumor cells but absent in normal cells.6 The VEGF receptor (VEGFR) is a protein tyrosine kinase highly expressed in tumor endothelial vessels but not normal tissue,7,8 and is a specific marker of tumor vascular endothelial cells. EphA2, one of the markers of tumor VM, is a specific ligand for ephrin A1 that is abnormally expressed at excessive levels in malignant tumors, but absent in normal tissue.9 Immunotoxins are considered the most potent anticancer-drug candidates for killing tumor cells at picomolar concentrations.10 Bispecific immunotoxins (BITs) are single-chain molecules with two distinct targeting ligands fused to a single toxin. The double-targeted immunotoxin has obvious advantages over monospecific immunotoxins, enhancing the killing effect on tumor tissue.11–13
The clinical application of immunotoxins has several limitations, including 1) difficulties in penetrating and specifically locating tumors and reaching the desired concentration, and 2) nonspecific associated neural toxicity, which reduces resistance and narrows the therapeutic time window.14,15 Therefore, new approaches of immunotoxin delivery to overcome these obstacles are critical. Human mesenchymal stem cells (hMSCs) have shown promise as potential vehicles for delivering therapeutic genes to treat brain tumors.16 These cells possess tropism for experimental tumors, including gliomas, following intra-arterial or intracranial injection. Moreover, hMSCs can be obtained from patients without ethical concerns, easily expanded in vitro, and genetically modified with viral vectors for the delivery of antitumor substances in vivo.17,18 Several preclinical trials of genetically modified hMSCs expressing IFNβ, IL-2, and TRAIL have revealed a significant antitumor effect in glioma models.19 Accordingly, we used hMSCs as a carrier for delivery of our novel immunotoxin against malignant glioma.
In a previous study, we examined the antiangiogenic effect of the immunotoxin VEGF-PE38, targeting VEGFR in glioma vascular endothelial cells, and ephrin A1-PE38, targeting EphA2-expressing tumor cells, which were correlated with VM in malignant cells and effectively inhibited gliomas. MSCs are effectively used as cell carriers for immunotoxin gene therapy.20,21 In the current investigation, we designed and synthesized a BIT, VEGF165-ephrin A1-PE38KDEL, consisting of truncated Pseudomonas exotoxin (PE) and two different ligands, specifically, in which VEGF165 targeted the VEGFR and ephrin A1 targeted the EphA2 receptor. Our main aim was to assess the anticancer effect of VEGF165-ephrin A1-PE38KDEL, potentially blocking both vascular endothelial and vascular mimicry, upon delivery by hMSCs in a mouse xenograft brain-tumor model. Human glioma U87 cells were genetically marked with the firefly luciferase reporter gene, facilitating the monitoring of intracranial tumor growth in real time using bioluminescent imaging.
Materials and methods
Cell culture
The human glioma cell lines U251 and U87 were obtained from the Cell Bank of Type Culture Collection of the Chinese Academy of Sciences (Shanghai, People’s Republic of China [PRC]). hMSCs were isolated and cultured as described previously.22 All cell lines were maintained in Dulbecco’s Modified Eagle’s Medium supplemented with 10% fetal bovine serum (Gibco, CA, USA). Cells were grown at 37°C and 5% CO2. At confluence, cells were trypsinized (0.25% trypsin with 0.1% ethylenediaminetetraacetic acid), and cells were passaged at a ratio of ~1:3. U87 was genetically altered via transfection with a reporter gene encoding firefly luciferase, creating the U87-Luc cell line for imaging. The line was subcloned using flow-cytometric cell sorting to obtain stable transfectants that were highly bioluminescent.
Construction of VEGF165-ephrin A1-PE38KDEL
The synthesis and assembly of hybrid genes encoding single-chain VEGF165-ephrin A1-PE38KDEL were accomplished using deoxyribonucleic acid (DNA) shuffling and cloning techniques. The fully assembled fusion gene (from the 5′ to 3′ end) consisted of an NcoI restriction site, an ATG initiation codon, genes for human VEGF165 and human ephrin A1, a 4GS linker for VEGF165 and ephrin A1, a KASGGPE amino acid linker for ephrin A1 and PE38KDEL, 362 residues of PE38 with the COOH terminus replaced with the endoplasmic reticulum (ER)-retention sequence Lys-Asp-Glu-Leu (KDEL), and a NotI restriction site at the 3′ end (shown in Figure 1A). The fragment of 2,230 bp between two restriction-site recognition regions was spliced into the GV218 lentivirus vector (GeneChem, Shanghai, PRC). DNA-sequencing analysis (Biomedical Genomics Center, University of Fudan, PRC) was used to confirm the gene sequence and in-frame cloning. Genes for monospecific cytotoxic VEGF-PE38KDEL and ephrin A1-PE38KDEL were generated using the same method.
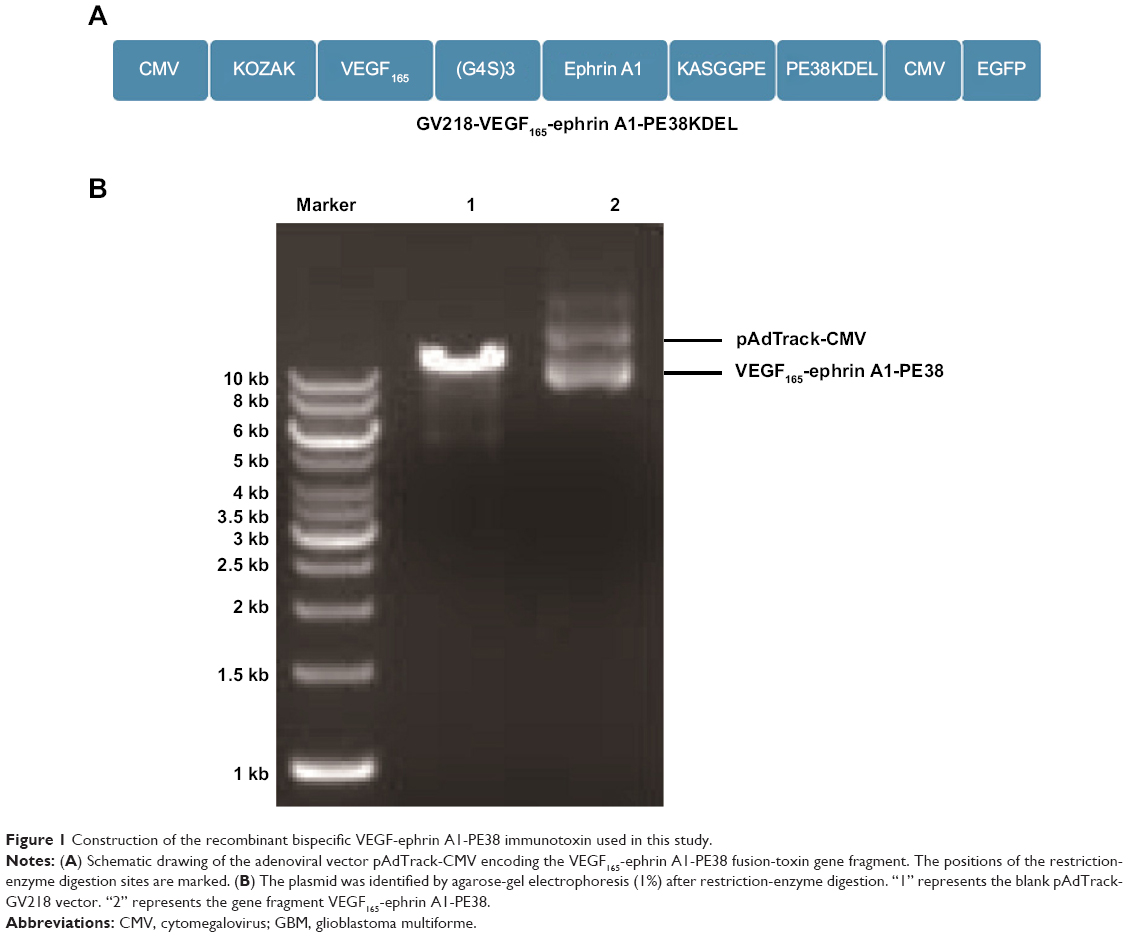 | Figure 1 Construction of the recombinant bispecific VEGF-ephrin A1-PE38 immunotoxin used in this study. |
Lentiviral vectors and ex vivo gene transduction
Lentivirus was packaged in 293 cells using the Lentiviral Vector System following the manufacturer’s protocol (GeneChem). Virus titer was determined by infection of 293 cells with serially diluted vector stock, followed by observation of green fluorescence protein (GFP)-positive cells. After three cycles of amplification and purification via density-gradient centrifugation, high-titer recombinant VEGF165-ephrin A1-PE38KDEL-containing lentiviral particles were harvested and stored at −80°C until use. For ex vivo gene transduction, 2×105 of hMSCs were plated in a 24-well plate 1 day before lentiviral infection. Cells were infected with VEGF165-ephrin A1-PE38KDEL at 100 MOI (multiplicity of infection) for 6 hours. Viral supernatants were subsequently replaced with fresh medium. Transduction efficiency was confirmed using fluorescence microscopy.
Detection of transgene expression in hMSCs
VEGF165-ephrin A1-PE38 transgene expression in transduced hMSC cells was confirmed using reverse-transcription polymerase chain reaction (RT-PCR). Briefly, total ribonucleic acid was purified using Trizol reagent. RT-PCR was carried out using One Step RT-PCR kit (Qiagen, Valencia, CA, USA) with primers for β-actin (5′-TGACTTCAACAGCGACACCCA-3′and 5′-CACCCTGTTGCTGTAGCCA AA-3′) and VEGF165-ephrin A1-PE38KDEL (5′-GACAAGAAAATCCCTGTGGG-3′ and 5′-CGTTTAACTCAAGCTGCCTC-3′). PCR conditions consisted of initial denaturation at 94°C for 4 minutes, followed by 30 cycles of denaturation at 94°C for 30 seconds, annealing at 52°C for 30 seconds, and extension at 72°C for 30 seconds. Amplified products were detected with 2% agarose-gel electrophoresis.
Quantitation of expression of VEGF165-ephrin A1-PE38KDEL in vitro
Secreted VEGF165-ephrin A1-PE38KDEL and VEGF165-PE38KDEL were measured using a VEGF enzyme-linked immunosorbent assay (ELISA) kit. Ephrin A1-PE38KDEL levels were assessed with an ephrin A1 ELISA kit (USCN Life Science, Wuhan, China). Absorbance was quantified at 450 nm using an ELISA reader (Molecular Devices, Sunnyvale, CA, USA) and the data analyzed. To quantify recombinant immunotoxin released in medium in vitro, hMSC cells (2×105/well) were plated in a 24-well plate and transduced with virus at 100 MOI. Culture supernatants were collected and fresh medium replaced every 3 days.
Cell proliferation
To determine the effects of the immunotoxin on various cancer cell lines, U251 and U87 cells were plated at a density of 1×104 cells/well in 96-well plates and cultured with increasing amounts of supernatant (25, 30, 40, 50, 75, and 100 μL) harvested from VEGF-ephrin A1-PE38KDEL hMSCs. Supernatant fractions from untransduced hMSCs were used as the control. The experiment was terminated 48 hours after treatment, and cell proliferation assessed using a Cell Counting Kit (CCK)-8 (Dojindo Molecular Technologies, Kumamoto, Japan). For blocking assays, supernatant fractions containing VEGF165-ephrin A1-PE38KDEL were preincubated with anti-ephrin A1 or anti-VEGF antibody (Santa Cruz Biotechnology, Inc., Dallas, TX, USA) for 1 hour at 37°C before addition to U251 cells. After further incubation for 72 hours, cell proliferation was measured as described earlier. Proliferation data were calculated as a percentage of untreated control cells. All experiments were conducted in triplicate.
In vivo efficacy studies
BALB/c nude mice (4–6 weeks old) weighing 17–19 g were purchased from Vital River Laboratory Animal Technology (Shanghai, People’s Republic of China). All animal-research procedures were performed according to local guidelines on the ethical use of animals, and the National Institutes of Health Guide for the Care and Use of Laboratory Animals.23 To determine the efficacy of VEGF165-ephrin A1-PE38KDEL against GBM, an intracranial xenograft tumor model was generated. Briefly, mice were anesthetized with isoflurane gas and immobilized in a stereotactic head frame (David Kopf Instruments, Tujunga, CA, USA). A middle incision was made on the skull and a burr hole placed 0.5 mm anterior to the bregma and 2.5 mm laterally to the midline using a drill (Foredom Electric, Bethel, CT, USA). With the stereotactic frame, a 25-gauge needle attached to 10 μL phosphate-buffered saline (PBS) was used to deliver tumor cells. The needle tip was inserted into the brain 3 mm deep relative to the skull surface and maintained at this depth for 2 minutes before injection of tumor cells. Under sterile conditions, 10 μL solution containing 4×105 U87 mCherry-flu cells was injected into the brain over a period of 5 minutes. After infusion, the needle was left in place for 5 minutes before slow withdrawal. The burr hole was sealed using sterile bone wax, and the wound was closed with surgical glue. All surgical procedures were performed under sterile conditions.
Mice were imaged in real time, and images were captured using the Ivis system (IVIS Lumina II, Caliper Life Sciences, Hopkinton, MA, USA) and analyzed with IGOR Pro 4.09a software (WaveMetrics, Beijing, People’s Republic of China). Before imaging, mice were anesthetized using isoflurane gas. All mice received 100 μL of 30 mg/kg D-luciferin aqueous solution (Gold Biotechnology, Beijing, People’s Republic of China) as a substrate for luciferase 10 minutes before imaging. Images were obtained with 5 minutes’ exposure time, and all regions of interest are expressed as photons/s/cm2/sr.
Immunohistochemistry
Firstly, the tumor tissues were fixed in 10% buffered formalin, and dehydrated and embedded in paraffin using routine protocols as previously reported.16 For immunohistochemical staining, formalin-fixed, paraffin-embedded tissue was sectioned, dried overnight at 65°C and deparaffinized in xylene. Sections were rehydrated through graded alcohol into water. Endogenous peroxidase was blocked with 3% hydrogen peroxide in 50% methanol for 10 minutes at room temperature. After rehydration, sections were washed with PBS and pretreated with citrate buffer (0.01 M citric acid, pH 6.0) for 20 minutes at 100°C in a microwave oven. Slides were rinsed with PBS, and incubated overnight at 4°C with primary polyclonal antibodies (antimouse CD34, 1:100). After being washed with PBS, sections were incubated with secondary antibody for 30 minutes at 37°C. Visualization was performed using a DAB Kit (DC 10; Boster Biological Technology, Wuhan, PRC) under a microscope. Nuclei were counterstained with hematoxylin, followed by dehydration and coverslip mounting. Following immunohistochemical staining, sections were exposed to 1% sodium periodate for 10 minutes, rinsed with distilled water for 5 minutes, and incubated with periodic acid–Schiff (PAS) for 15 minutes. Finally, all sections were counterstained with hematoxylin, and dehydrated and mounted under the microscopes. Normal human stomach mucous membrane was used as the positive control.
Quantification analysis of microvessel density and VM
The antibody of anti-CD34 was used as an endothelial marker to highlight intratumoral microvessels. Tissue sections were viewed at 200× magnification, and images were captured with a digital camera (Leica Q500MC; Berlin, Germany). Four fields per section were analyzed, excluding peripheral connective tissue and necrotic regions. Areas of CD34-positive samples were quantified using Image Pro Plus version 6.0. The microvessel area in each field was calculated as (area of CD34-positive object/measured tissue area) ×200. Mean values of microvessel-positive areas in each group were calculated from five tumor samples. CD34-PAS double staining was used to distinguish VM and endothelial-dependent vessels. We counted PAS-positive and CD34-negative vessels (×400) in four areas per sample. Mean values of the VM channel in each group were calculated from five tumor samples.
Statistical analysis
All experiments were conducted at least three times, with reproducible results. Results from representative experiments are presented. Where applicable, data are expressed as mean values ± standard error. The statistical significance of differences between the test and control groups was analyzed with SPSS 13.0 software. Unpaired Student t-tests were applied for comparison of two groups and one-way analysis of variance (ANOVA) for evaluation of multiple groups. Counts of blood vessels in the treatment and control groups were evaluated with one-way ANOVA. P-values <0.05 were considered statistically significant.
Results
Generation of lentivirus encoding ephrin A1-PE38
The construction of the pAdTrack-cytomegalovirus (CMV) vector encoding the fusion immunotoxin VEGF165-ephrin A1-PE38 is shown in Figure 1A. This fusion immunotoxin-expression vector contained a signal sequence and GFP. The pAdTrack-CMV vector ligated with the VEGF165-ephrin A1-PE38 complementary DNA (cDNA) was digested with restriction enzymes. The electrophoresed 1% agarose gel (Figure 1B) and DNA sequencing analysis indicated that the sequence of the ephrin A1-PE38 cDNA was correct.
Expression of VEGF165-ephrin A1-PE38KDEL in genetically modified hMSCs
In order to determine whether the recombinant immunotoxin could be successfully expressed in hMSCs via lentiviral transduction, cells were incubated with VEGF165-ephrin A1-PE38KDEL lentivirus at 100 MOI, and untransduced hMSCs were used as the control. At 72 hours after transduction, GFP expression was observed in nearly 90% of hMSCs, as shown in Figure 2A. Transduction with lentivirus expressing VEGF165-ephrin A1-PE38KDEL did not alter the morphology of hMSCs compared to untransduced cells. hMSCs expressing EGF165-ephrin A1-PE38KDEL presented a long fusiform, fibroblast-like shape similar to the morphological appearance of hMSCs. RT-PCR analysis revealed overexpression of VEGF165-ephrin A1-PE38KDEL in transduced hMSCs, in contrast to untransduced hMSCs (P=0.001514), as shown in Figure 2B. To confirm further whether VEGF165-ephrin A1-PE38KDEL was secreted from transduced hMSCs, ELISA was performed on collected medium to determine the concentrations of secreted immunotoxin at different time points after transduction. High levels of recombinant VEGF165-ephrin A1-PE38KDEL were detected on day 7, which persisted to 12 days, before decline (Figure 2C). As expected, VEGF-ephrin A1-PE38KDEL was not detected in supernatants from untransduced hMSCs by ELISA. Our results clearly showed that the ephrin A1-PE38KDEL was expressed and secreted from transduced hMSCs to the culture medium.
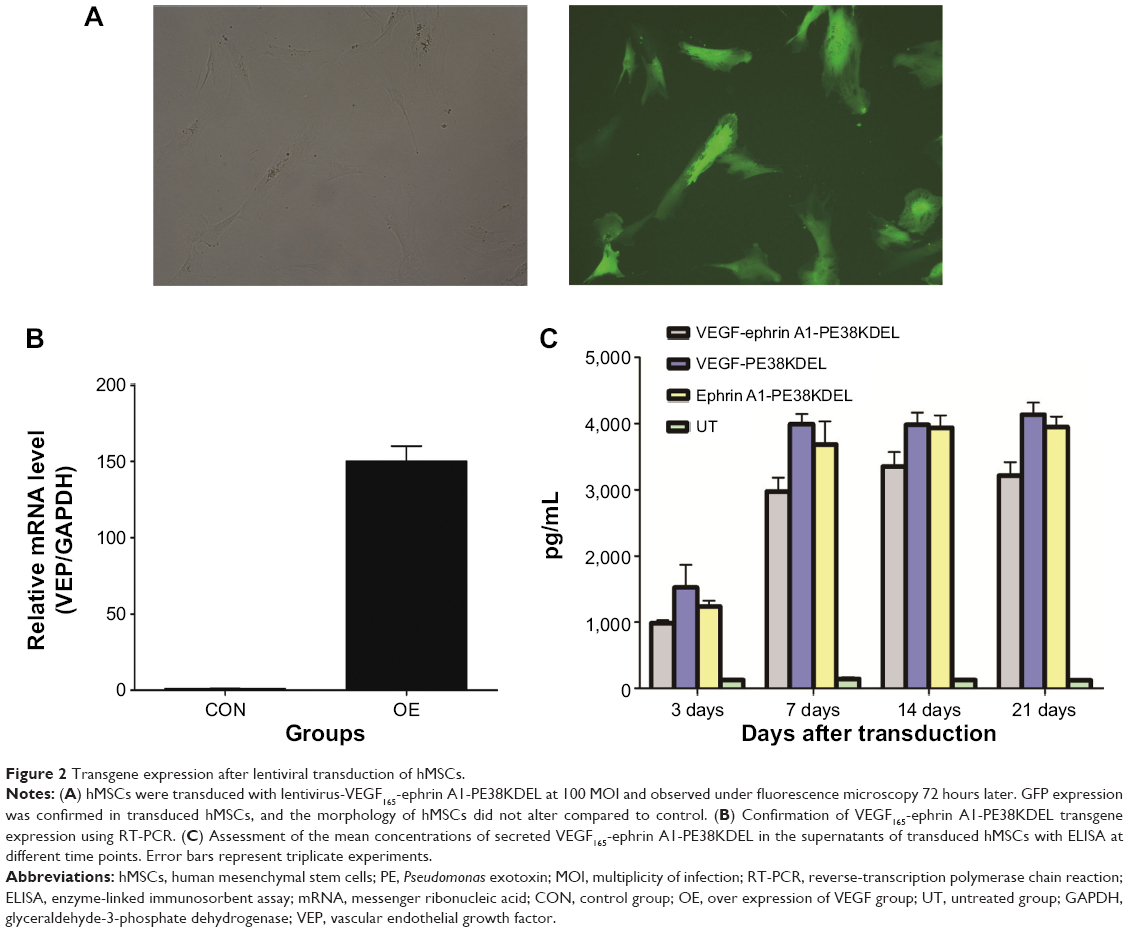 | Figure 2 Transgene expression after lentiviral transduction of hMSCs. |
Effect and specificity of recombinant VEGF165-ephrin A1-PE38KDEL
VEGF165-ephrin A1-PE38KDEL secreted from hMSC cells was tested to evaluate its killing effects against the U251 cells. We observed a clear dose-dependent killing effect by the supernatant fractions from VEGF165-ephrin A1-PE38KDEL-transduced hMSCs (Figure 3A), whereas untransduced hMSC supernatants did not induce cell death. Both bispecific VEGF165-ephrin A1-PE38KDEL and monospecific ephrin A1-PE38KDEL induced U251 and U87 cell death, while monospecific VEGF165-PE38KDEL did not affect cell viability (Figure 3B). These findings indicated that bispecific VEGF165-ephrin A1-PE38KDEL does not improve its monospecific immunotoxin in vitro, and its effect depended on the receptor recognizing targeting cells. To confirm that VEGF and ephrin A1 ligands on the VEGF165-ephrin A1-PE38KDEL molecule are both active, anti-VEGF or ephrin A1 antibodies were used to block the ligands, and the effect of killing of U251 cells by VEGF165-ephrin A1-PE38KDEL (Figure 3C) was examined. When added to 50 μL supernatant of VEGF165-ephrin A1-PE38KDEL-transduced hMSCs, ephrin A1 antibody blocked 85% of the cytotoxic effect, while anti-VEGF had no blocking effect, possibly because when one ligand was blocked, the other remained active. Our data collectively indicate that VEGF165-ephrin A1-PE38KDEL from transduced hMSCs can effectively induce cell death of GBM cells in vitro.
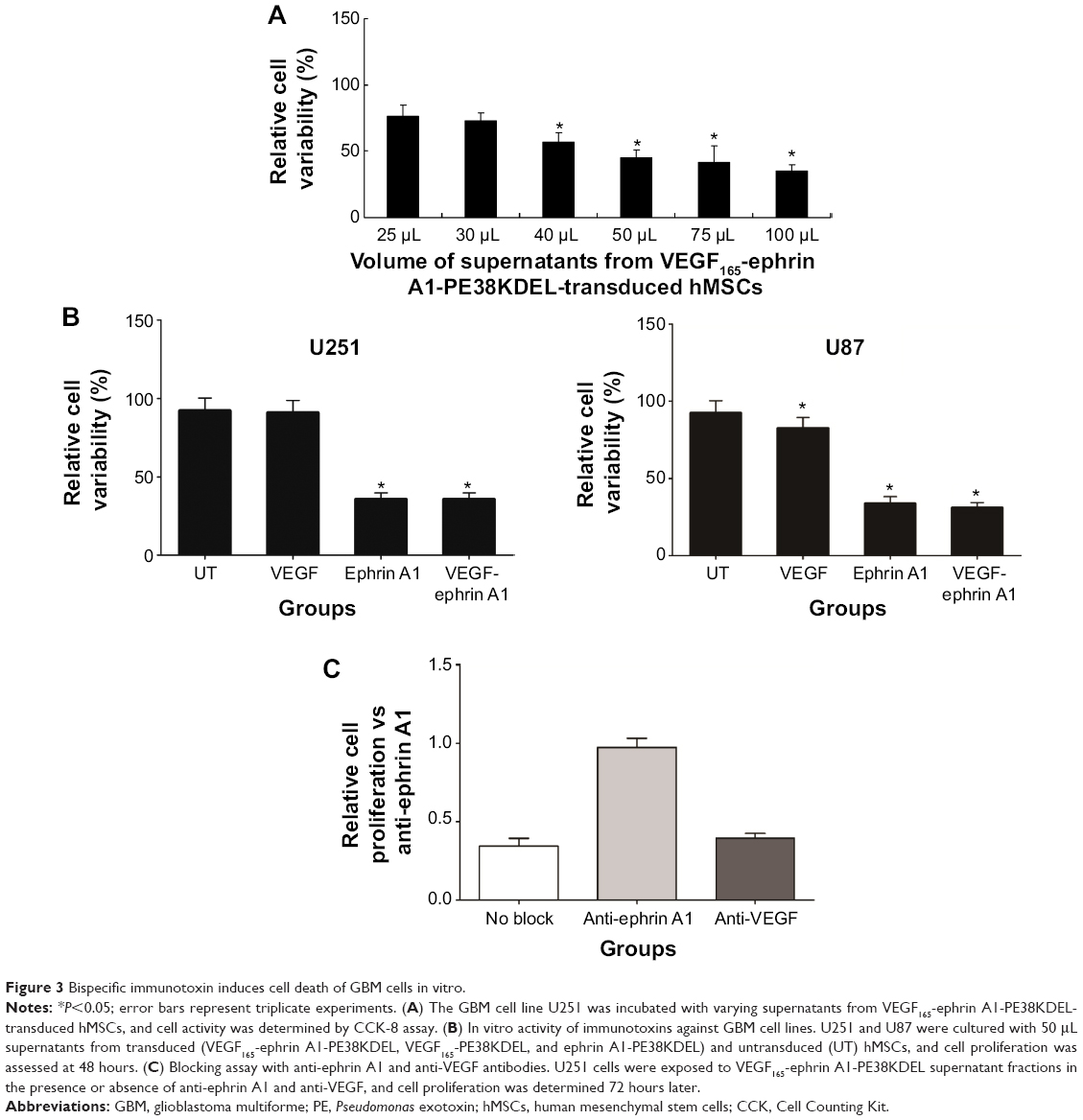 | Figure 3 Bispecific immunotoxin induces cell death of GBM cells in vitro. |
Antitumor effect of VEGF165-ephrin A1-PE38KDEL in vivo
To determine whether VEGF165-ephrin A1-PE38KDEL mediates an antitumor effect in vivo, a GBM-bearing mouse model was developed. In this GBM-bearing mouse model, U87 cells were transfected with a luciferase reporter gene and intracranially injected into the nude mice. Twenty-five xenografted mice were randomly divided into five groups, and the treatment was initiated on day 7. Mice were administered with 105 cells of VEGF165-ephrin A1-PE38KDEL-transduced, VEGF165-PE38KDEL-transduced, ephrin A1-PE38KDEL-transduced, or untransduced hMSCs. Control mice were given PBS. Individual data from each group are shown in Figure 4A. Significant reduction in tumors over time, determined as a decrease in total bioluminescent activity, was observed in the VEGF165-ephrin A1-PE38KDEL-transduced hMSC-treated group (denoted M1–M5). Mice from the VEGF165-PE38KDEL- and ephrin A1-PE38KDEL-transduced hMSC-treated groups showed moderate reduction in tumors (denoted M6–M10 and M11–M15). In contrast, no tumor response was observed in five mice administered untransduced hMSCs (denoted M16–M20). Tumors in untreated controls (denoted M21–M25) progressed in an aggressive manner. The mean total bioluminescent activity for each group was recorded and analyzed (Figure 4B). Curves were significantly different on day 28. Three of five (60%) VEGF165-ephrin A1-PE38KDEL-treated mice were long-term tumor-free survivors at day 65. Survival analysis revealed significantly extended median survival of mice treated with VEGF165-ephrin A1-PE38KDEL-transduced hMSCs relative to VEGF165-PE38KDEL or ephrin A1-PE38KDEL-transduced hMSCs (34 days versus 25 or 27 days, Figure 4B; P=0.006). Kaplan–Meier survival curves (Figure 4C) demonstrated a marked increase in survival time for VEGF165-ephrin A1-PE38KDEL-transduced hMSC-treated mice, compared to those treated with VEGF165-PE38KDEL- and ephrin A1-PE38KDEL-transduced hMSCs and negative controls (P<0.01). In order to confirm the antitumor effect was induced by immunotoxin, we further determined the concentrations of immunotoxin in tumors at different time points. The immunotoxins in tumors were secreted by hMSCs slowly, as concentrations increased slightly at the first week. Then, the immunotoxins kept at a stable level (60–80 pg/mL) to inhibit the growth of tumor cells during the following 2 weeks. The bioluminescent activity of tumor-bearing mice significantly declined from day 28 after administration of transduced hMSCs, which was synchronous with the concentration of immunotoxins (Figure 4D). These results suggested that BITs can be delivered by hMSCs to target and inhibit tumor growth in vivo.
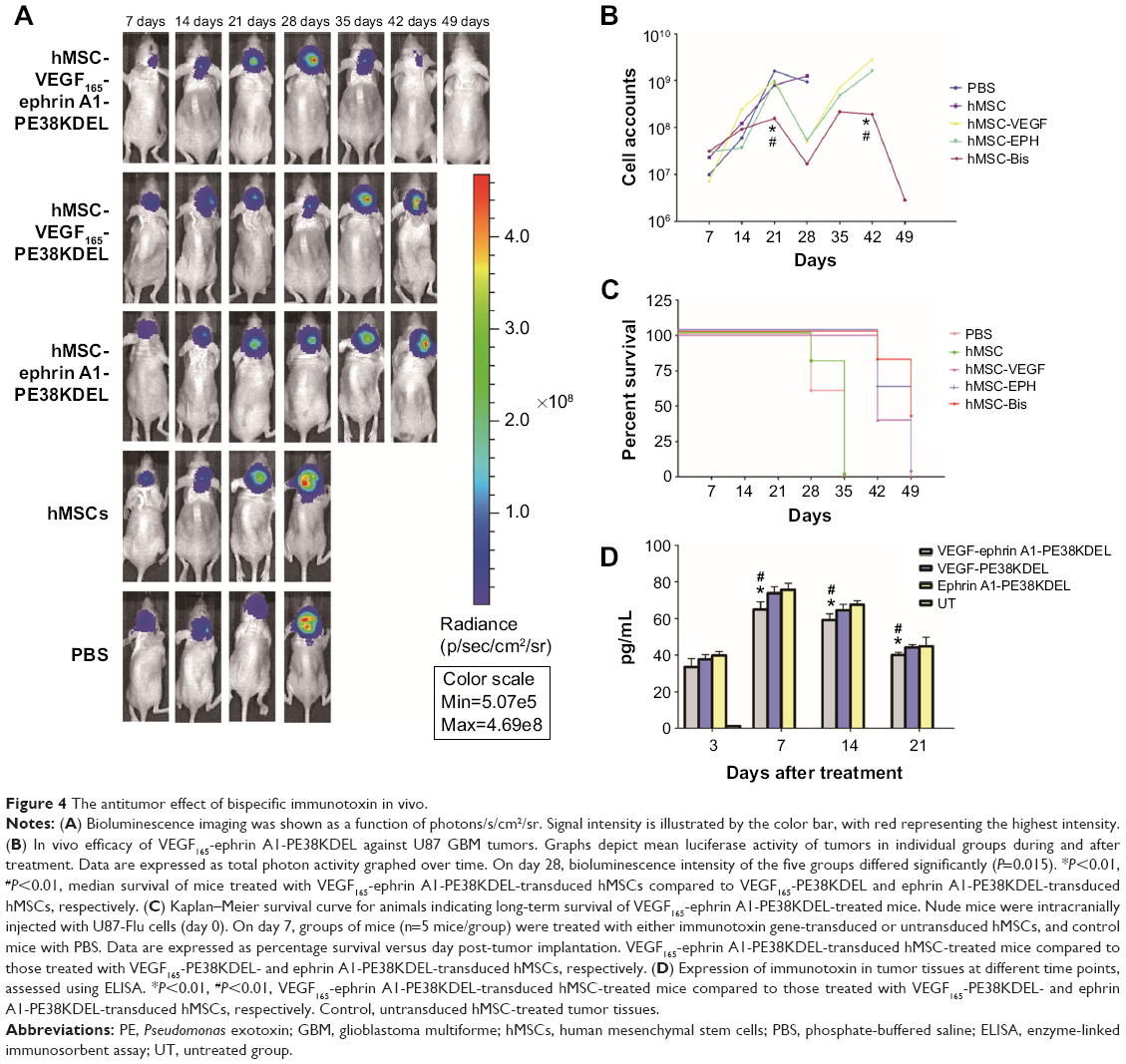 | Figure 4 The antitumor effect of bispecific immunotoxin in vivo. |
Immunohistochemistry analysis
The immunohistochemistry assay was performed to investigate the target of immunotoxins. CD34 is a specific endothelial cell marker. Tumors were stained with antibodies directed against CD34. Notably, tumors from mice treated with VEGF165-ephrin A1-PE38KDEL and VEGF165-PE38KDEL displayed decreased CD34 expression compared with the other groups of mice (Figure 5A). In quantitative immunohistochemical analyses of tumor-microvessel density (MVD; Figure 5B), tumor specimens from mice treated with VEGF165-ephrin A1-PE38KDEL or VEGF165-PE38KDEL-transduced hMSCs had significantly lower tumor MVD compared with specimens from the other groups.
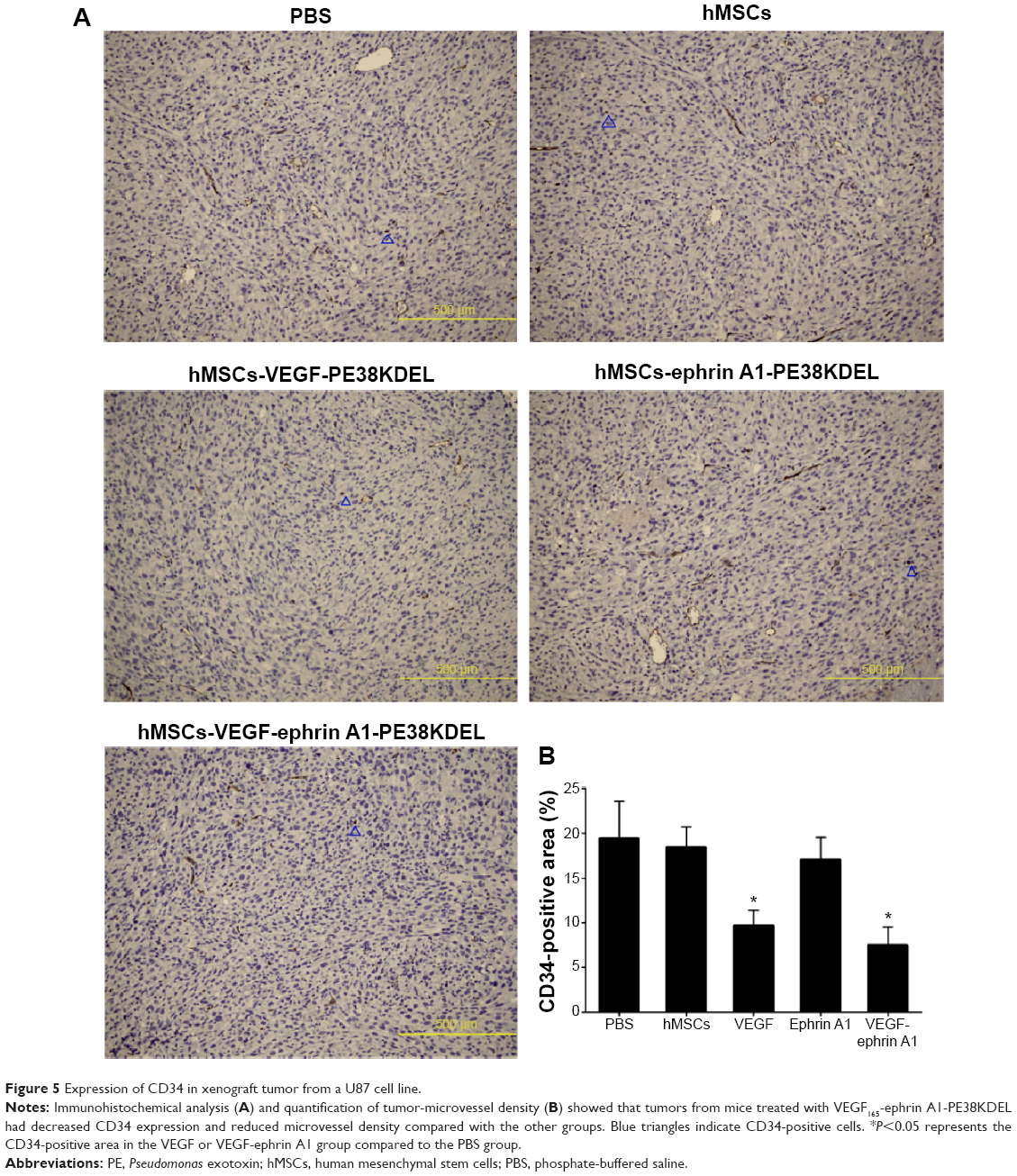 | Figure 5 Expression of CD34 in xenograft tumor from a U87 cell line. |
Ephrin A1-PE38KDEL-transduced hMSCs display decreased VM
VM is an alternate mechanism of vascularization in malignant tumors by tumor cells instead of endothelial cells. Double-immunofluorescence staining assay with CD34 and PAS results showed that tumors from mice treated with VEGF165-ephrin A1-PE38KDEL- or ephrinA1-PE38KDEL-transduced hMSCs displayed decreased VM compared with the other groups (Figure 6).
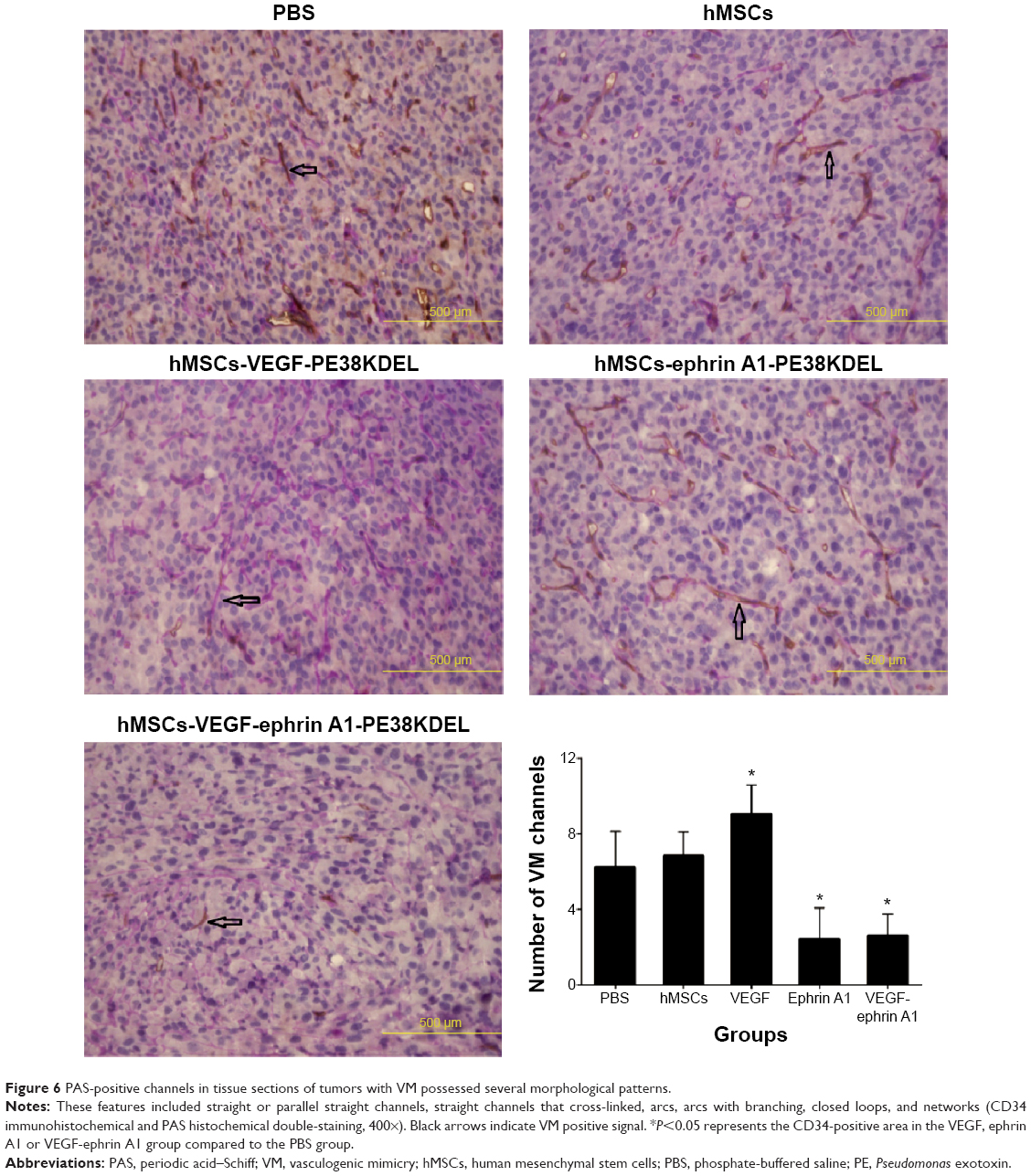 | Figure 6 PAS-positive channels in tissue sections of tumors with VM possessed several morphological patterns. |
Discussion
Immunotoxins are the proteins used to treat cancer, and are composed of an antibody fragment linked to a toxin. The immunotoxin is a hybrid molecule that consists of a targeting and toxic moiety. BITs contain a single-chain molecule with two distinct targeting ligands fused to a single toxin. Several studies have reported a potent anti-tumor effect of BITs. Earlier leukemia research showed that a BIT-designated DT2219ARL sensitized human Daudi B lymphoma cells. DT2219ARL displayed 1,000-fold more potency than DT22 or DT19 alone or an equal mixture of DT22 and DT19.11 Similarly, another BIT, DTEGF13, exerted 32- to 2,860-fold greater cytotoxicity in a variety of epithelial cancer cells lines than DTEGF or DTIL13 alone or an equal mixture of DTEGF and DTIL13. The cell lines tested included prostate carcinoma, lung carcinoma, glioblastoma, and pancreatic carcinoma.12 An earlier study by Stish et al13 tested the antitumor effect of BIT cotargeting human IL-13 and EGF receptors in a mouse glioblastoma xenograft model, with promising results. Immunotoxins can target the overexpressed antigens on the tumor-cell surface. The specificity of immunotoxin therapy relies on the presence of a unique receptor or antigen present on tumor cells, but not normal tissue. Application of BIT represents an important advancement in the field, because only certain combinations of ligands can enhance activity to this extent.
The VEGF family of growth factors and receptor tyrosine kinases mediate vasculogenesis in solid tumors, and have been a significant focus of research attention to date. The major mediator of tumor angiogenesis is VEGF-A, including VEGF121, VEGF145, VEGF165, VEGF189, and VEGF206. Among all of these isoforms, VEGF165 has the strongest ability to recognize the VEGFR. VEGF signals mainly via VEGFR-2, which is expressed at elevated levels by endothelial cells engaged in tumor angiogenesis.24 EphA2, a member of the Eph-receptor tyrosine-kinase family, is overexpressed in GBM, and represents a novel potential molecular target for therapeutic approaches, such as targeted drug delivery.25
In our previous studies, we constructed the recombinant monospecific immunotoxins ephrin A1-PE38 and VEGF-PE38, which exerted a significant antitumor effect in GBM by targeting EphA2 receptor-overexpressing GBM cells and tumor endothelial cells. In this study, VEGF165 and ephrin A1 were linked to a truncated PE A molecule to create the BIT VEGF165-ephrin A1-PE38KDEL. The VEGF165 gene of recombinant VEGF165-ephrin A1-PE38KDEL contains 27-residue signal sequence, which facilitates secretion into cell-culture supernatants. We fused the genes encoding human VEGF165 and human ephrin A1, a 4GS linker for VEGF165 and ephrin A1, the seven-amino acid KASGGPE linker for ephrin A1 and PE38KDEL, and 362 residues of PE38, where the COOH terminus is replaced with the ER-retaining sequence KDEL, which prevents the secretion of luminal ER proteins, leading to increased ER accumulation and enhanced potency of targeted toxins.26 Intracellular expression and subsequent secretion of the recombinant immunotoxin from transduced hMSCs were achieved through infection with lentivirus encoding VEGF165-ephrin A1-PE38KDEL. Genetically engineered hMSCs expressing recombinant immunotoxin were secreted persistently into the culture medium. In vitro experiments showed that supernatants containing VEGF165-ephrin A1-PE38KDEL and ephrin A1-PE38KDEL are capable of killing GBM cells. In contrast, VEGF165-PE38KDEL-transduced hMSCs expressed immunotoxin, but did not exert a cell-killing effect.
Owing to their tumor-tropic migratory capacity, hMSCs have recently emerged as promising delivery vehicles of therapeutic agents for malignant tumors, including GBM. hMSCs can be obtained in relatively large numbers via standard bone marrow aspiration, are easily expanded in culture, and are capable of being transduced to high levels with adenoviral and lentiviral vectors. Their administration can be autologous, given that they may be immunoprivileged.18 These characteristics make hMSCs an excellent vehicle for the delivery of immunotoxins if induced to secrete immunotoxins via gene transduction.
An important aspect of this study was the use of a luciferase reporter gene model that permitted the assessment of systemic tumor development in real time. The results disclosed the remarkable antitumor potency of the BIT. Specifically, intratumoral administration of hMSCs transduced with VEGF165-ephrin A1-PE38KDEL lentivirus resulted in a significant antitumor effect in a U87 intracranial tumor model. VEGF165-PE38KDEL and ephrin A1-PE38KDEL-transduced hMSC-treated groups showed moderate reduction in tumors, while tumors of untransduced hMSCs and untreated control groups progressed in an aggressive manner. The mean total bioluminescent activity showed significantly different curves on day 28. Survival analysis additionally revealed that the median survival of mice treated with VEGF165-ephrin A1-PE38KDEL-transduced hMSCs was significantly extended, relative to those treated with VEGF165-PE38KDEL- or ephrin A1-PE38KDEL-transduced hMSCs.
Glioblastomas are highly angiogenic and characterized by microvascular proliferation.27 However, the clinical effects of anti-blood vessel endothelium in glioblastoma remain unsatisfactory.28 While current antiangiogenic strategies are mainly directed against tumor endothelial cells, tumors not only rely on host blood vessels for nourishment but also form their own vasculature. VM, an endothelium-independent microcirculation pattern, exists in many malignant tumor types, and thus presents an additional target in antiangiogenesis strategies to treat solid tumors.29 The presence of VM correlates with increased risk of metastasis and poor clinical outcome.30,31 Several key molecules, such as EphA2, have been implicated in VM.32 The tumor microenvironment, including hypoxia, ischemia, and acidosis, plays a major role in trans-endothelial differentiation of aggressive tumor cells. Dedifferentiation of tumor cells is the key to VM-channel formation. Epithelial cell kinase (EphA2), a tyrosine-kinase receptor, is specifically expressed in highly aggressive melanoma cells. Inhibitors of tyrosine-kinase activity33 and transient knockout of EphA2 hinder VM-channel formation.
Our immunohistochemical findings showed that treatment of malignant gliomas with hMSC-VEGF165-ephrin A1-PE38KDEL and hMSC-VEGF165-PE38KDEL led to decreased expression of CD34 compared with the hMSC-ephrin A1-PE38KDEL and control groups. In quantitative immunohistochemical analyses of malignant gliomas, MVD in tumor specimens from mice treated with hMSC-VEGF165-ephrin A1-PE38KDEL and hMSC-VEGF165-PE38KDEL was significantly lower compared with that of control-treated mice. The results of double-immunofluorescence staining (CD34 and PAS) showed decreased VM in tumors from mice treated with hMSC-VEGF165-ephrin A1-PE38KDEL and hMSC-ephrin A1-PE38KDEL compared with the other groups. These results confirm our hypothesis that the newly generated BIT VEGF165-ephrin A1-PE38KDEL targets both tumor endothelial and EphA2 receptor-overexpressing GBM cells.
The significance of this study is in the development of VEGF165-ephrin A1-PE38KDEL, a novel anti-GBM agent with potential for clinical development. By linking VEGF165 and ephrin A1 to a truncated PE A molecule, we created a BIT targeting both endothelial cells and tumor VM-like cells. VEGF165-ephrin A1-PE38KDEL induced a significant decrease in tumor burden in an intracranial GBM tumor model. We further demonstrated successful production of the recombinant immunotoxin in hMSCs via lentivirus transduction, and showed potent killing activity of VEGF165-ephrin A1-PE38KDEL-containing hMSCs in GBM cells in vitro. Administration of hMSCs transduced with immunotoxin-expressing virus additionally resulted in a significant in vivo anticancer effect against U87 gliomas.
In conclusion, data from the current study have confirmed that the BIT VEGF165-ephrin A1-PE38KDEL has greater therapeutic efficacy than either monospecific VEGF165-PE38KDEL or Ephrin A1-PE38KDEL in vivo. This novel immunotoxin effectively destroys the tumor vascular system and the formation of tumor VM in a U87 intracranial tumor model. Our findings support the application of immunotoxins secreted from hMSCs as a novel strategy to improve treatment options for malignant glioma.
Acknowledgments
The present work received grants from the Nature Science Foundation of Guangdong (S2012010009088) and the National Nature Science Foundation of China (81272806).
Disclosure
The authors report no conflicts of interest in this work.
References
Braun K, Wiessler M, Ehemann V, et al. Treatment of glioblastoma multiforme cells with temozolomide-BioShuttle ligated by the inverse Diels-Alder ligation chemistry. Drug Des Devel Ther. 2009;2:289–301. | ||
Maniotis AJ, Folberg R, Hess A, et al. Vascular channel formation by human melanoma cells in vivo and in vitro: vasculogenic mimicry. Am J Pathol. 1999;155(3):739–752. | ||
Folberg R, Maniotis AJ. Vasculogenic mimicry. APMIS. 2004;112(7–8):508–525. | ||
Wolf P, Elsasser-Beile U. Pseudomonas exotoxin A: from virulence factor to anti-cancer agent. Int J Med Microbiol. 2009;299(3):161–176. | ||
Kreitman RJ, Squires DR, Stetler-Stevenson M, et al. Phase I trial of recombinant immunotoxin RFB4(dsFv)-PE38 (BL22) in patients with B-cell malignancies. J Clin Oncol. 2005;23(27):6719–6729. | ||
Pastan I, Hassan R, FitzGerald DJ, Kreitman RJ. Immunotoxin treatment of cancer. Annu Rev Med. 2007;58(1):221–237. | ||
Hardee ME, Zagzag D. Mechanisms of glioma-associated neovascularization. Am J Pathol. 2012;181(4):1126–1141. | ||
Chimote G, Sreenivasan J, Pawar N, Subramanian J, Sivaramakrishnan H, Sharma S. Comparison of effects of anti-angiogenic agents in the zebrafish efficacy-toxicity model for translational anti-angiogenic drug discovery. Drug Des Devel Ther. 2014;8:1107–1123. | ||
Hess AR, Seftor EA, Gardner LM, et al. Molecular regulation of tumor cell vasculogenic mimicry by tyrosine phosphorylation: role of epithelial cell kinase (Eck/EphA2). Cancer Res. 2001;61(8):3250–3255. | ||
Kreitman RJ, Pastan I. Accumulation of a recombinant immunotoxin in a tumor in vivo: fewer than 1000 molecules per cell are sufficient for complete responses. Cancer Res. 1998;58(5):968–975. | ||
Vallera DA, Chen H, Sicheneder AR, Panoskaltsis-Mortari A, Taras EP. Genetic alteration of a bispecific ligand-directed toxin targeting human CD19 and CD22 receptors resulting in improved efficacy against systemic B cell malignancy. Leuk Res. 2009;33(9):1233–1242. | ||
Stish BJ, Chen H, Shu Y, Panoskaltsis-Mortari A, Vallera DA. A bispecific recombinant cytotoxin (DTEGF13) targeting human interleukin-13 and epidermal growth factor receptors in a mouse xenograft model of prostate cancer. Clin Cancer Res. 2007;13(21):6486–6493. | ||
Stish BJ, Oh S, Vallera DA. Anti-glioblastoma effect of a recombinant bispecific cytotoxin cotargeting human IL-13 and EGF receptors in a mouse xenograft model. J Neurooncol. 2008;87(1):51–61. | ||
Kreitman RJ. Immunotoxins for targeted cancer therapy. AAPS J. 2006;8(3):E532–E551. | ||
Rustamzadeh E, Vallera DA, Todhunter DA, Low WC, Panoskaltsis-Mortari A, Hall WA. Immunotoxin pharmacokinetics: a comparison of the anti-glioblastoma bi-specific fusion protein (DTAT13) to DTAT and DTIL13. J Neurooncol. 2006;77(3):257–266. | ||
Hall B, Dembinski J, Sasser AK, Studeny M, Andreeff M, Marini F. Mesenchymal stem cells in cancer: tumor-associated fibroblasts and cell-based delivery vehicles. Int J Hematol. 2007;86(1):8–16. | ||
Nakamizo A, Marini F, Amano T, et al. Human bone marrow-derived mesenchymal stem cells in the treatment of gliomas. Cancer Res. 2005;65(8):3307–3318. | ||
Lee DH, Ahn Y, Kim SU, et al. Targeting rat brainstem glioma using human neural stem cells and human mesenchymal stem cells. Clin Cancer Res. 2009;15(15):4925–4934. | ||
Nakamura K, Ito Y, Kawano Y, et al. Antitumor effect of genetically engineered mesenchymal stem cells in a rat glioma model. Gene Ther. 2004;11(14):1155–1164. | ||
Hu CC, Ji HM, Chen SL, et al. Investigation of a plasmid containing a novel immunotoxin VEGF165-PE38 gene for antiangiogenic therapy in a malignant glioma model. Int J Cancer. 2010;127(9): 2222–2229. | ||
Sun XL, Xu ZM, Ke YQ, Hu CC, Wang SY, Ling GQ. Molecular targeting of malignant glioma cells with an EphA2-specific immunotoxin delivered by human bone marrow-derived mesenchymal stem cells. Cancer Lett. 2011;312(1):168–177. | ||
Hu CC, Ke YQ, Sun XL, et al. Human mesenchymal stem cells-like cells as cellular vehicles for delivery of immunotoxin in vitro. Biotechnol Lett. 2009;31(2):181–189. | ||
The National Academies Press. Guide for the care and use of laboratory animals. Washington, DC: The National Academies Press; 1996. Available from: http://www.nap.edu/openbook.php?record_id=5140&page=R1. Accessed May 14, 2015. | ||
Ellis LM, Hicklin DJ. VEGF-targeted therapy: mechanisms of anti-tumour activity. Nat Rev Cancer. 2008;8(8):579–591. | ||
Hatano M, Eguchi J, Tatsumi T, et al. EphA2 as a glioma-associated antigen: a novel target for glioma vaccines. Neoplasia. 2005;7(8): 717–722. | ||
Kreitman RJ, Pastan I. Importance of the glutamate residue of KDEL in increasing the cytotoxicity of Pseudomonas exotoxin derivatives and for increased binding to the KDEL receptor. Biochem J. 1995;307(Pt 1):29–37. | ||
Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359(5):492–507. | ||
Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. 2008;8(8):592–603. | ||
van der Schaft DW, Hillen F, Pauwels P, et al. Tumor cell plasticity in Ewing sarcoma, an alternative circulatory system stimulated by hypoxia. Cancer Res. 2005;65(24):11520–11528. | ||
Sharma N, Seftor RE, Seftor EA, et al. Prostatic tumor cell plasticity involves cooperative interactions of distinct phenotypic subpopulations: role in vasculogenic mimicry. Prostate. 2002;50(3):189–201. | ||
Shirakawa K, Wakasugi H, Heike Y, et al. Vasculogenic mimicry and pseudo-comedo formation in breast cancer. Int J Cancer. 2002;99(6):821–828. | ||
Zhang S, Zhang D, Sun B. Vasculogenic mimicry: current status and future prospects. Cancer Lett. 2007;254(2):157–164. | ||
Zhang M, Drummen GP, Luo S. Anti-insulin-like growth factor-IIP3 DNAzymes inhibit cell proliferation and induce caspase-dependent apoptosis in human hepatocarcinoma cell lines. Drug Des Devel Ther. 2013;7:1089–1102. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.