")
Back to Journals » OncoTargets and Therapy » Volume 14
A Novel Biological Activity of the STAT3 Inhibitor Stattic in Inhibiting Glutathione Reductase and Suppressing the Tumorigenicity of Human Cervical Cancer Cells via a ROS-Dependent Pathway
Authors Xia Y, Wang G, Jiang M, Liu X, Zhao Y, Song Y, Jiang B, Zhu D, Hu L, Zhang Z, Cao T , Wang JM, Hu J
Received 7 April 2021
Accepted for publication 17 June 2021
Published 5 July 2021 Volume 2021:14 Pages 4047—4060
DOI https://doi.org/10.2147/OTT.S313507
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Arseniy Yuzhalin
Yuchen Xia,1,2 Guihua Wang,2 Manli Jiang,3 Xueting Liu,3 Yan Zhao,4 Yinghui Song,5 Binyuan Jiang,3 Demao Zhu,4 Ling Hu,6 Zhao Zhang,6 Ting Cao,6 Ji Ming Wang,7 Jinyue Hu3
1Graduate School, Hunan University of Chinese Medicine, Changsha, Hunan, People’s Republic of China; 2Department of Oncology, Changsha Central Hospital, University of South China, Changsha, Hunan, People’s Republic of China; 3Medical Research Center, Changsha Central Hospital, University of South China, Changsha, Hunan, People’s Republic of China; 4Department of Pathology, Changsha Central Hospital, University of South China, Changsha, Hunan, People’s Republic of China; 5Department of Hepatobiliary Surgery, Hunan Provincial People’s Hospital, Hunan Normal University, Changsha, Hunan, People’s Republic of China; 6Department of Clinical Laboratory, Changsha Central Hospital, University of South China, Changsha, Hunan, People’s Republic of China; 7Laboratory of Cancer Immunometabolism, Center for Cancer Research, National Cancer Institute at Frederick, Frederick, MD, 21702, USA
Correspondence: Jinyue Hu
Medical Research Center, Changsha Central Hospital, University of South China, Changsha, Hunan, 410004, People’s Republic of China
Tel +86-731-85667664
Fax +86-731-85668157
Email [email protected]
Guihua Wang
Department of Oncology, Changsha Central Hospital, University of South China, Changsha, Hunan, People’s Republic of China
Tel +86-731-85667664
Fax +86-731-85668157
Email [email protected]
Introduction: Glutathione reductase (GSR) provides reduced glutathione (GSH) to maintain redox homeostasis. Inhibition of GSR disrupts this balance, resulting in cell damage, which benefits cancer therapy. However, the effect of GSR inhibition on the tumorigenicity of human cervical cancer is not fully understood.
Materials and Methods: Tissue microarray analysis was employed to determine GSR expression in cervical cancer tissues by immunohistochemical staining. Cell death was measured with PI/FITC-annexin V staining. mRNA levels were measured via quantitative RT-PCR. Protein expression was measured by Western blotting and flow cytometry. STAT3 deletion was performed with CRISPR/Cas9 technology. GSR knockdown was achieved by RNA interference. Reactive oxygen species (ROS) levels were measured by DCF staining. GSR enzymatic activity was measured with a GSR assay kit. The effect of GSR inhibition on the growth of tumors formed by cervical cancer cells was investigated using a xenograft model.
Results: The expression of GSR was increased in human cervical cancer tissues, as shown by immunohistochemical staining. GSR knockdown by RNA interference in human cervical cancer cell lines resulted in cell death, suggesting the ability of GSR to maintain cancer cell survival. The STAT3 inhibitor 6-nitrobenzo[b]thiophene 1,1-dioxide (Stattic) also inhibited the enzymatic activity of GSR and induced the death of cervical cancer cells. More importantly, Stattic decreased the growth of xenograft tumors formed by cervical cancer cells in nude mice. Mechanistically, tumor cell death induced by Stattic-mediated GSR inhibition was ROS-dependent, since the ROS scavengers GSH and N-acetyl cysteine (NAC) reversed the effect of Stattic. In contrast, pharmacological and molecular inhibition of STAT3 did not induce the death of cervical cancer cells, suggesting a STAT3-independent activity of Stattic.
Conclusion: Stattic inhibits the enzymatic activity of GSR and induces STAT3-independent but ROS-dependent death of cervical cancer cells, suggesting its potential application as a therapeutic agent for human cervical cancers.
Keywords: Stattic, glutathione reductase, cell death, cervical cancer, reactive oxygen species, tumor growth
Introduction
Cervical cancer is a significant threat to the health of women. Screening for early detection and vaccination for HPV have reduced the incidence of cervical cancer in developed countries, but its mortality rates are 2-fold higher in developing countries, as the fourth leading cause of cancer-related death.1 Over the years, various treatment strategies, including surgery, radiotherapy, chemotherapy, and immunotherapy, have been developed for cervical cancer. Although chemotherapy is still one of the main treatment methods for cervical cancer, it is limited by the lack of tumor sensitivity to drugs, the potential side effects, and the development of multidrug resistance, leading to a decrease in the long-term efficacy of anticancer therapy.2 Therefore, it is important to discover novel agents for cervical cancer treatment.
Reactive oxygen species (ROS) are oxygen-containing free radicals that are produced through aerobic metabolism, and the mitochondrial respiratory chain, peroxisomes, and NADPH oxidase as the main endogenous sources.3 Under physiological conditions, there is a balance between the generation of ROS (pro-oxidants) and ROS elimination (antioxidant capacity), with low levels of ROS promoting cell proliferation.4 However, moderate levels of ROS cause either temporary or permanent growth arrest, while high levels of ROS cause cell death via apoptotic or necrotic mechanisms.5 In cancer cells, oxygen metabolism is aberrant because of hypoxia, leading to excessive accumulation of ROS to levels undesirable for cell survival.5 Under this selection pressure from excessive ROS, cancer cells have developed powerful antioxidant systems, including the production of nonenzymatic antioxidants, such as GSH, and enzymatic antioxidant proteins, such as superoxide dismutase (SOD), to neutralize the cytotoxic effect of ROS.5 By targeting these antioxidant molecules within tumors, various natural and synthetic compounds have been selected for cancer therapy.5–11
GSH is composed of glutamic acid, cysteine, and glycine and is widely distributed throughout the human body.12 As a crucial antioxidant, GSH exhibits antiproliferative, antiapoptotic, and anti-inflammatory effects. It maintains cellular homeostasis by inhibiting the intracellular level of ROS, thereby protecting cells from damage induced by oxidative stress. Glutathione reductase (GSR) is the key enzyme that catalyzes glutathione disulfide (GSSG) to reduced GSH.13 Hence, GSR may be an important target for cancer therapy.
Stattic (6-nitrobenzo[b]thiophene 1,1-dioxide) is a synthetic nonpeptide small molecule that selectively inhibits the activation of STAT3 and induces the apoptosis of breast cancer cells.14 Stattic also promotes chemosensitivity and/or radiosensitivity in multiple cancer types,15–18 suggesting that it may be a valuable clinical cancer therapeutic agent. However, the exact mechanisms of Stattic-induced cell death are not well understood.
In this study, we found that the expression of GSR was increased in human cervical cancer tissues. RNA interference of GSR induced the death of cervical cancer cells. In addition, Stattic inhibited the enzymatic activity of GSR and induced the death of human cervical cancer cells. Furthermore, Stattic decreased the growth of xenograft tumors formed by cervical cancer cells in nude mice. Mechanistically, we found that tumor cell death induced by Stattic-mediated inhibition of GSR was ROS-dependent but STAT3-independent. These results indicate a novel biological function of Stattic and suggest its potential application as a therapeutic agent for human cervical cancer.
Materials and Methods
Animals and Cell Lines
Female BALB/c nude mice (6–8 weeks old) were purchased from SLAC Laboratory Animal Center (Shanghai, China). All animal studies were carried out in accordance with the Guidelines for the Care and Use of Laboratory Animals issued by the National Institutes of Health and approved by the Animal Ethics Committee of the Changsha Central Hospital, University of South China (No. CCH-AEC-2020-02). Animals were maintained with standard rodent chow and free access to water under controlled conditions with a 12-h light and 12-h dark cycle and a temperature of 24 ± 2°C.
The human cervical cancer cell lines C33A, CaSki, SiHa, and HeLa were purchased from ATCC (Manassas, VA, USA). Cells were grown in DMEM containing 10% FCS, 100 units/mL penicillin, and 100 mg/mL streptomycin. All cells were cultured in a humidified atmosphere with 5% CO2 at 37°C.
Reagents
A mouse monoclonal anti-human GSR (sc-133245, 1:1000) antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Rabbit anti-human phosphorylated STAT3 (#9145, 1:1000), total STAT3 (#4904, 1:1000), and caspase 3 (#9662, 1:1000) antibodies were purchased from Cell Signaling Technology (Beverly, MA, USA). The STAT3 inhibitors Stattic (#2798) and niclosamide (#4079) were purchased from Tocris (Ellisville, MO, USA). A GSR assay kit (S0055), a ROS assay kit (S0033), the ROS scavengers N-acetyl cysteine (NAC, ST1546) and reduced glutathione (GSH, S0073), the pan-caspase inhibitor Z-vad-FMK (C1202), and a mouse monoclonal anti-human GAPDH antibody (AF5009) were purchased from Beyotime (Shanghai, China).
Western Blotting
Cervical cancer cells (1–2 × 106) were lysed in 200 µL of lysis buffer (20 mM Tris (pH 7.5), 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, 1 mM β-glycerophosphate, 1 mM sodium pyrophosphate, 1 mM Na3VO4, 1 mg/mL leupeptin). The cell lysate was centrifuged at 12,000 × g for 5 min at 4°C. Proteins were electrophoresed on 10% SDS-PAGE gels and transferred onto Immobilon P membranes (Millipore, Billerica, MA, USA). The membranes were blocked by incubation in 3% nonfat dry milk at room temperature for 1 h and then incubated with primary antibodies in PBS containing 0.01% Tween 20 at 4°C overnight. After incubation with a horseradish peroxidase-conjugated secondary antibody, protein bands were detected with SuperSignal chemiluminescent substrate-stable peroxide solution (Pierce Rockford, IL, USA) and BIOMAX-MR film (Eastman Kodak Co., Rochester, NY, USA). When necessary, membranes were stripped with Restore Western blotting stripping buffer (Pierce) and reprobed with antibodies against various cellular proteins.
Quantitative Reverse Transcription-Polymerase Chain Reaction (qRT-PCR)
qRT-PCR was performed as described by Sun et al.19 Briefly, total RNA was extracted from 1–2×106 cells with TRIzol (Invitrogen, Carlsbad, CA, USA) as described by the manufacturer. mRNA was reverse transcribed with RevertAid (MBI Fermentas, Burlington, Ontario, Canada) at 42°C for 60 min. cDNA was amplified with TaqMan Universal PCR master mix (Roche Applied Science) and a LightCycler 96 detection system (Roche Applied Science). The amplification levels of the target genes were normalized to the amplification level of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as the endogenous control. The PCR efficiency was tested by amplification of the target from serially diluted cDNA generated by reverse transcription of a stock set of human RNA. Data analysis and calculations were performed using the comparative 2−ΔΔCT method, as described by the manufacturer. Gene expression values are shown as the fold induction of the gene measured in Stattic-treated samples relative to its level in samples cultured in Stattic-free medium. The forward and reverse primer pairs are listed (5΄ to 3΄) as follows:
GAPDH-F, AATCCCATCACCATCTTCCA,
GAPDH-R, CCTGCTTCACCACCTTCTTG;
GSR-S: GGCTTTCCAAGTTGTGAGGG,
GSR-A: TATTCCTAAGCTGGCACCGG.
DCF Staining Assay for Measurement of ROS
Intracellular ROS levels were measured by ROS assay according to the manufacturers’ standard protocols. Briefly, 1–2 × 106 cells were cultured in FCS-free medium with 10 μM dichlorofluorescein diacetate (DCFH-DA) at 37°C for 30 min and were then washed three times with FCS-free medium prior to treatment with various reagents as described in the figure legends. Intracellular ROS cause oxidation of DCFH-DA, yielding the fluorescent product 2′,7′-dichlorofluorescein (DCF). The fluorescence of DCF was measured using a FACScan system (BD Bioscience, San Jose, CA). For each analysis, 10,000 events were recorded.
GSR Activity Assays
GSR activity was measured with a GSR assay kit (S0055, Beyotime, China) according to the manufacturer’s instructions. In the reaction mixture, GSR (or sample), glutathione disulfide (GSSG), NADPH (triphosphopyridine nucleotide), and DTNB (5,5ʹ-dithiobis-(2-nitrobenzoic acid) were included. In the presence of NADPH, GSSG is reduced to glutathione (GSH) via a reaction catalyzed by GSR. GSH reacts with DTNB to produce yellow TNB. Then, GSR activity was determined by measuring the production of TNB from the absorbance at 412 nm.
Flow Cytometric Analysis
Cell death was detected by propidium iodide (PI)/fluorescein isothiocyanate (FITC)-annexin V staining. Briefly, 1–2×106 cells were washed twice with PBS and then labeled with FITC-annexin V and PI in binding buffer according to the manufacturer’s instructions. The fluorescence signals were detected on a FACScan (BD Bioscience, San Jose, CA). The log of FITC-annexin V–fluorescence is displayed on the x-axis, and the log of PI fluorescence is displayed on the y-axis. For each analysis, 10,000 events were recorded.
For protein detection, cells were cultured in 6-well plates for 24 h and then harvested and washed with fluorescence-activated cell sorting buffer (5 mmol/L EDTA, 0.1% NaN3, and 1% FCS in Dulbecco’s PBS). After incubation with an antibody against human GSR for 30 min on ice, the cells were stained with a FITC-labeled secondary antibody, and protein expression was examined by flow cytometry (BD Bioscience, San Jose, CA).
Plasmid Transfection
Cells cultured in 6-well plates were transfected with 1 μg of plasmid using Lipofectamine 3000 (Invitrogen) according to the manufacturer’s instructions. The expression of proteins in the transfected cells was examined by Western blotting 48 h after transfection. For stable transfection, resistant cells were selected by incubation with 800 μg/mL G418 or 1 μg/mL puromycin for 2 weeks.
siRNA Transfection
The siRNA against human GSR (sc-44843) and silencer negative siRNA control (sc-37007) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). siRNA transfection reagent (sc-29528, Santa Cruz, CA, USA) was used to transfect siRNA into cervical cancer cells according to the manufacturer’s instructions. Briefly, 10–40 pmol siRNA and 6 μL of siRNA transfection reagent were used for each transfection (6-well plates, 0.5×106 cells/well). Forty-eight hours after siRNA transfection, knockdown of GSR was confirmed by Western blotting.
Knockout of STAT3 Using CRISPR/Cas9 Genome Editing Technology
To generate the STAT3 knockout cell line, CRISPR/Cas9 genome-editing was applied as reported.20 Guide RNA (gRNA; 5ʹ-GCAGCTTGACACACGGTACC-3ʹ) targeting STAT3 exon 3 was subcloned into the lentiCRISPR v2 vector (52961, Addgene). Plasmids containing gRNAs were transfected into cervical cancer cells using Lipofectamine 3000 (Invitrogen). Genome-edited colonies were selected on 96-well plates and verified by Western blotting. Positive colonies were further verified by DNA sequencing.
Tissue Microarray and Immunohistochemical Analyses
For immunohistochemical analysis, a tissue microarray was purchased from Bioaitech Company (F1081301, Xian, China); the microarray contained 40 samples of cervical adenocarcinoma, 60 samples of squamous cervical cancer, and 8 samples of nontumor tissues. The use of the human tissue microarrays was approved by the ethics committee of Changsha Central Hospital. Immunohistochemistry was performed to detect GSR expression as described in a previous study.21 The staining data were scored using the H-score method, which combined the staining intensity and percentage of tumor cells stained, as described in a report.22 Positive staining was evaluated in four random fields (100 cells) under a microscope at 400× magnification by two pathologists (Y.Z. and D.Z.). The staining intensity was scored as follows: 0 = no expression, 1 + = weak expression, 2 + = moderate expression, 3 + = strong expression, and 4 + = very strong expression. The final score was expressed as the immunohistochemical staining score (IHC score) and was obtained by multiplying the percentage of positive cells by the staining intensity.
Implantation of Cervical Cancer Cells in Nude Mice
Female BALB/c nude mice (6–8 weeks old) from SLAC Laboratory Animal Center (Shanghai, China) were used in all experiments. A total of 1×107 cervical cancer cells in 200 μL PBS were implanted by injection into the right flanks of the mice. At day 15 after initial implantation, 10 mice were randomly divided into a control group and an experimental group. In experimental group, 5 mice were injected i.p. with Stattic (400 nmol per mouse) every two days 14 times starting on day 15. In the control group, 5 mice were injected with vehicle. The growth of implanted tumors was examined every two days. Tumor sizes were calculated by the formula LW2/2, where L was the length of the tumors in centimeters and W was the width of the tumors in centimeters. On day 45, all mice were euthanized, and the tumor and mouse body weights were measured.
Statistical Analysis
All experiments were performed at least three times, and representative and reproducible results are shown. The results are expressed as the mean ± S.D. values. Differences between groups were examined for statistical significance using two-tailed Student’s t-test. P values equal to or less than 0.05 were considered statistically significant (n = 3 for each qRT-PCR analysis and ELISA).
Results
The Expression of GSR Was Increased in Clinical Cervical Cancer Tissues
GSR activity affects cell death in colorectal cancer and induces G(2)/M arrest in esophageal cancer cells.23,24 To determine the effect of GSR on human cervical cancer cells, the expression of GSR in clinical cervical cancer tissues was measured. A cervical cancer tissue microarray, which included 40 samples of cervical adenocarcinomas, 60 samples of squamous cell carcinomas, and 8 samples of nontumor tissues (3 samples of normal tissue and 5 samples of cervicitis tissue), was purchased (Bioaitech). Figure 1A shows the immunohistochemical (IHC) staining of GSR in the full tissue microarray. When observed at 400× magnification, GSR-positive staining in both the cytoplasm and nuclei of cells in cancer tissues (Figure 1B) was found. The predominant cytoplasmic staining (Figure 1C) or nuclear staining (Figure 1D) was also observed in cervical adenocarcinomas. In cervical squamous cell carcinomas, GSR negativity (Figure 1E) and positive staining in both the cytoplasm and nuclei (Figure 1F and G) were found. Figure 1H shows weak positive staining in cervicitis. Semiquantitative IHC score evaluation at 400× magnification showed that 98 of the 100 samples of cervical cancer were GSR-positive—12 of the 13 samples of well-differentiated cancers, 43 of the 44 samples of moderately differentiated cancers, and all 44 samples of poorly differentiated cancers. In nontumor tissues, 5 of the 8 samples were GSR-positive—2 of 3 samples of normal cervical tissue and 3 of the 5 samples of cervicitis tissue (Figure 1I). The IHC scores showed that GSR expression was not significantly different among grade I, II, and III cervical cancers. However, GSR expression in all three cancer grades was significantly increased compared with that in nontumor tissues (Figure 1I). Regarding the different cervical cancer types, there was no difference in the IHC score between adenocarcinoma and squamous cell carcinoma (Figure 1J). However, GSR expression in these two cancer groups was significantly higher than that in nontumor cases (Figure 1J). These results indicate that GSR expression is increased in cervical cancers.
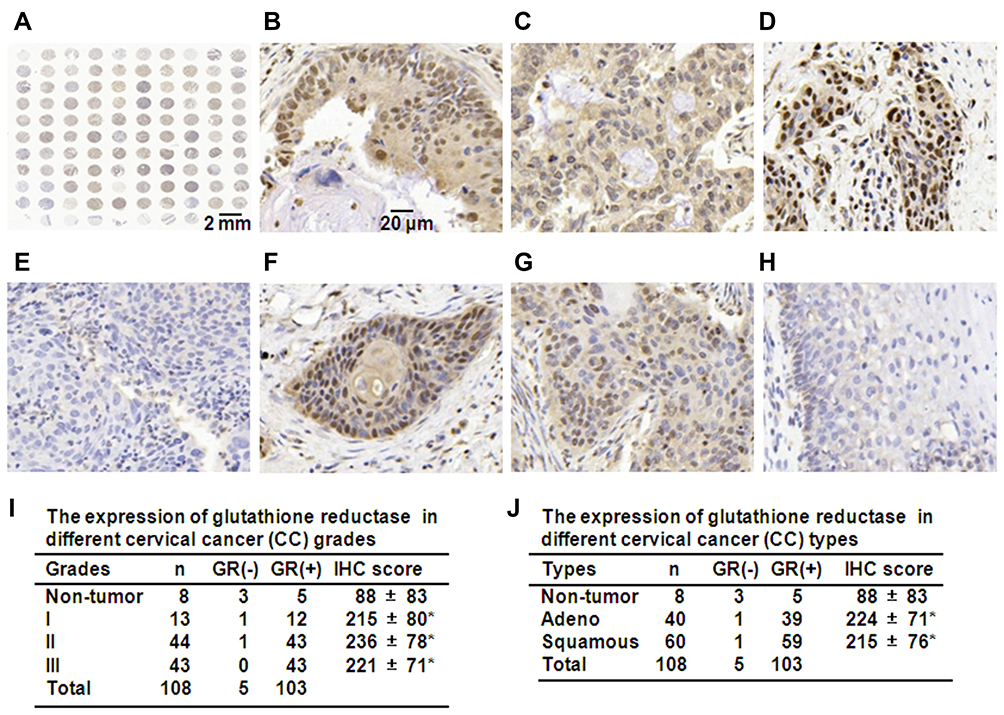 |
Figure 1 The expression of GSR in cervical cancers. (A) Full cervical cancer microarray examined by GSR IHC staining. (B–D) Representative GSR IHC staining in cervical adenocarcinomas. Nuclear and cytoplasmic positive staining (B), predominantly cytoplasmic positive staining (C), and predominantly nuclear positive staining (D). (E–G) Representative GSR IHC staining in squamous cell carcinomas. Negative staining (E), Nuclear and cytoplasmic positive staining (F and G). (H) Representative GSR IHC staining of cervicitis tissues. Weak positive staining in the cytoplasm. (I) IHC scores of cervical cancer of different grades. *P < 0.05 compared with nontumor tissues. (J) IHC scores of different cervical cancer types. *P < 0.05 compared with nontumor samples. |
GSR Knockdown Results in Cell Death
To examine the biological effect of increased GSR expression in cervical cancers, the expression of GSR in cancer cell lines was measured. FACS analysis showed that three cell lines, C33A, CaSki, and SiHa, expressed high levels of the GSR protein (Figure 2A). GSR expression at the mRNA and protein levels was inhibited by RNA interference, as shown by qRT-PCR (Figure 2B) and Western blotting (Figure 2C). PI/FITC-Annexin V staining showed that GSR knockdown resulted in cell death (Figure 2D and E). These results suggest that GSR supports the survival of cervical cancer cells.
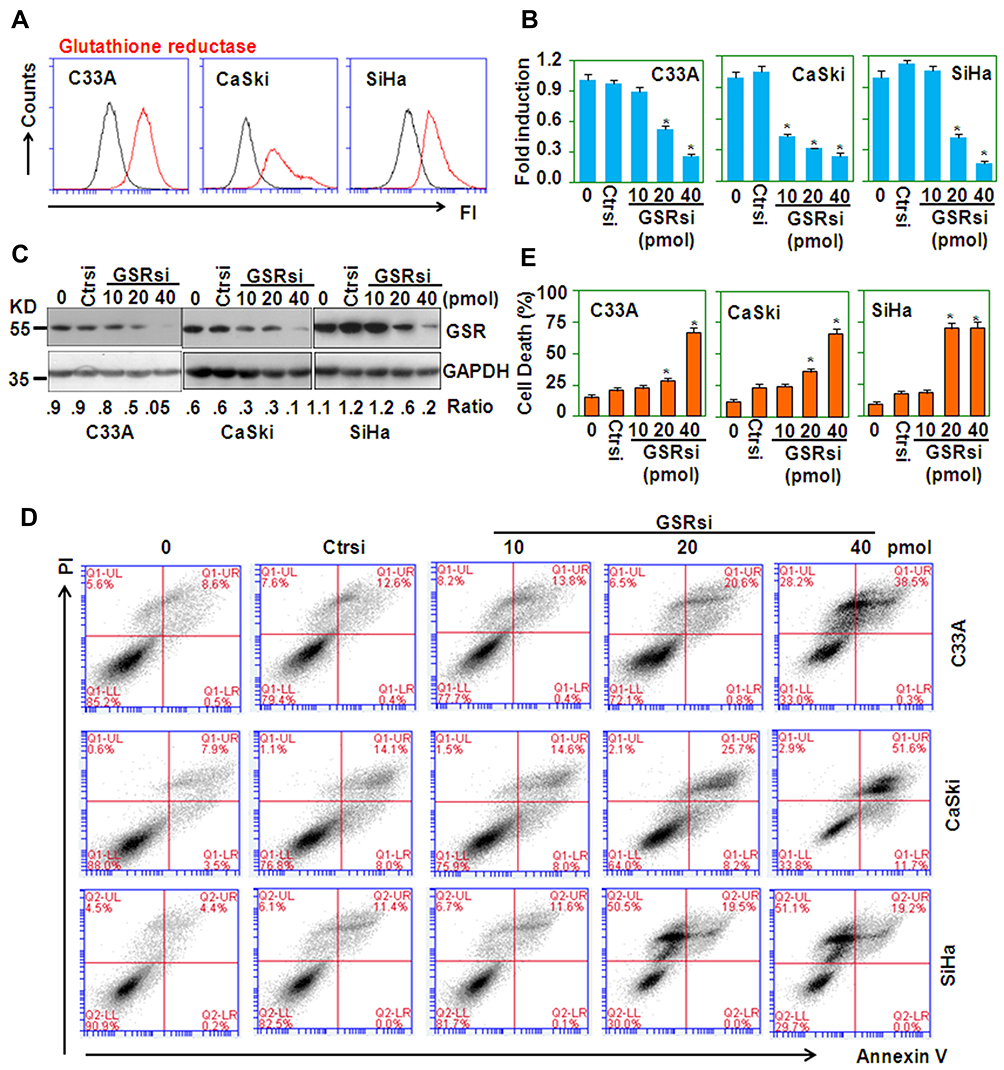 |
Figure 2 GSR RNA interference induces the death of cervical cancer cells. (A) FACS analysis of GSR protein expression in cervical cancer cells. (B) qRT-PCR analysis of GSR mRNA levels in cervical cancer cells. Tumor cells were treated with the indicated doses of GSR siRNA for 5 h and were then cultured for 48 h. *P < 0.05 compared with the control groups. (C) Western blot analysis of GSR protein levels in cervical cancer cells transfected with the indicated doses of GSR siRNA for 5 h and then cultured for 48 h. The GAPDH protein level was measured as the loading control. (D) PI/FITC-Annexin V staining of cervical cancer cells transfected with the indicated doses of GSR siRNA for 5 h and then cultured for 48 h. (E) Quantitative data from D. *P < 0.05 compared with the nonspecific siRNA (Ctrsi) groups. |
Stattic Inhibits GSR and Induces the Death of Cervical Cancer Cells
In THP-1 monocytic cells, Stattic inhibits the enzymatic activity of GSR and induces ROS-dependent production of the chemokine IL-8 (data not published). To determine the effect of Stattic on GSR activity in cervical cancer cells, C33A, CaSki, SiHa, and HeLa cells were treated with Stattic, and the protein levels of GSR were measured by Western blotting, which showed that Stattic did not change the GSR protein levels (Figure 3A). When GSR enzymatic activity was measured, Stattic treatment reduced the enzymatic activity of GSR in a dose- (Figure 3B) and time-dependent (Figure 3C) manner. PI/FITC-annexin V staining showed that Stattic significantly induced cell death in all 4 cervical cancer cell lines (Figure 3D–H). These results suggest that Stattic induces GSR-dependent death of cervical cancer cells.
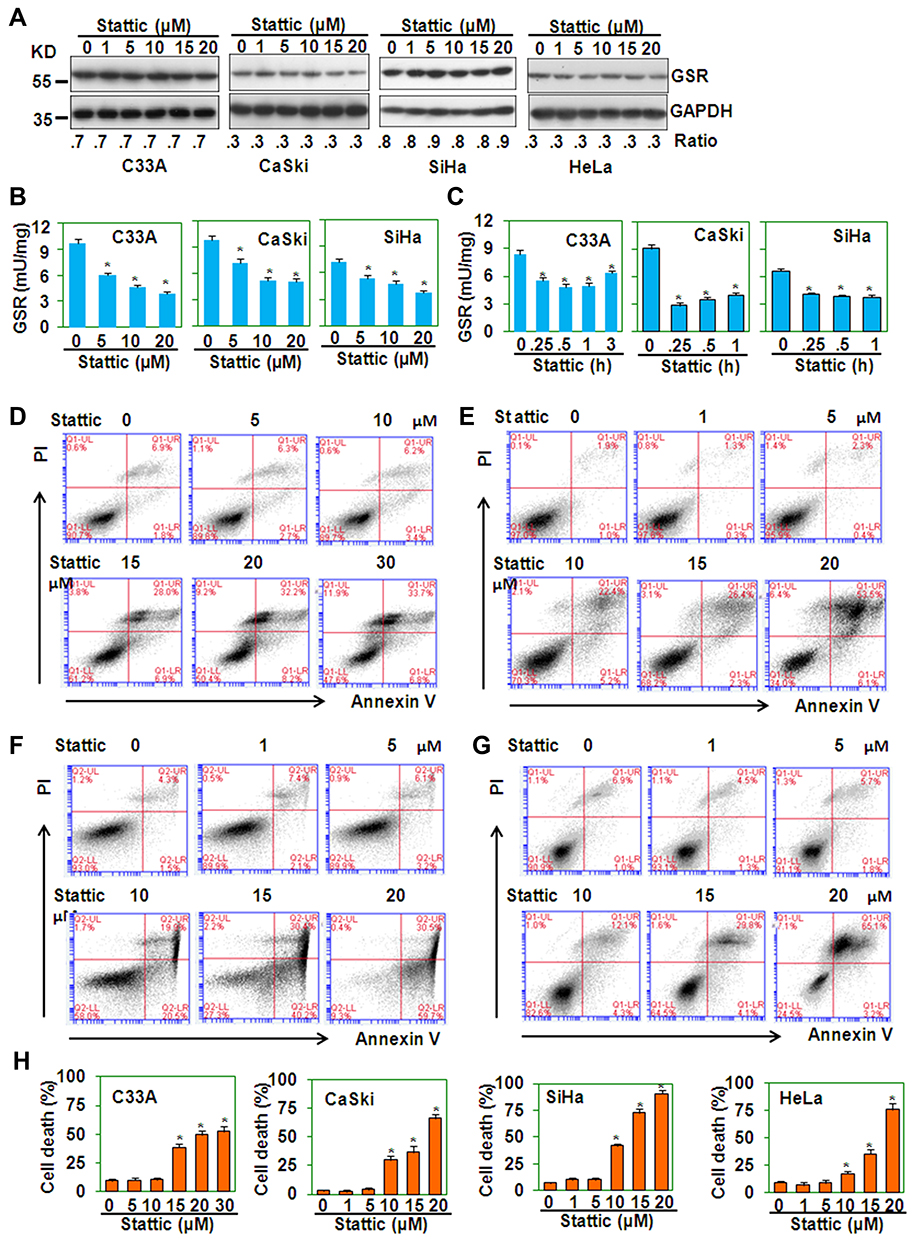 |
Figure 3 Stattic inhibits glutathione reductase activity and induces cell death. (A) Western blot analysis of the protein levels of GSR in cervical cancer cells treated with the indicated concentrations of Stattic for 24 h. The GAPDH protein level was measured as the loading control. (B) GSR activity in cervical cancer cells treated with the indicated concentrations of Stattic for 30 min. *P < 0.05 compared with the control groups. (C) GSR activity in cervical cancer cells treated with 20 µM Stattic for the indicated time periods. *P < 0.05 compared with the control groups. (D–G) PI/FITC-Annexin V staining of C33A (D), CaSki (E), SiHa (F), and HeLa (G) cells treated with the indicated concentrations of Stattic for 24 h. (H) Quantitative data from (D–G). *P < 0.05 compared with the medium groups. |
Stattic Decreases Tumor Growth in Nude Mice
To determine the effect of Stattic on tumor growth in vivo, CaSki cervical cancer cells were implanted s.c. into the flanks of nude mice (n = 10). On day 15 after implantation, the mice in the experimental group were treated with Stattic. Tumors in Stattic-treated mice grew more slowly than those in control mice (Figure 4A), as shown by the photographs in Figure 4B, as well as the weight of tumors shown in Figure 4C. However, there was no difference in the mouse body weight between the Stattic-treated and control groups (Figure 4D). These results suggest that Stattic inhibits tumor growth in vivo without causing side effects in mice.
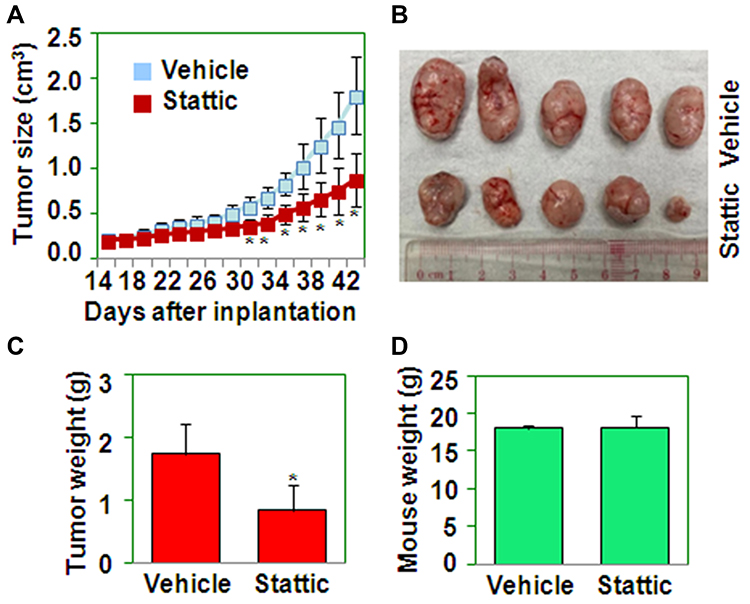 |
Figure 4 Stattic decreases tumor growth in nude mice. (A) Tumor growth curve for nude mice implanted with CaSki cells and treated with or without Stattic. *P < 0.05 compared with the vehicle groups. (B) Tumors from nude mice implanted with CaSki cells and treated with or without Stattic. (C) The weights of tumors from nude mice implanted with CaSki cells and treated with or without Stattic. *P < 0.05 compared with the vehicle group. (D) The weights of mice implanted with CaSki cells and treated with or without Stattic. |
Stattic-Induced Cell Death is Caspase-Independent
Caspases are an evolutionarily conserved family of cysteine proteases involved in cell death.25 To determine whether Stattic-induced cell death is dependent on caspases, the activation of caspase 3 was measured. Stattic treatment did not induce the activation of caspase 3. Cleaved fragments of caspase 3 were not observed (Figure 5A). Moreover, when cells were pretreated with the pan-caspase inhibitor Z-vad-FMK and then treated with Stattic, the level of cell death did not change (Figure 5B–D). These results suggest that Stattic-induced cell death is caspase-independent.
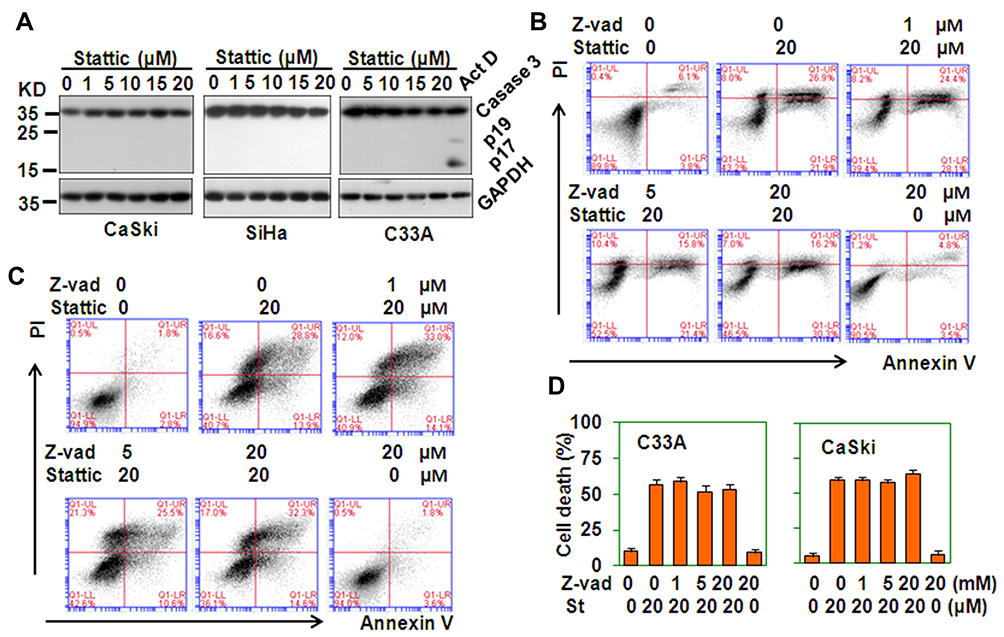 |
Figure 5 Caspases are not involved in Stattic-induced cell death. (A) Western blot analysis of the protein levels of caspase 3 in CaSki, SiHa, and C33A cells treated with the indicated concentrations of Stattic for 24 h. C33A cells treated with 5 μg/mL actinomycin D (Act D) were used as the positive control. The GAPDH protein level was measured as the loading control. (B and C) PI/FITC-Annexin V staining of C33A (B) and CaSki (C) cells pretreated with the indicated concentrations of Z-Vad-FMK (Z-vad, pan-Caspase inhibitor) for 1 h and retreated with 20 μM Stattic for 24 h. (D) The quantitative data from (B and C). |
Stattic-Induced Cell Death is ROS-Dependent
GSR regulates cell death via a ROS-dependent pathway.23 We measured ROS levels in Stattic-treated cervical cancer cells. Stattic dose- and time-dependently induced the production of ROS in 4 cervical cancer lines (Figure 6A). Two ROS scavengers, GSH and NAC, reversed the cell death induced by Stattic (Figure 6B–F). These results suggest that Stattic-induced cell death is ROS-dependent.
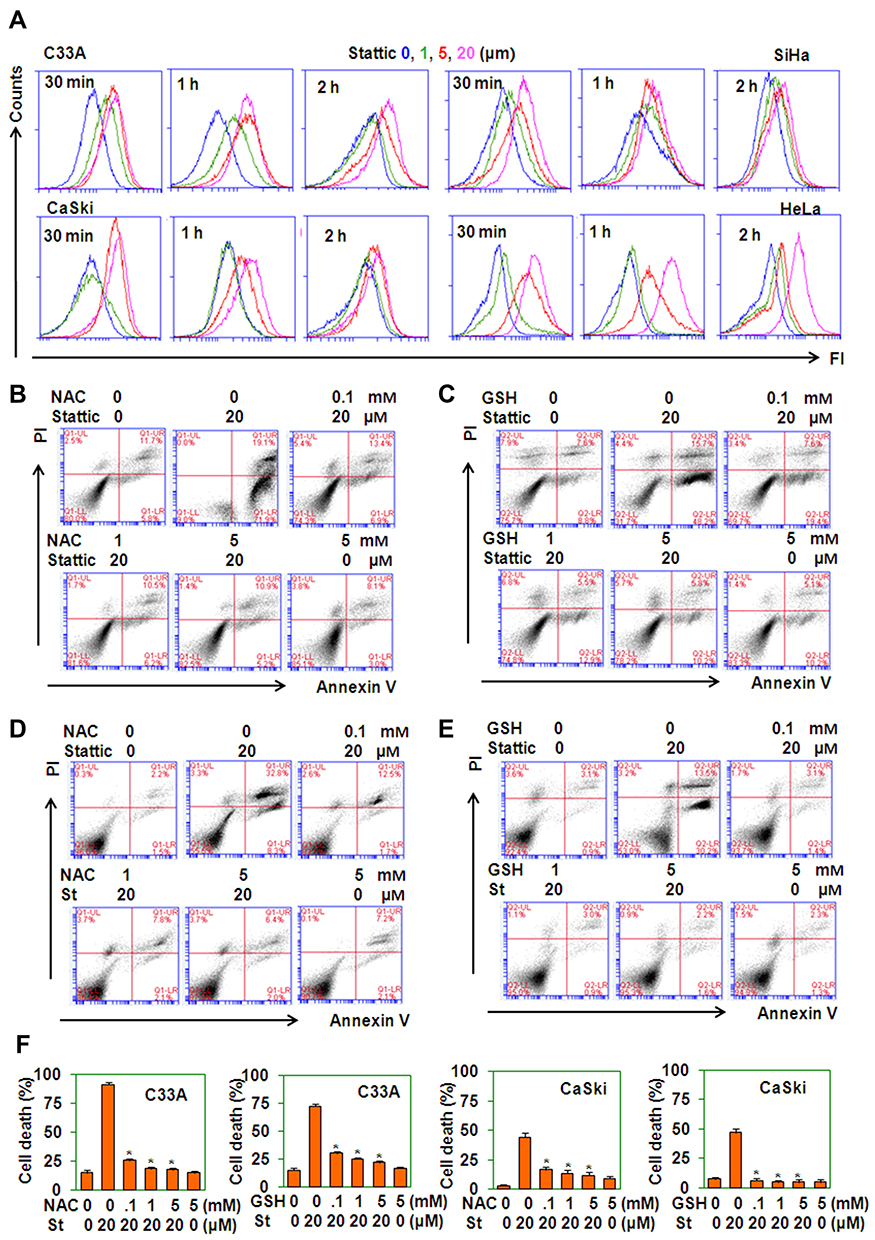 |
Figure 6 Stattic-induced cell death is ROS-dependent. (A) DCF staining of ROS levels in cervical cancer cells treated with the indicated concentrations of Stattic for the indicated time periods. (B–E) PI/FITC-Annexin V staining of C33A cervical cancer cells pretreated with the indicated concentrations of NAC (B) or GSH (C) for 1 h and of CaSki cervical cancer cells pretreated with the indicated concentrations of NAC (D) or GSH (E) for 1 h and retreated with 20 µM Stattic for 24 h. (F) The quantitative data from (B–E). |
Stattic-Induced Cell Death is STAT3-Independent
Stattic is a well known inhibitor of STAT3. To determine whether STAT3 inhibition contributes to Stattic-induced cell death, niclosamide, another STAT3 inhibitor, was tested for its ability to induce the death of cervical cancer cells. As expected, both Stattic and niclosamide decreased the level of phosphorylated STAT3 (Figure 7A). However, niclosamide did not induce the death of cervical cancer cells (Figure 7B–D). Further, when STAT3 was knocked down by CRISPR/Cas9 genome editing, it was fully deleted from the cervical cancer cell lines (Figure 7E). However, STAT3 deletion did not induce cell death (Figure 7F and G). These results suggest that Stattic induces cell death via a STAT3-independent mechanism.
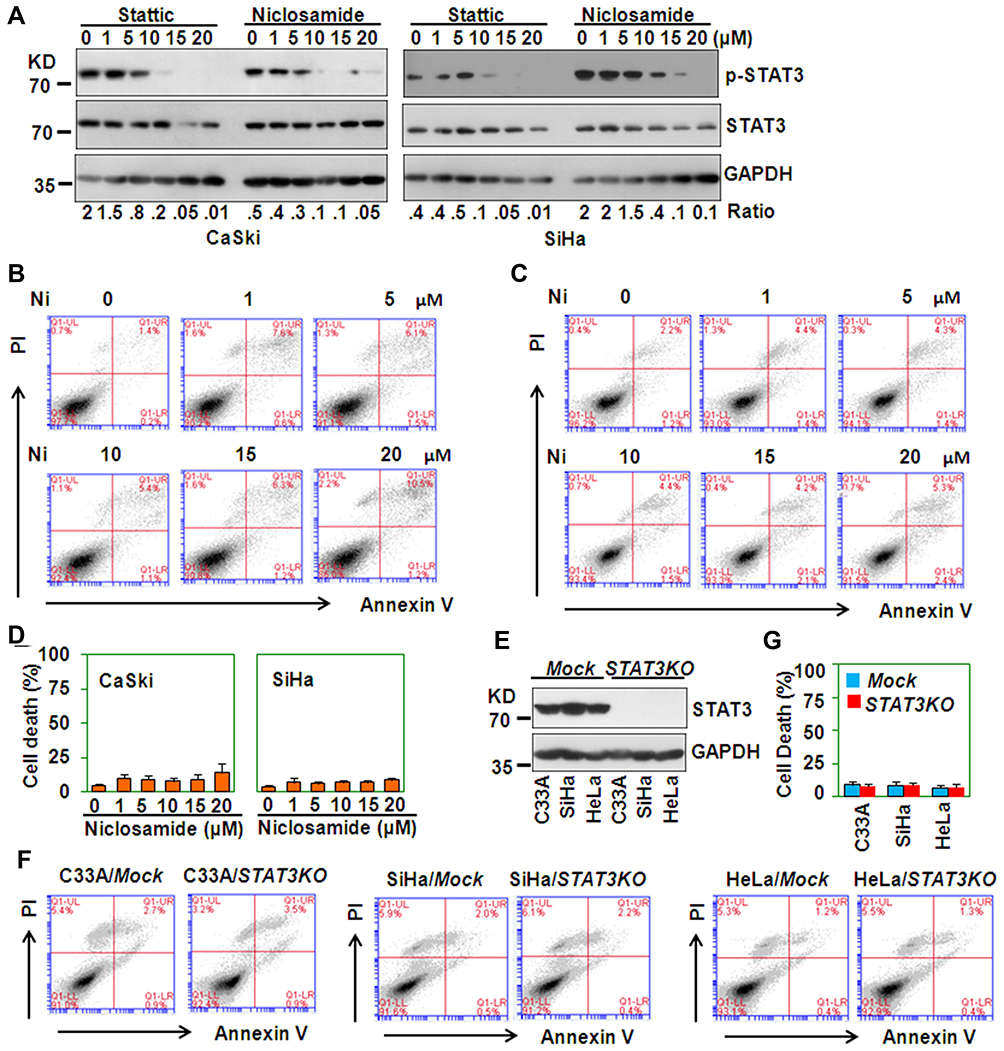 |
Figure 7 Stattic-induced cell death is STAT3-independent. (A) Western blot analysis of phosphorylated STAT3 in CaSki and SiHa cells treated with the indicated concentrations of Stattic or niclosamide for 8 h. The GAPDH protein level was measured as the loading control. (B and C) PI/FITC-Annexin V staining of CaSki (B) and SiHa (C) cells treated with the indicated concentrations of niclosamide (Ni) for 24 h. (D) The quantitative data from (B and C). (E) Western blot analysis of STAT3 protein levels in mock- and STAT3 CRISPR/Cas9-transfected C33A, CaSki, and SiHa cells. (F) PI/FITC-Annexin V staining of cervical cancer cells with STAT3 deletion. (G) The quantitative data from (F). |
Discussion
GSR is involved in cell survival regulation. In cancer cells, high levels of GSR are observed. Phosphorylation of GSR enhances its enzymatic activity, which confers a survival advantage on colorectal cancer cells.26 The increase in GSR activity also leads to a reduced cellular redox state and suppresses the death of colorectal cancer cells.23 Pharmacological inhibition of GSR induces ROS-mediated apoptosis in hepatocellular carcinoma cells and lung cancer cells.27 In our study, we found that in human cervical cancers, the expression of GSR was significantly upregulated. Stattic inhibited the enzymatic activity of GSR and induced ROS-dependent death of cervical cancer cells. Additionally, GSR knockdown by RNA interference induced cell death similar to that induced by pharmacological inhibition of GSR. Stattic also inhibited tumor growth in vivo.
GSR is also involved in cancer metastasis. GSR inhibition elicits oxidative stress and decreases the lung metastasis and subcutaneous growth of melanoma in vivo.28 In vitro, GSR downregulation inhibits the proliferation, colony formation, adhesion, migration and invasion of melanoma cells.28 Moreover, GSR inhibition-induced oxidative stress was found to block epithelial-to-mesenchymal transition (EMT) by reducing the expression of ERK1/2, Vimentin, and Snail and increasing the expression of E-cadherin.28 These observations indicate that GSR, as an effective regulator of oxidative stress, affects metastatic processes in melanoma cells.28
Stattic has been reported to activate the apoptotic pathway by increasing the levels of proapoptotic proteins, including Bax and cleaved caspase-3, and downregulating the antiapoptotic protein Bcl-2.29 In this study, Stattic did not induce activation of caspase 3, and the pan-caspase inhibitor did not reverse Stattic-induced cell death, suggesting that Stattic-induced cell death is caspase-independent.
GSR maintains redox homeostasis via GSH production. Inhibition of GSR results in an increase in ROS, which is associated with cell death.30 In this study, Stattic induced ROS production in four cervical cancer cell lines, and this effect was reversed by ROS scavengers, suggesting that Stattic-induced cell death is ROS-dependent.
As a STAT3 inhibitor, Stattic has been reported to promote chemo- and radiosensitivity in several cancer types, including melanoma,31 breast cancer,18,32 hepatocellular carcinoma,18 prostate cancer,17 glioblastoma,33 esophageal squamous cell carcinoma,16 and nasopharyngeal carcinoma15 in a STAT3-dependent manner. In our study, although Stattic induced the death of cervical cancer cells, neither treatment with another STAT3 inhibitor nor STAT3 deletion by CRISPR/Cas9 genome editing induced cell death. These observations indicate that STAT3 inhibition is insufficient for the induction of cell death and that Stattic-induced cell death is a STAT3-independent effect (STAT3 off-target effect). In addition, Stattic inhibited the enzymatic activity of GSR, leading to excessive production of ROS and finally inducing ROS-dependent cell death. Thus, the STAT3-independent anticancer biological activity of Stattic may be utilized as a novel therapeutic approach for human cervical cancer by targeting GSR.
Conclusion
Inhibition of GSR disrupts redox homeostasis, leading to cell damage, which benefits cancer therapy. The observations in this study showed that the expression of GSR was increased in human cervical cancer tissues. GSR knockdown by RNA interference induced the death of human cervical cancer cells. Similarly, Stattic, a STAT3 inhibitor, also inhibited the enzymatic activity of GSR and induced cell death via ROS-dependent but STAT3-independent mechanisms. Stattic also decreased the growth of xenograft tumors formed by cervical cancer cells. These results indicate a novel biological function of Stattic and suggest its potential application as a treatment for human cervical cancer. The results presented in this study are consistent with the model outlined in Figure 8.
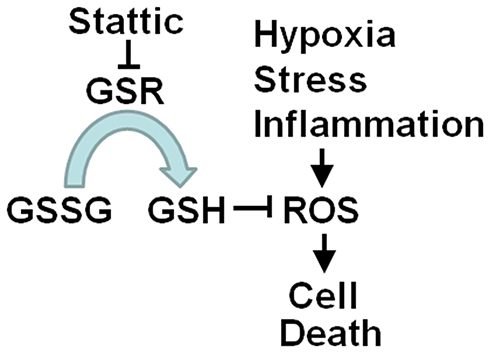 |
Figure 8 Model of the pathway by which Stattic induces ROS-dependent cell death. Stattic inhibits the enzymatic activity of GSR, resulting in a decrease in GSH, leading to accumulation of ROS and finally eliciting cell death. |
Abbreviations
IHC, immunohistochemistry; GSH, reduced glutathione; GSR, glutathione reductase; NAC, N-acetyl cysteine; ROS, reactive oxygen species; RT-PCR, reverse transcriptional-polymerase chain reaction; STAT3, signal transducer and activator of transcription 3; Stattic, 6-nitrobenzo[b]thiophene 1,1-dioxide.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81172042).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7–34. doi:10.3322/caac.21551
2. Venkatas J, Singh M. Cervical cancer: a meta-analysis, therapy and future of nanomedicine. Ecancermedicalscience. 2020;14:1111. doi:10.3332/ecancer.2020.1111
3. Schieber M, Chandel NS. ROS function in redox signaling and oxidative stress. Curr Biol. 2014;24:R453–462. doi:10.1016/j.cub.2014.03.034
4. Bhardwaj V, He J. Reactive oxygen species, metabolic plasticity, and drug resistance in cancer. Int J Mol Sci. 2020;21:3412. doi:10.3390/ijms21103412
5. Martindale JL, Holbrook NJ. Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol. 2002;192:1–15. doi:10.1002/jcp.10119
6. Ruiz-Torres V, Rodríguez-Pérez C, Herranz-López M, et al. Marine invertebrate extracts induce colon cancer cell death via ROS-mediated DNA oxidative damage and mitochondrial impairment. Biomolecules. 2019;9:771. doi:10.3390/biom9120771
7. Sunilkumar D, Drishya G, Chandrasekharan A, et al. Oxyresveratrol drives caspase-independent apoptosis-like cell death in MDA-MB-231 breast cancer cells through the induction of ROS. Biochem Pharmacol. 2020;173:113724. doi:10.1016/j.bcp.2019.113724
8. Lin X, Jia Y, Dong X, et al. Diplatin, a novel and low-toxicity anti-lung cancer platinum complex, activation of cell death in tumors via a ROS/JNK/p53-dependent pathway, and a low rate of acquired treatment resistance. Front Pharmacol. 2019;10:982. doi:10.3389/fphar.2019.00982
9. Teng JF, Qin DL, Mei QB, et al. Polyphyllin VI, a saponin from Trillium tschonoskii Maxim. induces apoptotic and autophagic cell death via the ROS triggered mTOR signaling pathway in non-small cell lung cancer. Pharmacol Res. 2019;147:104396. doi:10.1016/j.phrs.2019.104396
10. Zhang T, Zheng P, Shen X, et al. Curcuminoid WZ26, a TrxR1 inhibitor, effectively inhibits colon cancer cell growth and enhances cisplatin-induced cell death through the induction of ROS. Free Radic Biol Med. 2019;141:93–102. doi:10.1016/j.freeradbiomed.2019.06.005
11. Li Y, Tian X, Liu X, et al. Bufalin inhibits human breast cancer tumorigenesis by inducing cell death through the ROS-mediated RIP1/RIP3/PARP-1 pathways. Carcinogenesis. 2018;39:700–707. doi:10.1093/carcin/bgy039
12. Liu D, Qiu X, Xiong X, Chen X, Pan F. Current updates on the role of reactive oxygen species in bladder cancer pathogenesis and therapeutics. Clin Transl Oncol. 2020;22:1687–1697. doi:10.1007/s12094-020-02330-w
13. Fan HJ, Tan ZB, Wu YT, et al. The role of ginsenoside Rb1, a potential natural glutathione reductase agonist, in preventing oxidative stress-induced apoptosis of H9C2 cells. J Ginseng Res. 2020;44:258–266. doi:10.1016/j.jgr.2018.12.004
14. Schust J, Sperl B, Hollis A, et al. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem Biol. 2006;13:1235–1242. doi:10.1016/j.chembiol.2006.09.018
15. Pan Y, Zhou F, Zhang R, et al. Stat3 inhibitor Stattic exhibits potent antitumor activity and induces chemo- and radio-sensitivity in nasopharyngeal carcinoma. PLoS One. 2013;8:e54565. doi:10.1371/journal.pone.0054565
16. Zhang Q, Zhang C, He J, et al. STAT3 inhibitor stattic enhances radiosensitivity in esophageal squamous cell carcinoma. Tumour Biol. 2015;36:2135–2142. doi:10.1007/s13277-014-2823-y
17. Mohammadian J, Sabzichi M, Molavi O, et al. Combined treatment with stattic and docetaxel alters the Bax/Bcl-2 gene expression ratio in human prostate cancer cells. Asian Pac J Cancer Prev. 2016;17:5031–5035. doi:10.22034/APJCP.2016.17.11.5031
18. Xu G, Zhu L, Wang Y, et al. Stattic enhances radiosensitivity and reduces radio-induced migration and invasion in HCC cell lines through an apoptosis pathway. Biomed Res Int. 2017;2017:1832494. doi:10.1155/2017/1832494
19. Sun R, Zhang Y, Lv Q, et al. Toll-like receptor 3 (TLR3) induces apoptosis via death receptors and mitochondria by up-regulating the transactivating p63 isoform alpha (TAP63alpha). J Biol Chem. 2011;286:15918–15928. doi:10.1074/jbc.M110.178798
20. Ran FA, Hsu PD, Wright J, et al. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–2308. doi:10.1038/nprot.2013.143
21. Hu J, Deng X, Bian X, et al. The expression of functional chemokine receptor CXCR4 is associated with the metastatic potential of human nasopharyngeal carcinoma. Clin Cancer Res. 2005;11:4658–4665. doi:10.1158/1078-0432.CCR-04-1798
22. Wong SC, Lo ES, Lee KC, et al. Prognostic and diagnostic significance of beta-catenin nuclear immunostaining in colorectal cancer. Clin Cancer Res. 2004;10:1401–1408. doi:10.1158/1078-0432.CCR-0157-03
23. Oh BM, Lee SJ, Cho HJ, et al. Cystatin SN inhibits auranofin-induced cell death by autophagic induction and ROS regulation via glutathione reductase activity in colorectal cancer. Cell Death Dis. 2017;8:e2682. doi:10.1038/cddis.2017.100
24. Li X, Jiang Z, Feng J, et al. 2-Acetylamino-3-[4-(2-acetylamino-2-carboxyethylsulfanylcarbonylamino) phenyl carbamoylsulfanyl] propionic acid, a glutathione reductase inhibitor, induces G(2)/M cell cycle arrest through generation of thiol oxidative stress in human esophageal cancer cells. Oncotarget. 2017;8:61846–61860. doi:10.18632/oncotarget.18705
25. Van Opdenbosch N, Lamkanfi M. Caspases in cell death, inflammation, and disease. Immunity. 2019;50:1352–1364. doi:10.1016/j.immuni.2019.05.020
26. Wang YN, Lu YX, Liu J, et al. AMPKα1 confers survival advantage of colorectal cancer cells under metabolic stress by promoting redox balance through the regulation of glutathione reductase phosphorylation. Oncogene. 2020;39:637–650. doi:10.1038/s41388-019-1004-2
27. Hwang GH, Ryu JM, Jeon YJ, et al. The role of thioredoxin reductase and glutathione reductase in plumbagin-induced, reactive oxygen species-mediated apoptosis in cancer cell lines. Eur J Pharmacol. 2015;765:384–393. doi:10.1016/j.ejphar.2015.08.058
28. Li X, Wu J, Zhang X, et al. Glutathione reductase-mediated thiol oxidative stress suppresses metastasis of murine melanoma cells. Free Radic Biol Med. 2018;129:256–267. doi:10.1016/j.freeradbiomed.2018.07.025
29. Guha P, Gardell J, Darpolor J, et al. STAT3 inhibition induces Bax-dependent apoptosis in liver tumor myeloid-derived suppressor cells. Oncogene. 2019;38:533–548. doi:10.1038/s41388-018-0449-z
30. Volpe CMO, Villar-Delfino PH, Dos Anjos PMF, et al. Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell Death Dis. 2018;9:119. doi:10.1038/s41419-017-0135-z
31. Mohammadian J, Mahmoudi S, Pourmohammad P, et al. Formulation of Stattic as STAT3 inhibitor in nanostructured lipid carriers (NLCs) enhances efficacy of doxorubicin in melanoma cancer cells. Naunyn Schmiedebergs Arch Pharmacol. 2020;393:2315–2323. doi:10.1007/s00210-020-01942-x
32. Khaki-Khatibi F, Ghorbani M, Sabzichi M, et al. Adjuvant therapy with stattic enriches the anti-proliferative effect of doxorubicin in human ZR-75-1 breast cancer cells via arresting cell cycle and inducing apoptosis. Biomed Pharmacother. 2019;109:1240–1248. doi:10.1016/j.biopha.2018.10.183
33. Leidgens V, Proske J, Rauer L, et al. Stattic and metformin inhibit brain tumor initiating cells by reducing STAT3-phosphorylation. Oncotarget. 2017;8:8250–8263. doi:10.18632/oncotarget.14159
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.