")
Back to Journals » Nature and Science of Sleep » Volume 13
A Novel Application of Ketamine for Improving Perioperative Sleep Disturbances
Received 30 September 2021
Accepted for publication 4 December 2021
Published 25 December 2021 Volume 2021:13 Pages 2251—2266
DOI https://doi.org/10.2147/NSS.S341161
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ahmed BaHammam
Bijia Song, Junchao Zhu
Department of Anesthesiology, Shengjing Hospital of China Medical University, Shenyang, Liaoning, People’s Republic of China
Correspondence: Junchao Zhu
Department of Anesthesiology, Shengjing Hospital of China Medical University, Shenyang, Liaoning, People’s Republic of China
Email [email protected]
Abstract: Perioperative sleep disturbances are commonly observed before, during, and after surgery and can be caused by several factors, such as preoperative negative moods, general anesthetics, surgery trauma, and pain. Over the past decade, the fast-acting antidepressant effects of the N-methyl-D-aspartate (NMDA) receptor antagonist ketamine represent one of the most attractive discoveries in the field of psychiatry, such as antidepressant and anxiolytic effects. It is also widely used as a short-acting anesthetic and analgesic. Recent research has revealed new possible applications for ketamine, such as for perioperative sleep disorders and circadian rhythm disorders. Here, we summarize the risk factors for perioperative sleep disturbances, outcomes of perioperative sleep disturbances, and mechanism of action of ketamine in improving perioperative sleep quality.
Keywords: perioperative sleep disturbances, ketamine, antidepressant, anxiolytic, anti-inflammation
Introduction
The perioperative period refers to the duration from when the patient decides to receive surgery until the basic recovery period after surgery; this includes the time before, during, and after surgery. High sleep quality can accelerate incision healing, enhance immunity, and promote recovery and plays an important role in ensuring the quality of life of perioperative patients. However, due to numerous factors, patients can experience issues of varying degrees during the perioperative period.1,2 A previous study reported that more than 40% of patients complained of poor sleep quality the first night before surgery and that their sleep problems usually last a few days after surgery.3 Ruyi4 compared the sleep conditions of perioperative patients and healthy volunteers and found that the incidence of sleep disturbances in perioperative patients was 17% higher than that in healthy volunteers. Preoperative negative moods, hormone levels, personality characteristics, general anesthetics, surgical trauma, and pain are all factors affecting sleep conditions and circadian rhythm in perioperative patients.5–9 Long-term sleep disorders are closely related to many diseases, especially in elderly patients, such as chronic pain or pain sensitivity and postoperative cardiovascular events.10,11 In addition, sleep deprivation caused by psychological problems may lead to neuronal apoptosis in cognition-related brain regions through neuroinflammation, changes in neurotransmitter activity (such as adenosine), and brain hypoxia or hypoperfusion injury.12 The clinical use of the N-methyl-D-aspartate (NMDA) receptor antagonist ketamine began in the 1970s. Since then, there has been increasing interest in ketamine for perioperative anesthesia, analgesia, and antidepressants.13–15 Recent intensive research has shed light on new potential applications of this drug, such as for perioperative sleep disturbances and circadian dysregulation. Here, we summarize the risk factors for perioperative sleep disturbances, outcomes of perioperative sleep disturbances (Table 1), and mechanism of action of ketamine in improving perioperative sleep quality (Figures 1–3).
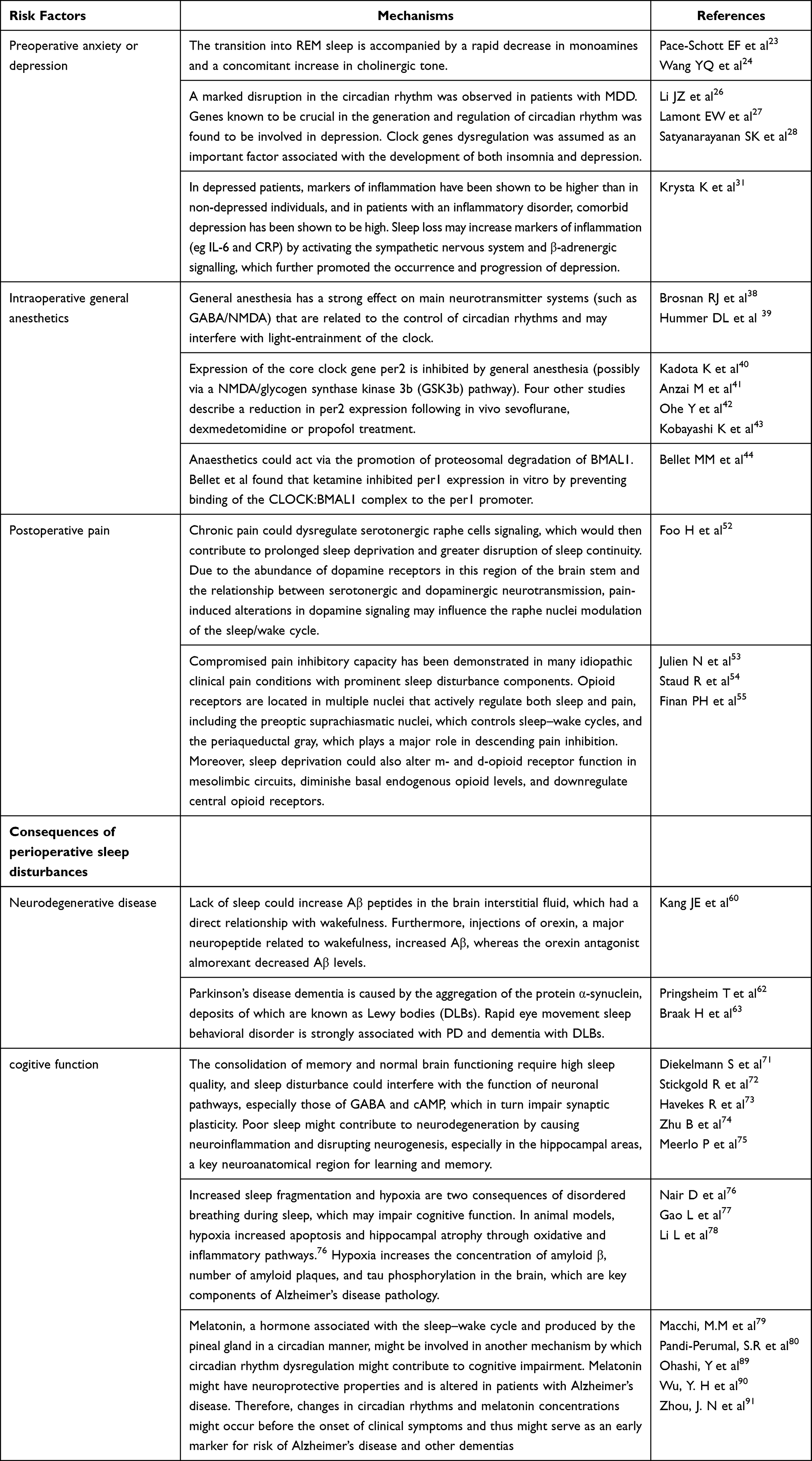 |
Table 1 The Risk Factors and Consequences of Perioperative Sleep Disturbances |
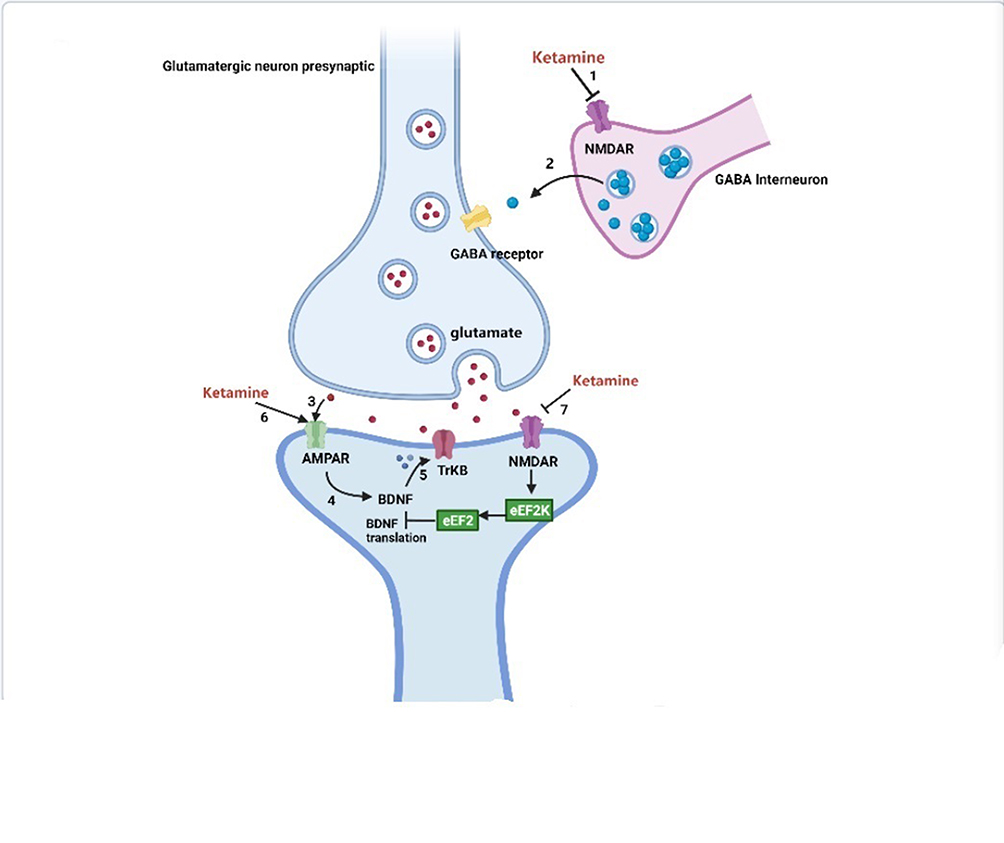 |
Figure 1 Antidepressant mechanisms of ketamine and potential biochemical biomarkers. |
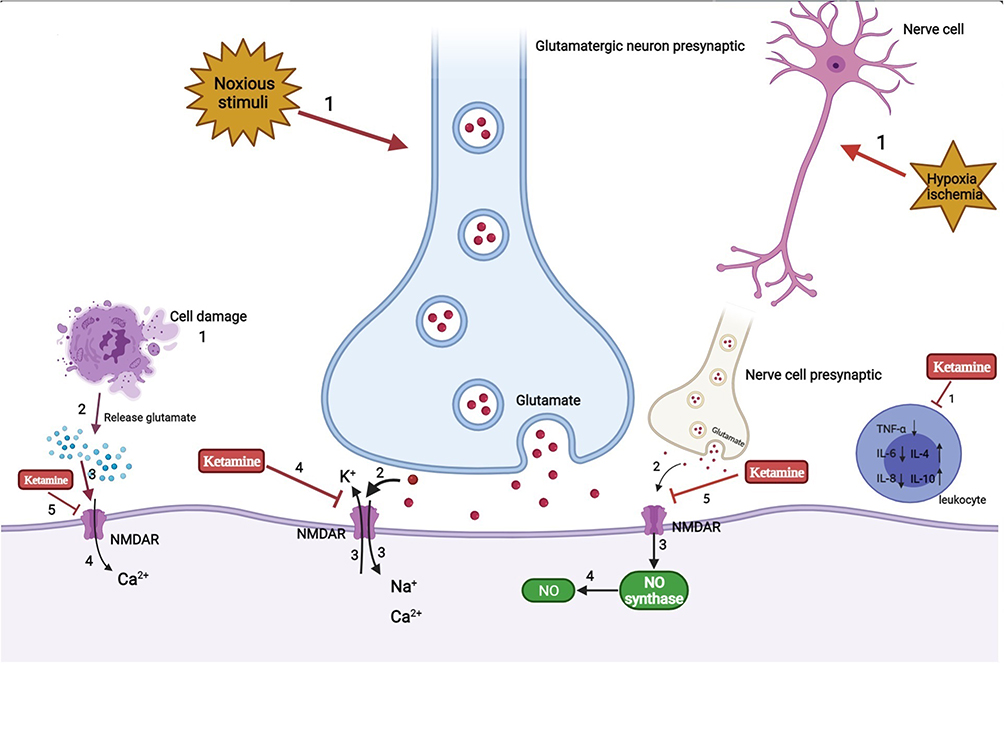 |
Figure 2 Analgesia and anti-inflammation mechanism of ketamine. |
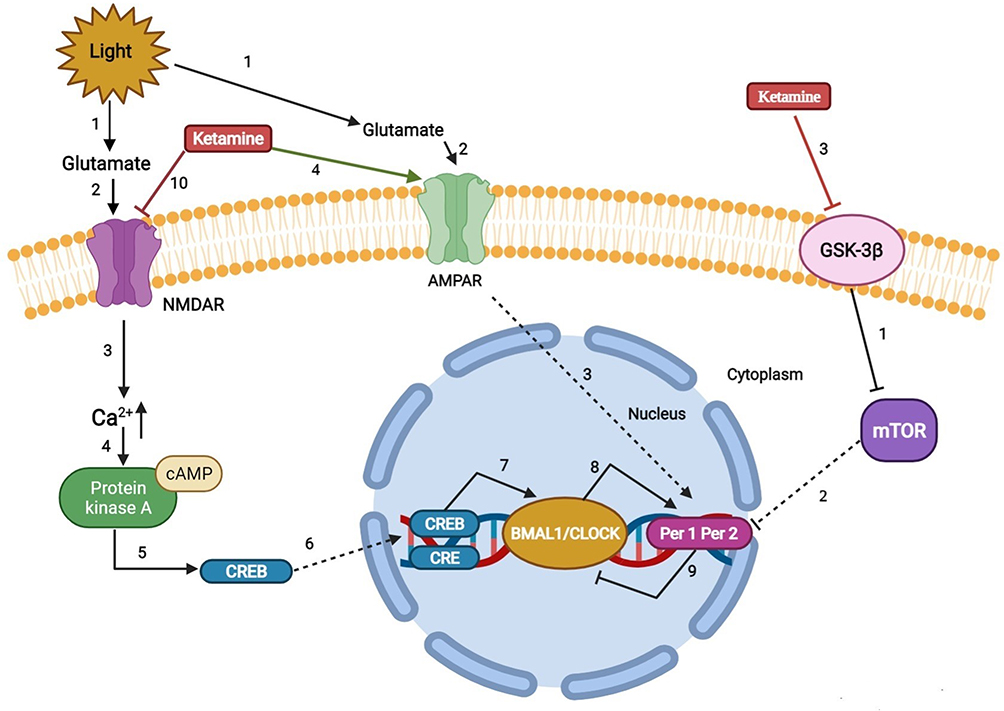 |
Figure 3 Regulation mechanism of ketamine on sleep and circadian system. |
Risk Factors for Perioperative Sleep Disturbances
Effect and Mechanism of Preoperative Anxiety or Depression on Sleep Disturbances
Anxiety or depression of patients planning to undergo various surgical procedures has always been a concern for patients’ health professionals. With over 312.9 million operations being performed each year worldwide,16 the individual patient’s perception of surgery and outcomes should be better assessed. It is estimated that between 25% and 80% of patients admitted to the hospital for surgery experience preoperative anxiety, which can negatively influence patient recovery.17–19 Preoperative anxiety or depression has been recognized as a potential and preventable risk factor for postoperative complications. Changes in sleep neurophysiology are often observed in patients with depression. In many cases, sleep impairment is the main complaint of depression, which is often characterized by decreased sleep efficiency and a longer time of rapid eye movement (REM) sleep.20,21 Moreover, preoperative sleep disturbances could enhance surgery-induced neuroinflammation, neuronal damage, blood–brain barrier disruption, and memory impairment 24 h after surgery, which may also cause or aggravate postoperative sleep disturbances and cognitive function.22 However, the relationship between depression and insomnia is complex and bidirectional rather than a cause-effect relationship. The underlying mechanism between depression and sleep disturbances may be as follows: 1) Major depression is related to the interruption of REM. The transition to REM sleep is accompanied by a rapid decrease in monoamines and an increase in cholinergic tone.23,24 The release disorder of these monoamine neurotransmitters that cause REM sleep abnormalities is also related to the manifestations of depression.25 2) The circadian rhythm is a 24-h physiological and behavioral rhythm controlled by the molecular clock in the suprachiasmatic nucleus. It plays an important role in regulating the sleep/wake cycle, including sleep duration, continuity, and structure. Biological clock imbalance is an important factor leading to insomnia and depression.26–28 Many studies have identified a strong correlation between single nucleotide polymorphisms of clock genes and depression. For example, the single nucleotide polymorphism rs2287161 in cryptochrome may indicate a higher susceptibility to circadian dysregulation and major depression.29,30 3) A strong relationship has been observed between inflammation and depression. Inflammatory markers were reported to be higher in patients with depression than in those without depression. Furthermore, comorbid depression has been shown to be high in patients with inflammatory diseases. Sleep deprivation was confirmed to increase inflammatory markers (eg, interleukin‐6 and C-reactive protein) through activation of the sympathetic nervous system and β‐adrenergic signaling, which further promoted the occurrence and progression of depression.31
Effect and Mechanism of Intraoperative General Anesthetics on the Circadian Clock and Sleep
General anesthesia is a pharmacological state involving amnesia, immobility, unconsciousness, and analgesia. Its purpose is to deprive senses to prevent a motor response to stimuli and induce amnesia.32 Two kinds of general anesthetics are widely used: halogenated gases (for example, halothane, desflurane, isoflurane, and sevoflurane) and intravenous anesthetics (for example ketamine, opioids, etomidate, and propofol). Most general anesthetics act on γ-aminobutyric acid (GABA)A receptors, and their mechanism and location of action in GABAergic transmission differ.33 Ozone et al34 showed that propofol anesthesia for 1 hour between 14:00 and 15:00 may cause the sleep structure of healthy volunteers to be disturbed at night after anesthesia. Sleep latency was significantly increased, and stage 2 sleep latency was significantly decreased. The sleep quality of children aged 4 to 6 months who use sevoflurane or propofol to repair cleft lip and palate surgery is also disturbed at night after anesthesia. The effect of propofol on sleep is more significant than that of sevoflurane.35 The effects of general anesthesia on sleep might possibly be due to anesthesia-induced clock disruption.36 Circadian rhythm and sleep quality dysfunction have harmful effects on mood, cognitive function, inflammation, and immune function.37 Therefore, circadian rhythm disorders caused by general anesthesia may hinder postoperative recovery on multiple levels. This is potentially because 1) general anesthesia has a strong effect on the main neurotransmitter systems (such as GABA/NMDA) that are related to the control of circadian rhythms and may interfere with light entrainment of the clock. Since the retinal hypothalamic tract is NMDA receptor-dependent, isoflurane, sevoflurane, and ketamine are NMDA receptor antagonists,38 which may inhibit hypothalamic retinal signal transduction, therefore inhibiting the optical entrainment of the biological clock. Continuous activation of GABA receptors can also change the clock phase and inhibit light-induced phase advances in rhythms.39 2) The expression of the core clock gene per2 is inhibited by general anesthesia (possibly through the NMDA/glycogen synthase kinase 3b pathway). Kadota et al found that administration of sevoflurane during the rising phase of the per2 expression cycle resulted in lower per2 mRNA levels in murine suprachiasmatic nuclei than in controls.40 Consistent with the above findings, four other studies also described the decrease in per2 expression after administration of sevoflurane, dexmedetomidine or propofol.41–43 3) Similar to the mechanism of sevoflurane-mediated inhibition of per2, Bellet et al found that ketamine was found to inhibit the expression of per1 in vitro by preventing the binding of the CLOCK:BMAL1 complex to the per1 promoter.44 The histone acetyltransferase activity of CLOCK acetylates both histones 3 and 4, preventing CLOCK:BMAL1 binding to the per1 promoter, which may result in a decrease in the level of acetylated histones in the promoter region. This will eventually lead to circadian dysfunction.45
Effect and Mechanism of Postoperative Pain on Sleep Disturbances
Although meaningful progress has been made in understanding pain mechanisms and the development of analgesics and anesthetics, postoperative acute pain control remains a challenge for approximately one-third of surgical patients.46 A large Dutch cohort study of 1490 surgical patients who received postoperative pain treatment reported that patients still experienced moderate to severe pain on the day of surgery, which continued in 15% of patients at 4 days after surgery.47 Acute postoperative pain is also accompanied by chronic pain, and 2–10% of postoperative patients experience severe pain.48 Chronic pain is highly comorbid with sleep disturbances such as decreased total sleep time and increased sleep arousals.49 The biobehavioral mechanisms of the association between sleep and pain are as follows: 1) dopamine is the principal neurotransmitter of the forebrain reward system and underlies the human behavioral drive to pursue pleasure. Dopamine is integral to the promotion and maintenance of arousal states and is intimately tied to the regulation of sleep and wake.50,51 Foo and Mason theorized that chronic pain could dysregulate serotonergic raphe cell signaling, which would then contribute to prolonged sleep deprivation and greater disruption of sleep continuity. Due to the abundance of dopamine receptors in this region of the brain stem and the relationship between serotonergic and dopaminergic neurotransmission, pain-induced alterations in dopamine signaling may influence raphe nuclei modulation of the sleep/wake cycle.52 2) Previous studies have found that opioid peptides play a key mediating role in the downward pain regulation system. Studies have shown that prominent sleep disorders appear when the ability to inhibit pain is impaired.53,54 Opioid receptors are located in multiple nuclei that actively regulate both sleep and pain, including the preoptic suprachiasmatic nuclei, which control sleep-wake cycles, and the periaqueductal gray, which plays a major role in decreasing pain inhibition. Moreover, sleep deprivation could also alter mu and delta opioid receptor function in mesolimbic circuits, diminish basal endogenous opioid levels, and downregulate central opioid receptors.55
Outcomes of Perioperative Sleep Disturbances
Perioperative Sleep Disturbances in Neurodegenerative Disorders
Neurodegenerative disorders are characterized by progressive loss of selectively vulnerable populations of neurons, which can be broadly classified by their clinical presentations, with extrapyramidal and pyramidal movement disorders and cognitive or behavioral disorders being the most common. Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS) are three of the major neurodegenerative diseases.56 Malhotra RK et al demonstrated that common sleep disturbances that may occur in most neurodegenerative conditions include insomnia, sleep apnea, restless legs syndrome, and circadian rhythm disorders. Conversely, in terms of duration and quality, lack of sleep could also increase the neurodegenerative process and aggravate the underlying clinical condition.57 Several studies have demonstrated that one night of sleep deprivation or interruption of nonrapid eye movement sleep in healthy volunteers may increase the levels of Aβ1-42 and Aβ1-40 in the cerebrospinal fluid.58,59 In rats, lack of sleep could increase Aβ peptides in the brain interstitial fluid, which had a direct relationship with wakefulness. Furthermore, injections of orexin, a major neuropeptide related to wakefulness, increased Aβ, whereas the orexin antagonist almorexant decreased Aβ levels.60 There may also be a significant relationship between sleep interruption and tau pathology. For example, tau protein metabolism and synaptic integrity were observed during sleep deprivation in a mouse model of AD.61 In addition, PD is a progressive multisystem neurodegenerative disease that mainly affects people later in life. It is the second most common neurodegenerative disease in the world. With changes in population structure, its incidence and prevalence are increasing.62 PD has unique neuropathic brain changes, whose formation of abnormal protein spheres is called Lewy bodies.63 In patients with PD, both the sleep macrostructure (manifesting, for instance, as sleep fragmentation and a relative increase in superficial sleep) and sleep microstructure, manifesting as disturbed integrity of certain sleep stages (eg, disturbed sleep spindles and Kcomplexes, or insufficient muscle atonia during REM sleep), are affected.64–66
Perioperative Sleep Disturbances on Cognitive Function
In general, short-term total sleep deprivation has a deleterious effect across most cognitive domains, including attention, working memory, processing speed, short-term memory, and reasoning, with smaller effects on tasks of greater complexity.67 There is increasing evidence that sleep time may predict cognitive outcomes in older adults.68 Elderly women who reported a short sleep time were at an increased risk of cognitive impairment; however, there was no association with cognitive changes within 2 years, which may be due to the short follow-up time.69 In addition, the circadian rhythm reflects changes in biological processes that oscillate and regulate several physiological processes in a 24-hour period, including sleep. In a prospective study of elderly women living in the community for more than 5 years, changes in circadian rhythms, such as decreased amplitude, less robust rhythm, and delayed peak activity, were all related to dementia or mild cognitive impairment.70 Several mechanisms could explain the relationship between poor sleep quality and cognitive impairment. First, the consolidation of memory and normal brain functioning require high sleep quality,71,72 and sleep disturbance could interfere with the function of neuronal pathways, especially those of GABA and cAMP, which in turn impair synaptic plasticity.73 Poor sleep might contribute to neurodegeneration by causing neuroinflammation and disrupting neurogenesis, especially in the hippocampal areas, a key neuroanatomical region for learning and memory.74,75 Second, increased sleep fragmentation and hypoxia are two consequences of disordered breathing during sleep, which may impair cognitive function. In animal models, hypoxia increased apoptosis and hippocampal atrophy through oxidative and inflammatory pathways.76 Hypoxia increases the concentration of amyloid β, number of amyloid plaques, and tau phosphorylation in the brain, which are key components of Alzheimer’s disease pathology.77,78 Third, melatonin is a hormone produced by the pineal gland and related to the sleep-wake cycle. Renewed attention has been given to the role of melatonin in modulating behaviour, the immune system, and responses to stress, cancer and ageing.79 Additionally, exogenous melatonin can affect circadian rhythm/sleep disorders, insomnia, cancer, neurodegenerative diseases, immune function disorders and oxidative damage.80 Sedative, anxiolytic, anticonvulsant, antinociceptive and antidepressant actions have also been described.81–84 Melatonin has been shown to increase the numbers of GABAA receptors and reduce 5-HT2A receptor transmission, which may contribute to its antidepressant-like action.85,86 In addition, it has been reported that melatonin levels are disturbed in some neurological conditions, such as stroke, AD and PD, which indicates its involvement in the pathophysiology of these diseases. Its properties qualify it to be a promising potential therapeutic neuroprotective agent, with no side effects, for some neurological disorders.87,88 Clinically, many studies have reported that compared with healthy people, the level of melatonin in AD patients is reduced. After death, the level of melatonin in the CSF of the cerebral ventricle is negatively correlated with the Braak and modified Braak staging in the human cortex. Therefore, the level of melatonin is considered to be a marker of the progression of AD neuropathology. In the “preclinical” stages of AD Braak stages I and II, due to norepinephrine dysfunction and monoamine oxidase production, the melatonin circadian rhythm disappears.89–91 In a transgenic rat model of Alzheimer’s disease, long-term application of melatonin (approximately two months) reduced the deposition of immunoreactive antibodies in the hippocampus and cortex by 43% and 37%, respectively.92 Melatonin in the active stage of disease progression can reduce amyloid deposition in the hippocampus (β1-42 and β1-40) and frontal cortex (β1-42), reduce hippocampal degenerative changes, prevent mitochondrial dysfunction, and delay anxiety and cognitive impairment in a sporadic rat model of Alzheimer’s disease.93 Therefore, changes in the circadian rhythm and melatonin concentration may serve as early markers for the risk of Alzheimer’s disease and other dementias.
The Effects of Ketamine in Improving Perioperative Sleep Quality
The mechanisms by which ketamine improves sleep quality may be due to its antidepressant efficacy, anti-inflammatory properties, analgesic efficacy, interaction with the circadian system (Figures 1–3) and neurocognitive and anxiolytic effects.
Antidepressant Mechanisms of Ketamine and Potential Biochemical Biomarkers
Placebo-controlled trials have provided strong evidence for the rapid-acting (within hours) and sustained (lasting up to 7 days) antidepressant effects of a single administration of a subanesthetic dose of the noncompetitive NMDA receptor antagonist ketamine in treatment-resistant patients with depression.94–96 Moreover, the antidepressant effects of ketamine have been demonstrated in many antidepressant-relevant tests in experimental animals.97–99 The antidepressant mechanisms related to ketamine are listed as follows (Figure 1): 1) The cluster of electrical signals in the “anti-reward center” of the lateral habenula strengthened the inhibition of the “reward center” of the downstream cerebral monoamine nucleus, which causes depression. Blocking the cluster discharge of lateral habenular neurons can relieve the excessive inhibition of the “reward center” and play an antidepressant role. Cluster discharge of the lateral habenula depends on the NMDA receptor in the brain. Ketamine has long been recognized as a noncompetitive antagonist at NMDARs, which may explain its rapid antidepressant effects.100,101 Ketamine can selectively block NMDA receptors expressed on GABA inhibitory interneurons, resulting in decreased activity of GABAergic interneurons and deinhibition of pyramidal neurons, which further increases excitatory neurotransmitter glutamate released from the synaptic cleft, activates the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR), and increases the level of brain-derived neurotrophic factor.102 .2) The changes in brain-derived neurotrophic factor levels caused by ketamine are related to its antidepressant effects, the increase in early sleep slow wave activity during nonrapid eye movement sleep, and the improvement of sleep quality in patients with treatment-resistant depression. Brain-derived neurotrophic factor is a well-known marker of depression, and the magnitude of increase has been shown to predict the acute mood response to ketamine.103,104 The level of brain-derived neurotrophic factor depends on the regulation of eukaryotic elongation factor 2. Phosphorylated eukaryotic elongation factor 2 can inhibit the translation of brain-derived neurotrophic factor. Ketamine promotes the translation of brain-derived neurotrophic factors by reducing eukaryotic elongation factor 2 kinase activity and inhibiting the phosphorylation of eukaryotic elongation factor 2. Tropomyosin-related kinase B is the receptor of brain-derived neurotrophic factor, and activation of the brain-derived neurotrophic factor-tropomyosin-related kinase B pathway plays an important role in the treatment of depression.105,106 3) AMPAR is an ionotropic glutamate receptor responsible for rapid synaptic nerve conduction. Ketamine can either directly activate AMPAR or indirectly activate AMPAR by antagonizing NMDA receptors to promote the release of glutamate, which has antidepressant effects. After ketamine activates AMPAR, it causes the opening of L-type voltage-dependent calcium ion channels, stimulates the release of brain-derived neurotrophic factor, and enhances its antidepressant effects.107 4) Ketamine rapidly activates the mammalian target of rapamycin (mTOR) pathway, leading to an increase in synaptic signaling protein in the prefrontal cortex of rats and an increase in the number and function of new spinous synapses, which represents the mechanism of rapid antidepressant effects of ketamine. In addition, in the depression model, the blockade of mTOR signaling completely blocked the induction of synaptic and behavioral responses by ketamine.108 5) Ketamine may significantly increase the release of monoamine transmitters in the central nervous system, promote angiogenesis and synaptic regeneration, and enhance neuronal activity, which may be associated with its antidepressant effects.109,110
Analgesia and Anti-Inflammation Mechanism of Ketamine
The use of ketamine has been limited due to its psychodysleptic effects. At first, it was mainly used for anesthetic purposes. After United States Food and Drug Administration approval in 1970, ketamine began to be used for analgesic purposes.111 Human studies have reported that adding ketamine to opioid treatment of acute pain can prevent opioid-induced respiratory depression and hyperalgesia.112 Using the smallest dose of S-ketamine during perioperative anesthesia, lower than normal low-dose racemic ketamine, could reduce postoperative opioid consumption and hyperalgesia and reduce the risk of postoperative delirium.113 The use of esketamine in the perioperative period can relieve pain by 20%~25% at 48 h after surgery while reducing the total dosage of analgesics required by 30%~50%, reducing the adverse effects experienced by patients, such as nausea and vomiting.114 The potential mechanism may be as follows (Figure 2): 1) The emergence of noxious stimuli causes the release of the nerve presynaptic membrane excitatory transmitter glutamate. Then, activation of the NMDA receptor in the central and peripheral nervous systems causes voltage-dependent sodium and calcium ions to enter the cell and potassium ions to exit the cell. As a noncompetitive antagonist of NMDA receptors, ketamine blocks the transmission of glutamate by shortening the opening time of these receptor channels and reducing the frequency of receptor channel opening, thereby exerting anesthetic and analgesic effects. In addition, ketamine also induces a sustained blocking effect by reducing the rate of separation from the receptor.115 2) NO is an important factor in tissue damage and inflammation. Hypoxia and ischemia can activate NMDA receptors to produce NO. Ketamine can reduce the production of NO by inhibiting NO synthase, thereby inhibiting inflammatory pain.116 3) In the case of ischemia, hypoxia, and trauma, nerve cells release a large amount of excitatory amino acids such as glutamate, which act on NMDA receptors and cause nerve cell damage. Ketamine can inhibit the activation of NMDA receptors and reduce the concentration of glutamate to protect nerve cells. Cell injury also leads to the release of excitatory amino acids that bind to the receptor, resulting in a large concentration of calcium ions entering the cell. Ketamine blocks the NMDA receptor, thereby stabilizing intracellular calcium ion levels and inducing neuroprotective effects.117 4) Ketamine can inhibit the activation of leukocytes during the inflammatory response, reduce the production of inflammatory cytokines such as tumor necrosis factor-α, interleukin-6, and interleukin-8, and stimulate the secretion of anti-inflammatory factors such as interleukin-4 and interleukin-10 to relieve the nerve damage caused by inflammation.117
Regulatory Mechanism of Ketamine on Sleep and the Circadian System
REM reduction, slow wave sleep, and sleep fragmentation are important clinical biomarkers of perioperative sleep disturbance and circadian rhythm asynchrony.118 Given the beneficial effects of ketamine on sleep, an early rat study showed that ketamine stimulates slow-wave activity during non-REM sleep.119 The results of Ahnaou et al further extend these observations. In particular, ketamine prolongs the average duration of deep sleep, indicating that deep sleep is maintained and consolidated.120 In addition, ketamine-induced brain-derived neurotrophic factor, a modulator of neuroplasticity, induced stage N3 sleep.121,122 Furthermore, data from human, animal, and neuronal cell studies also suggested that low-dose ketamine can modulate circadian rhythms by regulating clock genes.123 These findings suggest that the rapid antidepressant effect of ketamine may also be related to its effect on clock gene-related molecules, leading to changes in the circadian timing of the central clock and/or its effect on the entrainment circuit that synchronizes the central clock with the external light cycle.103 The possible reasons may be as follows (Figure 3): 1) ketamine’s effects on circadian rhythms include attenuating phase-shifting responses to light, altering the diurnal rhythms of the widespread NMDA receptors and AMPARs in the suprachiasmatic nucleus, and increasing the expression of the core clock genes per1 and per2 via its actions on glutamate receptors.124 2) Recent work has implicated mammalian target of rapamycin as a potential site for ketamine’s rapid antidepressant actions. Glycogen synthase kinase 3β is a potent inhibitor of mammalian target of rapamycin C1 (rapamycin complex 1). Blocking mammalian target of rapamycin significantly attenuates the light-induced expression of the circadian PER1 and PER2 proteins. Furthermore, rodent studies demonstrated that inhibition of glycogen synthase kinase 3β may play a role in the antidepressant actions of low-dose ketamine.125–129 2) Ketamine downregulates the period circadian regulator 2 gene by changing its inhibitory actions on the heterodimer CLOCK/BMAL1. In addition, ketamine can alter the Notch signaling pathway and the MAPK signaling pathway, which responds to excitatory glutamatergic signaling that controls synaptic plasticity and higher brain processes.130 3) The lateral habenula is a region of the brain connected with the suprachiasmatic nucleus and could be a potential candidate for the circadian effects induced by ketamine. The lateral habenula appears to function as an individual circadian oscillator in the central nervous system. In particular, it exhibits inhibitory control over connecting dopaminergic, noradrenergic, and serotonergic areas. The increased activity of lateral habenular nucleus neurons may promote stronger inhibitory control in these areas and have a negative impact on mood regulation. A recent rodent study showed that the burst activity of lateral habenular neurons in depressive animals increased, and ketamine reversed this phenomenon.131 Moreover, Kushikata T et al also found that ketamine and propofol had opposite effects on the activity of the sleep–wakefulness-related endogenous substances orexin (OX) and melanin-concentrating hormone (MCH). Ketamine has a greater effect on OX activity during the perianaesthetic period, while propofol causes a greater increase in MCH activity after the anaesthesia period and has a smaller effect on OX activity. These differences are related to the different effects of the two drugs on the sleep structure after anaesthesia. Ketamine immediately enhanced wakefulness after anesthesia on the first day after anesthesia, and NREMS rebounded. Their research shows that anesthetics can affect various sleep-related endogenous substances; however, the modulation mode may depend on the type of anesthetic. In other words, the process of sleep disturbance after anesthesia may be drug-specific.132
Neurocognitive and Anxiolytic Effects of Ketamine
Major depressive disorder (MDD) is the most common mental disorder, affecting more than 16% of adults throughout their lives. Although standard antidepressant treatment is usually effective, approximately 30% of MDD patients do not respond adequately to established medications, psychotherapy, or physical therapy, which may be because symptoms of MDD, such as low mood, anhedonia, cognitive dysfunctions and somatic manifestations, are very heterogeneous.133 Convincing reports indicate that the most important susceptibility factors for MDD are chronic stress and hypothalamic–pituitary–adrenal (HPA) axis dysfunction. Although the mechanisms by which glucocorticoids contribute to depressive symptoms have not been entirely established, strong pieces of evidence have reported that chronic exposure to glucocorticoids causes structural defects in the dentategyrus (DG) area of the hippocampus, including reduction of dendritic arbor and spine density, concomitant with the onset of depressive symptoms.134 An animal study by Fraga DB et al showed that ketamine can reverse corticosterone-induced loss of dendritic branches in the ventral and dorsal DG regions, which are associated with mood regulation and cognitive function, respectively. This study provides new evidence that a single dose of ketamine can rescue the damage to glucocorticoid receptors and dendritic branches in the hippocampus of mice receiving chronic corticosterone administration. These effects may be related to their rapid antidepressant response.135 In addition, a single dose of S-ketamine, instead of R-ketamine, can cause the loss of paralbumin (PV)-positive cells in the medial prefrontal cortex and the anterior DG area.136 Moreover, anxiety disorders, currently the most prevalent psychiatric disorder, cause a high social impact and economic burden.137 They are often comorbid with major depressive disorder, which has an overall rate of 50–60% for the occurrence of any anxiety disorder. Patients with anxious forms of depression are more likely to have severe depressive symptoms, including fatigue, thoughts of guilt, and worthlessness, and are more resistant to treatments.138 Recent literature proves that ketamine may be a potential treatment option for patients with refractory generalized anxiety disorder/social anxiety disorder. A subanesthetic dose of ketamine induces anxiety with a long lasting time in 12 patients (up to 1 week).139–141 Animal models also show different dose-dependent effects of ketamine on cognition, depression, and anxiety. The main hypothetical mechanism is to change BDNF levels. Subanaesthetic doses of ketamine are considered to have a positive effect on the level of BDNF in the hippocampus. Wu C et al found that a single infusion of subanaesthetic doses of ketamine (0.5 mg/kg) increased hippocampal volume (usually a representative of increased BDNF). This is also true in a small group of MDD patients who are not receiving medication.142 An animal study confirmed that an anesthetic dose reduced BDNF expression in the hippocampus, correlating with depressive-like behaviors, anxiety-like behaviors and cognitive impairment.143 Human studies have shown that infusion of higher analgesic doses (8–20 mg/h) in healthy volunteers also shows significant cognitive deficits, which indicates that acute ketamine is potentially dose-dependent on cognitive and cognitive symptoms.144 Despite this, several studies have shown that the dose of anesthetics obviously lacks any form of long-term side effects, and the dose of subanesthetics seems to have a very low risk in clinical trials.145 de Souza I et al’s study also demonstrated that a single injection of ketamine can cause impaired episodic memory, but this may be an acute finding and not related to long-term cognitive dysfunction.146 Another recent study on intranasal esketamine administration in healthy volunteers showed that there was significant cognitive dysfunction at 40 min, but no cognitive dysfunction was found at 2, 4, and 6 h postdose.147 In conclusion, the dose of ketamine used to treat resistant depression seems to have an overall cognitive effect, which may be the basis of its rapid effectiveness. The negative cognitive side effects of ketamine may appear transiently during acute administration of ketamine but only persist when ketamine is used in large quantities for a long time and appear to be reversible.
Conclusions and Future Directions
Perioperative sleep disturbances are commonly observed before, during, and after surgery. They are usually ignored and may increase the risk of postoperative neurocognitive disorders. Thus, improving perioperative sleep may reduce the incidence of postoperative delirium and postoperative cognitive dysfunction. Ketamine, as a nonselective, noncompetitive antagonist of the NMDA receptor, has made remarkable progress in the last 5 years in terms of identification of the molecular and cellular mechanisms critical to its rapid antidepressant effects. In particular, combining two markers of synaptic plasticity, brain-derived neurotrophic factor levels and EEG sleep slow waves, was demonstrated to be an effective approach for identifying ketamine’s capacity to increase synaptic strength. Meanwhile, ketamine could also improve symptoms of anxiety in patients with treatment refractory generalized anxiety disorder or social anxiety disorder who are not currently depressed and is safe and well tolerated. Recent intensive research has shed light on new possible applications of this drug. As indicated in this review paper, studies on the mechanism of action of ketamine in improving perioperative sleep quality have resulted in some interesting findings worthy of further research. Further research is needed to identify other signs of plasticity and expand emotional responses.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
The present study was funded by the Joint Plan of Key R&D Plan of Liaoning Provincial Science and Technology Department (2020JH2/10300123), 345 Talent Project and the Support Plan for Innovative Talents in Liaoning Higher Education Institution (grant no. 201834).
Disclosure
The authors declare no competing interests in this work.
References
1. Cok OY, Seet E, Kumar CM, Joshi GP. Perioperative considerations and anesthesia management in patients with obstructive sleep apnea undergoing ophthalmic surgery. J Cataract Refract Surg. 2019;45:1026–1031. doi:10.1016/j.jcrs.2019.02.044
2. Rhon DI, Snodgrass SJ, Cleland JA, Cook CE. Comorbid insomnia and sleep apnea are associated with greater downstream health care utilization and chronic opioid use after arthroscopic hip surgery. Pain Physician. 2019;22:E351–60. doi:10.36076/ppj/2019.22.E351
3. Chouchou F, Khoury S, Chauny JM, Denis R, Lavigne GJ. Postoperative sleep disruptions: a potential catalyst of acute pain? Sleep Med Rev. 2014;18:273–282. doi:10.1016/j.smrv.2013.07.002
4. Ruyi Z. Relationship between sleep condition and negative psychology in perioperative patients. Chin J Practical Neurodiseases. 2014;17(17):93.
5. Trbojević-Stanković J, Stojimirović B, Bukumirić Z, et al. Depression and quality of sleep in maintenance hemodialysis patients. Srp Arh Celok Lek. 2014;142(7–8):437–443. doi:10.2298/SARH1408437T
6. Mansano-Schlosser TC, Ceolim MF. Factors associated with poor sleep quality in women with cancer. Rev Lat Am Enfermagem. 2017;25:e2858. doi:10.1590/1518-8345.1478.2858
7. Nie A, Wang C, Song Y, Xie X, Yang H, Chen H. Prevalence and factors associated with disturbed sleep in outpatients with ankylosing spondylitis. Clin Rheumatol. 2018;37(8):2161–2168. doi:10.1007/s10067-018-4190-3
8. Herrero Babiloni A, De Koninck BP, Beetz G, De Beaumont L, Martel MO, Lavigne GJ. Sleep and pain: recent insights, mechanisms, and future directions in the investigation of this relationship. J Neural Transm. 2020;127(4):647–660. doi:10.1007/s00702-019-02067-z
9. Tamisier R, Fabre F, O’Donoghue F, Lévy P, Payen JF, Pépin JL. Anesthesia and sleep apnea. Sleep Med Rev. 2018;40:79–92. doi:10.1016/j.smrv.2017.10.006
10. Yaremchuk K. Sleep Disorders in the Elderly. Clin Geriatr Med. 2018;34(2):205–216. doi:10.1016/j.cger.2018.01.008
11. Javaheri S, Barbe F, Campos-Rodriguez F, et al. Sleep Apnea: types, Mechanisms, and Clinical Cardiovascular Consequences. J Am Coll Cardiol. 2017;69(7):841–858. doi:10.1016/j.jacc.2016.11.069
12. de Almondes KM, Costa MV, Malloy-Diniz LF, Diniz BS. The relationship between sleep complaints, depression, and executive functions on older adults. Front Psychol. 2016;7:1–8. doi:10.3389/fpsyg.2016.01547
13. Kaur U, Pathak BK, Singh A, Chakrabarti SS. Esketamine: a glimmer of hope in treatment-resistant depression. Eur Arch Psychiatry Clin Neurosci. 2021;271(3):417–429. doi:10.1007/s00406-019-01084-z
14. Pham TH, Gardier AM. Fast-acting antidepressant activity of ketamine: highlights on brain serotonin, glutamate, and GABA neurotransmission in preclinical studies. Pharmacol Ther. 2019;199:58–90. doi:10.1016/j.pharmthera.2019.02.017
15. Barrett W, Buxhoeveden M, Dhillon S. Ketamine: a versatile tool for anesthesia and analgesia. Curr Opin Anaesthesiol. 2020;33(5):633–638. doi:10.1097/ACO.0000000000000916
16. Weiser TG, Haynes AB, Molina G, et al. Size and distribution of the global volume of surgery in 2012. Bull World Health Organ. 2016;94:201–9F. doi:10.2471/BLT.15.159293
17. Ruis C, Wajer IH, Robe P, van Zandvoort M. Anxiety in the preoperative phase of awake brain tumor surgery. Clin Neurol Neurosurg. 2017;157:7–10. doi:10.1016/j.clineuro.2017.03.018
18. Hellstadius Y, Lagergren J, Zylstra J, et al. Prevalence and predictors of anxiety and depression among esophageal cancer patients prior to surgery. Dis Esophagus. 2016;29:1128–1134. doi:10.1111/dote.12437
19. Britteon P, Cullum N, Sutton M. Association between psychological health and wound complications after surgery. Br J Surg. 2017;104:769–776. doi:10.1002/bjs.10474
20. Wichniak A, Wierzbicka A, Jernajczyk W. Sleep as a biomarker for depression. Int Rev Psychiatry. 2013;25(5):632–645. doi:10.3109/09540261.2013.812067
21. Olbrich S, Arns M. EEG biomarkers in major depressive disorder: discriminative power and prediction of treatment response. Int Rev Psychiatry. 2013;25(5):604–618. doi:10.3109/09540261.2013.816269
22. Ni P, Dong H, Zhou Q, et al. Preoperative Sleep Disturbance Exaggerates Surgery-Induced Neuroinflammation and Neuronal Damage in Aged Mice. Mediators Inflamm. 2019;2019:8301725. doi:10.1155/2019/8301725
23. Pace‐Schott EF, Hobson JA. The neurobiology of sleep: genetics, cellular physiology and subcortical networks. Nat Rev Neurosci. 2002;3(8):591–605. doi:10.1038/nrn895
24. Wang YQ, Li R, Zhang MQ, Zhang Z, Qu WM, Huang ZL. The neurological mechanisms and treatments of REM sleep disturbances in depression. Curr Neuropharmacol. 2015;13(4):543–553. doi:10.2174/1570159X13666150310002540
25. Caspi A, Hariri AR, Holmes A, Uher R, Moffitt TE. Genetic sensitivity to the environment: the case of the serotonin transporter gene and its implications for studying complex diseases and traits. Am J Psychiatry. 2010;167(5):509–527. doi:10.1176/appi.ajp.2010.09101452
26. Li JZ, Bunney BG, Meng F, et al. Circadian patterns of gene expression in the human brain and disruption in major depressive disorder. Proc Natl Acad Sci USA. 2013;110(24):9950–9955. doi:10.1073/pnas.1305814110
27. Lamont EW, Legault‐Coutu D, Cermakian N, Boivin DB. The role of circadian clock genes in mental disorders. Dialogues Clin Neurosci. 2007;9(3):333.
28. Satyanarayanan SK, Su H, Lin YW, Su KP. Circadian Rhythm and Melatonin in the Treatment of Depression. Curr Pharm Des. 2018;24(22):2549–2555. doi:10.2174/1381612824666180803112304
29. Li SX, Liu LJ, Xu LZ, et al. Diurnal alterations in circadian genes and peptides in major depressive disorder before and after escitalopram treatment. Psychoneuroendocrinology. 2013;38(11):2789–2799. doi:10.1016/j.psyneuen.2013.07.009
30. Patke A, Murphy PJ, Onat OE, et al. Mutation of the human circadian clock gene CRY1 in familial delayed sleep phase disorder. Cell. 2017;169(2):203–215.e13. doi:10.1016/j.cell.2017.03.027
31. Krysta K, Krzystanek M, Bratek A, Krupka-Matuszczyk I. Sleep and inflammatory markers in different psychiatric disorders. J Neural Transm. 2017;124(Suppl 1):179–186. doi:10.1007/s00702-015-1492-3
32. Jiang-Xie LF, Yin L, Zhao S, et al. A Common Neuroendocrine Substrate for Diverse General Anesthetics and Sleep. Neuron. 2019;102(5):1053–1065.e4. doi:10.1016/j.neuron.2019.03.033
33. Luo M, Song B, Zhu J. Electroacupuncture: a New Approach for Improved Postoperative Sleep Quality After General Anesthesia. Nat Sci Sleep. 2020;12:583–592. doi:10.2147/NSS.S261043
34. Ozone M, Itoh H, Yamadera W, et al. Changes in subjective sleepiness, subjective fatigue and nocturnal sleep after anaesthesia with propofol. Psychiatry Clin Neurosci. 2000;54:317–318. doi:10.1046/j.1440-1819.2000.00694.x
35. Steinmetz J, Holm-Knudsen R, Eriksen K, Marxen D, Rasmussen LS. Quality differences in postoperative sleep between propofol-remifentanil and sevoflflurane anesthesia in infants. Anaesth Analg. 2007;104:779–783. doi:10.1213/01.ane.0000255694.00651.5b
36. Dispersyn G, Touitou Y, Coste O, et al. Desynchronization of daily rest-activity rhythm in the days following light propofol anesthesia for colonoscopy. Clin Pharmacol Ther. 2009;85(1):51e5. doi:10.1038/clpt.2008.179
37. Kincheski GC, Valentim IS, Clarke JR, et al. Chronic sleep restriction promotes brain inflammation and synapse loss, and potentiates memory impairment induced by amyloid-β oligomers in mice. Brain Behav Immun. 2017;64:140–151. doi:10.1016/j.bbi.2017.04.007
38. Brosnan RJ, Thiesen R. Increased NMDA receptor inhibition at an increased Sevoflurane MAC. BMC Anesthesiol. 2012;12:12. doi:10.1186/1471-2253-12-12
39. Hummer DL, Ehlen JC. Sustained activation of GABAA receptors in the suprachiasmatic nucleus mediates light-induced phase delays of the circadian clock: a novel function of ionotropic receptors. Eur J Neurosci. 2015;42(2):1830e8. doi:10.1111/ejn.12918
40. Kadota K, Iijima N, Ohe-Hayashi Y, et al. Timedependent repression of mPer2 expression in the suprachiasmatic nucleus by inhalation anesthesia with sevoflflurane. Neurosci Lett. 2012;528(2):153e8. doi:10.1016/j.neulet.2012.07.061
41. Anzai M, Iijima N, Higo S, et al. Direct and specific effect of sevoflurane anesthesia on rat Per2 expression in the suprachiasmatic nucleus. PLoS One. 2013;8(3):e59454. doi:10.1371/journal.pone.0059454
42. Ohe Y, Iijima N, Kadota K, Sakamoto A, Ozawa H. The general anesthetic sevoflurane affects the expression of clock gene mPer2 accompanying the change of NAD(þ) level in the suprachiasmatic nucleus of mice. Neurosci Lett. 2011;490(3):231e6. doi:10.1016/j.neulet.2010.12.059
43. Kobayashi K, Takemori K, Sakamoto A. Circadian gene expression is suppressed during sevoflurane anesthesia and the suppression persists after awakening. Brain Res. 2007;1185:1e7. doi:10.1016/j.brainres.2007.09.011
44. Bellet MM, Vawter MP, Bunney BG, Bunney WE, Sassone-Corsi P. Ketamine influences CLOCK: BMAL1 function leading to altered circadian gene expression. PLoS One. 2011;6(8):e23982. doi:10.1371/journal.pone.0023982
45. Doi M, Hirayama J, Sassone-Corsi P. Circadian regulator CLOCK is a histone acetyltransferase. Cell. 2006;125(3):497e508. doi:10.1016/j.cell.2006.03.033
46. Wu CL, Raja SN. Treatment of acute postoperative pain. Lancet. 2011;377:2215e25. doi:10.1016/S0140-6736(11)60245-6
47. Sommer M, de Rijke JM, van Kleef M, et al. The prevalence of postoperative pain in a sample of 1490 surgical inpatients. Eur J Anaesthesiol. 2008;25:267e74. doi:10.1017/S0265021507003031
48. Kehlet H, Jensen TS, Woolf CJ. Persistent postsurgical pain: risk factors and prevention. Lancet. 2006;367:1618e25. doi:10.1016/S0140-6736(06)68700-X
49. Haack M, Simpson N, Sethna N, Kaur S, Mullington J. Sleep deficiency and chronic pain: potential underlying mechanisms and clinical implications. Neuropsychopharmacology. 2020;45(1):205–216. doi:10.1038/s41386-019-0439-z
50. Perogamvros L, Schwartz S. The roles of the reward system in sleep and dreaming. Neurosci Biobehav Rev. 2012;36:1934–1951. doi:10.1016/j.neubiorev.2012.05.010
51. Qiu MH, Liu W, Qu WM, Urade Y, Lu J, Huang ZL. The role of nucleus accumbens core/shell in sleep-wake regulation and their involvement in modafinil-induced arousal. PLoS One. 2012;7:e45471. doi:10.1371/journal.pone.0045471
52. Foo H, Mason P. Brainstem modulation of pain during sleep and waking. Sleep Med Rev. 2003;7:145–154. doi:10.1053/smrv.2002.0224
53. Julien N, Marchand S. Endogenous pain inhibitory systems activated by spatial summation are opioid-mediated. Neurosci Lett. 2006;401:256–260. doi:10.1016/j.neulet.2006.03.032
54. Staud R, Robinson ME, Vierck CJ
55. Finan PH, Goodin BR, Smith MT. The association of sleep and pain: an update and a path forward. J Pain. 2013;14(12):1539–1552. doi:10.1016/j.jpain.2013.08.007
56. Dugger BN, Dickson DW. Pathology of Neurodegenerative Diseases. Cold Spring Harb Perspect Biol. 2017;9(7):a028035. doi:10.1101/cshperspect.a028035
57. Malhotra RK. Neurodegenerative Disorders and Sleep. Sleep Med Clin. 2018;13(1):63–70. doi:10.1016/j.jsmc.2017.09.006
58. Ju YS, Ooms SJ, Sutphen C, et al. Slow wave sleep disruption increases cerebrospinal fluid amyloid-beta levels. Brain. 2017;140:2104–2111. doi:10.1093/brain/awx148
59. Ooms S, Overeem S, Besse K, Rikkert MO, Verbeek M, Claassen JA. Effect of 1 night of total sleep deprivation on cerebrospinal fluid beta-amyloid 42 in healthy middle-aged men: a randomized clinical trial. JAMA Neurol. 2014;71:971–977. doi:10.1001/jamaneurol.2014.1173
60. Kang JE, Lim MM, Bateman RJ, et al. Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science. 2009;326:1005–1007. doi:10.1126/science.1180962
61. Di MA, Joshi YB, Pratico D. Sleep deprivation impairs memory, tau metabolism, and synaptic integrity of a mouse model of Alzheimer’s disease with plaques and tangles. Neurobiol Aging. 2014;35:1813–1820. doi:10.1016/j.neurobiolaging.2014.02.011
62. Pringsheim T, Jette N, Frolkis A, et al. The prevalence of Parkinson0 s disease: a systematic review and meta-analysis. Mov Disord. 2014;29:1583–1590. doi:10.1002/mds.25945
63. Braak H, Ghebremedhin E, Rub U, Bratzke H. and Del Tredici K. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 2004;318:121–134. doi:10.1007/s00441-004-0956-9
64. Christensen JA, Nikolic M, Warby SC, et al. Sleep spindle alterations in patients with Parkinson’s disease. Front Hum Neurosci. 2015;9:233. doi:10.3389/fnhum.2015.00233
65. Pont-Sunyer C, Iranzo A, Gaig C, et al. Sleep Disorders in Parkinsonian and Nonparkinsonian LRRK2 Mutation Carriers. PLoS One. 2015;10:e0132368. doi:10.1371/journal.pone.0132368
66. O’Dowd S, Galna B, Morris R, et al. Poor Sleep Quality and Progression of Gait Impairment in an Incident Parkinson’s Disease Cohort. J Parkinson’s Dis. 2017;7:465–470. doi:10.3233/JPD-161062
67. Ma Y, Liang L, Zheng F, Shi L, Zhong B, Xie W. Association Between Sleep Duration and Cognitive Decline. JAMA Netw Open. 2020;3(9):e2013573. doi:10.1001/jamanetworkopen.2020.13573
68. Liu Y, Wheaton AG, Chapman DP, Croft JB. Sleep duration and chronic diseases among US adults age 45 years and older: evidence from the 2010 behavioral risk factor surveillance system. Sleep. 2013;36:1421–1427. doi:10.5665/sleep.3028
69. Tworoger SS, Lee S, Schernhammer ES, Grodstein F. The association of self-reported sleep duration, difficulty sleeping, and snoring with cognitive function in older women. Alzheimer Dis Assoc Disord. 2006;20:41–48. doi:10.1097/01.wad.0000201850.52707.80
70. Tranah GJ, Blackwell T, Stone KL, et al. Circadian activity rhythms and risk of incident dementia and mild cognitive impairment in older women. Ann Neurol. 2011;70:722–732. doi:10.1002/ana.22468
71. Diekelmann S, Born J. The memory function of sleep. Nat Rev Neurosci. 2010;11:114–126.
72. Stickgold R. Sleep-dependent memory consolidation. Nature. 2005;437:1272–1278. doi:10.1038/nature04286
73. Havekes R, Vecsey CG, Abel T. The impact of sleep deprivation on neuronal and glial signaling pathways important for memory and synaptic plasticity. Cell Signal. 2012;24:1251–1260. doi:10.1016/j.cellsig.2012.02.010
74. Zhu B, Dong Y, Xu Z, et al. Sleep disturbance induces neuroinflammation and impairment of learning and memory. Neurobiol Dis. 2012;48:348–355. doi:10.1016/j.nbd.2012.06.022
75. Meerlo P, Mistlberger RE, Jacobs BL, Craig Heller H, McGinty D. New neurons in the adult brain: the role of sleep and consequences of sleep loss. Sleep Med Rev. 2009;13:187–194. doi:10.1016/j.smrv.2008.07.004
76. Nair D, Dayyat EA, Zhang SX, Wang Y, Gozal D. Intermittent hypoxia-induced cognitive deficits are mediated by NADPH oxidase activity in a murine model of sleep apnea. PLoS One. 2011;6:e19847. doi:10.1371/journal.pone.0019847
77. Gao L, Tian S, Gao H, Xu Y. Hypoxia increases Aβ-induced tau phosphorylation by calpain and promotes behavioral consequences in AD transgenic mice. J Mol Neurosci. 2013;51:138–147. doi:10.1007/s12031-013-9966-y
78. Li L, Zhang X, Yang D, Luo G, Chen S, Le W. Hypoxia increases Aβ generation by altering β- and γ-cleavage of APP. Neurobiol Aging. 2009;30:1091–1098. doi:10.1016/j.neurobiolaging.2007.10.011
79. Macchi MM, Bruce JN. Human pineal physiology and functional significance of melatonin. Front Neuroendocrinol. 2004;25:177–195. doi:10.1016/j.yfrne.2004.08.001
80. Pandi-Perumal SR, Srinivasan V, Maestroni GJ, Cardinali DP, Poeggeler B, Hardeland R. Melatonin: nature’s most versatile biological signal? FEBS J. 2006;273:2813–2838. doi:10.1111/j.1742-4658.2006.05322.x
81. Golombek DA, Pevet P, Cardinali DP. Melatonin effects on behavior: possible mediation by the central GABAergic system. Neurosci Biobehav Rev. 1996;20:403–412. doi:10.1016/0149-7634(95)00052-6
82. Mantovani M, Pertile R, Calixto JB, Santos AR, Rodrigues AL. Melatonin exerts an antidepressant-like effect in the tail suspension test in mice: evidence for involvement of N-methyl-D-aspartate receptors and the L-arginine-nitric oxide pathway. Neurosci Lett. 2003;343:1–4. doi:10.1016/S0304-3940(03)00306-9
83. Mantovani M, Pértile R, Calixto JB, Santos AR, Rodrigues AL. Melatonin exerts an antidepressant-like effect in the tail suspension test in mice: evidence for involvement of N-methyl-D-aspartate receptors and the L-arginine-nitric oxide pathway. Neurosci Lett. 2003;343(1):1–4. doi:10.1016/s0304-3940(03)00306
84. Sanacora G, Saricicek A. GABAergic contributions to the pathophysiology of depression and the mechanism of antidepressant action. CNS Neurol Disord Drug Targets. 2007;6:127–140. doi:10.2174/187152707780363294
85. Detanico BC, Piato AL, Freitas JJ, et al. Antidepressant-like effects of melatonin in the mouse chronic mild stress model. Eur J Pharmacol. 2009;607(1–3):121–125. doi:10.1016/j.ejphar.2009.02.037
86. Raghavendra V, Kaur G, Kulkarni SK. Anti-depressant action of melatonin in chronic forced swimming-induced behavioral despair in mice, role of peripheral benzodiazepine receptor modulation. Eur Neuropsychopharmacol. 2000;10:473–481. doi:10.1016/S0924-977X(00)00115-2
87. Alghamdi BS. The neuroprotective role of melatonin in neurological disorders. J Neurosci Res. 2018;96(7):1136–1149. doi:10.1002/jnr.24220
88. Wu YH, Swaab DF. The human pineal gland and melatonin in aging and Alzheimer’s disease. J Pineal Res. 2005;38:145–152. doi:10.1002/jnr.24220
89. Ohashi Y, Okamoto N, Uchida K, Iyo M, Mori N, Morita Y. Daily rhythm of serum melatonin levels and effect of light exposure in patients with dementia of the Alzheimer’s type. Biol Psychiatry. 1999;45(12):1646–1652. doi:10.1016/S0006-3223(98)00255-8
90. Wu YH, Feenstra MG, Zhou JN, et al. Molecular changes underlying reduced pineal melatonin levels in Alzheimer disease: alterations in preclinical and clinical stages. J Clin Endocrinol Metab. 2003;88(12):5898–5906. doi:10.1210/jc.2003-030833
91. Zhou JN, Liu RY, Kamphorst W, Hofman MA, Swaab DF. Early neuropathological Alzheimer’s changes in aged individuals are accompanied by decreased cerebrospinal fluid melatonin levels. J Pineal Res. 2003;35(2):125–130. doi:10.1034/j.1600-079X.2003.00065.x
92. Olcese JM, Cao C, Mori T, et al. Protection against cognitive deficits and markers of neurodegeneration by long-term oral administration of melatonin in a transgenic model of Alzheimer disease. J Pineal Res. 2009;47(1):82–96. doi:10.1111/j.1600-079X.2009.00692.x
93. Rudnitskaya EA, Muraleva NA, Maksimova KY, Kiseleva E, Kolosova NG, Stefanova NA. Melatonin Attenuates Memory Impairment, Amyloid-beta Accumulation, and Neurodegeneration in a Rat Model of Sporadic Alzheimer’s Disease. J Alzheimer’s Dis. 2015;47(1):103–116. doi:10.3233/jad-150161
94. Price RB, Iosifescu DV, Murrough JW, et al. Effects of ketamine on explicit and implicit suicidal cognition: a randomized controlled trial in treatment-resistant depression. Depress Anxiety. 2014;31:335–343. doi:10.1002/da.22253
95. DiazGranados N, Ibrahim LA, Brutsche NE, et al. Rapid resolution of suicidal ideation after a single infusion of an N-methylD-aspartate antagonist in patients with treatment-resistant major depressive disorder. J Clin Psychiatry. 2010;71:1605–1611. doi:10.4088/JCP.09m05327blu
96. Lapidus KA, Levitch CF, Perez AM, et al. A randomized controlled trial of intranasal ketamine in major depressive disorder. Biol Psychiatry. 2014;76:970–976. doi:10.1016/j.biopsych.2014.03.026
97. Zanos P, Gould TD. Mechanisms of ketamine action as an antidepressant. Mol Psychiatry. 2018;23(4):801–811. doi:10.1038/mp.2017.255
98. Zanos P, Piantadosi SC, Wu HQ, et al. The prodrug 4- chlorokynurenine causes ketamine-like antidepressant effects, but not side effects, by NMDA/glycineB-site inhibition. J Pharmacol Exp Ther. 2015;355:76–85. doi:10.1124/jpet.115.225664
99. Zanos P, Thompson SM, Duman RS, Zarate CA, Gould TD. Gould TD Convergent mechanisms underlying rapid antidepressant action. CNS Drugs. 2018;32:197–227. doi:10.1007/s40263-018-0492-x
100. Yang Y, Cui Y, Sang K, et al. Ketamine blocks bursting in the lateral habenula to rapidly relieve depression. Nature. 2018;554(7692):317–322. doi:10.1038/nature25509
101. Matveychuk D, Thomas RK, Swainson J, et al. Ketamine as an antidepressant: overview of its mechanisms of action and potential predictive biomarkers. Ther Adv Psychopharmacol. 2020;10:2045125320916657. doi:10.1177/2045125320916657
102. Aleksandrova LR, Phillips AG, Wang YT. Antidepressant effects of ketamine and the roles of AMPA glutamate receptors and other mechanisms beyond NMDA receptor antagonism. J Psychiatry-Neurosci. 2017;42(4):222–229. doi:10.1503/jpn.160175
103. Duncan WC
104. Bjorkholm C, Monteggia LM. BDNF: a key transducer of antidepressant effects. Neuropharmacology. 2016;102:72–79. doi:10.1016/j.neuropharm.2015.10.034
105. Kadriu B, Musazzi L, Henter ID, et al. Glu-tamatergic neurotransmission: pathway to developing novel rapid-acting antidepressant treatments. Int J Neuropsychopharmacol. 2019;22(2):119–135. doi:10.1093/ijnp/pyy094
106. Zhang JC, Yao W, Hashimoto K. Brain-derived neurotrophic factor(BDNF)-TrkB signaling in inflammation-related depression and potential therapeutic targets. Curr Neuropharmacol. 2016;14(7):721–731. doi:10.2174/1570159X14666160119094646
107. Derkach VA, Oh MC, Guire ES, et al. mechanisms of AMPA receptors in synaptic plas-ticity. Nat Rev Neurosci. 2007;8(2):101–113. doi:10.1038/nrn2055
108. Li N, Lee B, Liu RJ, et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010;329(5994):959–964. doi:10.1126/science.1190287
109. Ago Y, Tanabe W, Higuchi M, et al. (R)-Ketamine Induces a Greater Increase in Prefrontal 5-HT Release Than (S)-Ketamine and Ketamine Metabolites via an AMPA Receptor-Independent Mechanism. Int J Neuropsychopharmacol. 2019;22(10):665–674. doi:10.1093/ijnp/pyz041
110. Ardalan M, Wegener G, Rafati AH, Nyengaard JR. S-Ketamine Rapidly Reverses Synaptic and Vascular Deficits of Hippocampus in Genetic Animal Model of Depression. Int J Neuropsychopharmacol. 2017;20(3):247–256. doi:10.1093/ijnp/pyw098
111. Vadivelu N, Schermer E, Kodumudi V, Belani K, Urman R, Kaye A. Role of ketamine for analgesia in adults and children. J Anaesthesiol Clin Pharmacol. 2016;32:298–306. doi:10.4103/0970-9185.168149
112. Nesher N, Ekstein MP, Paz Y, Marouani N, Chazan S, Weinbroum AA. Morphine with adjuvant ketamine vs higher dose of morphine alone for immediate postthoracotomy analgesia. Chest. 2009;136:245–252. doi:10.1378/chest.08-0246
113. Bornemann-Cimenti H, Wejbora M, Michaeli K, Edler A, Sandner-Kiesling A. The effects of minimal-dose versus low-dose S-ketamine on opioid consumption, hyperalgesia, and postoperative delirium: a triple-blinded, randomized, active- and placebo-controlled clinical trial. Minerva Anestesiol. 2016;82:1069–1076.
114. Thiruvenkatarajan V, Wood R, Watts R, et al. The intraoperative use of non-opioid adjuvant analgesic agents: a survey of anaesthetists in Australia and New Zealand. BMC Anesthesiol. 2019;19(2):152–153. doi:10.1186/s12871-019-0857-9
115. Vyklicky V, Korinek M, Smejkalova T, et al. Structure, function, and pharmacology of NMDA receptor channels. Physiol Res. 2014;63(Suppl 1):S191–203. doi:10.33549/physiolres.932678
116. Fan W, Liu Q, Zhu X, et al. Regulatory effects of anesthetics on nitric oxide. Life Sci. 2016;151:76–85. doi:10.1016/j.lfs.2016.02.094
117. Wang CQ, Ye Y, Chen F, et al. Posttraumatic administration of a sub-anesthetic dose of ketamine exerts neuroprotection via attenuating inflammation and autophagy. Neuroscience. 2017;343:30–33. doi:10.1016/j.neuroscience.2016.11.029
118. Masllow GA, Lipinski WJ, Matlen LB, et al. Isoflurane anesthesia does not satisfy the homeostatic need for rapid eye movement sleep. Anesth Analg. 2010;110:1283.
119. Feinberg I, Campbell IG. Ketamine administration during waking increases delta EEG intensity in rat sleep. Neuropsychopharmacology. 1993;9:41–48. doi:10.1038/npp.1993.41
120. Ahnaou A, Huysmans H, Biermans R, Manyakov NV, Drinkenburg WHIM. Ketamine: differential neurophysiological dynamics in functional networks in the rat brain. Transl Psychiatry. 2017;7(9):e1237. doi:10.1038/tp.2017.198
121. Duncan WC, Sarasso S, Ferrarelli F, et al. Concomitant BDNF and sleep slow wave changes indicate ketamine-induced plasticity in major depressive disorder. Int J Neuropsychopharmacol. 2013;16(2):301–311. doi:10.1017/S1461145712000545
122. Faraguna U, Vyazovskiy VV, Nelson AB, Tononi G, Cirelli C. A causal role for brain-derived neurotrophic factor in the homeostatic regulation of sleep. J Neurosci. 2008;28(15):4088–4095. doi:10.1523/JNEUROSCI.5510-07.2008
123. Bunney BG, Li JZ, Walsh DM, et al. Circadian dysregulation of clock genes: clues to rapid treatments in major depressive disorder. Mol Psychiatry. 2015;20:48–55. doi:10.1038/mp.2014.138
124. Paul KN, Fukuhara C, Karom M, et al. AMPA/kinase receptor antagonist DNQX blocks the acute increase of Per2 mRNA levels in most but not all areas of the SCN. Brain Res Mol Brain Res. 2005;139:129–136. doi:10.1016/j.molbrainres.2005.05.017
125. Li N, Lee B, Liu RJ, et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010;329:959–964. doi:10.1126/science.1190287
126. Cao R, Lee B, Cho HY, Saklayen S, Obrietan K. Photic regulation of the mTOR signaling pathway in the suprachiasmatic circadian clock. Mol Cell Neurosci. 2008;38:312–324. doi:10.1016/j.mcn.2008.03.005
127. Cao R, Li A, Cho HY, Lee B, Obrietan K. Mammalian target of rapamycin signaling modulates photic entrainment of the suprachiasmatic circadian clock. J Neurosci. 2010;30:6302–6314. doi:10.1523/JNEUROSCI.5482-09.2010
128. Maiese K, Chong ZZ, Shang YC, Wang S. mTOR: on target for novel therapeutic strategies in the nervous system. Trends Mol Med. 2013;19:51–60. doi:10.1016/j.molmed.2012.11.001
129. Liu RJ, Fuchikami M, Dwyer JM, Lepack AE, Duman RS, Aghajanian GK. GSK-3 inhibition potentiates the synaptogenic and antidepressant-like effects of subthreshold doses of ketamine. Neuropsychopharmacology. 2013;38:2268–2277. doi:10.1038/npp.2013.128
130. Orozco-Solis R, Montellier E, Aguilar-Arnal L, et al. A circadian genomic signature common to ketamine and sleep deprivation in the anterior cingulate cortex. Biol Psychiatry. 2017;82(5):351–360. doi:10.1016/j.biopsych.2017.02.1176
131. Mendez-David I, Guilloux J-P, Papp M, et al. S 47445 Produces Antidepressant- and Anxiolytic-Like Effects through Neurogenesis Dependent and Independent Mechanisms. Front Pharmacol. 2017;8:462. doi:10.3389/fphar.2017.00462
132. Kushikata T, Sawada M, Niwa H, et al. Ketamine and propofol have opposite effects on postanesthetic sleep architecture in rats: relevance to the endogenous sleep-wakefulness substances orexin and melanin-concentrating hormone. J Anesth. 2016;30(3):437–443. doi:10.1007/s00540-016-2161-x
133. Fried EI, Nesse RM. Depression is not a consistent syndrome: an investigation of unique symptom patterns in the STAR*D study. J Affect Disord. 2015;172:96–102. doi:10.1016/j.jad.2014.10.010
134. Wang G, Cheng Y, Gong M, et al. Systematic correlation between spine plasticity and the anxiety/depression-like phenotype induced by corticosterone in mice. NeuroReport. 2013;24:682–687. doi:10.1097/WNR.0b013e32836384db
135. Fraga DB, Camargo A, Olescowicz G, et al. Ketamine, but not fluoxetine, rapidly rescues corticosterone-induced impairments on glucocorticoid receptor and dendritic branching in the hippocampus of mice. Metab Brain Dis. 2021;36(8):2223–2233. doi:10.1007/s11011-021-00743-2
136. Yang C, Shirayama Y, Zhang JC, et al. R-ketamine: a rapid-onset and sustained antidepressant without psychotomimetic side effects. Transl Psychiatry. 2015;5(9):e632. doi:10.1038/tp.2015.136
137. Kessler RC. The global burden of anxiety and mood disorders: putting the European Study of the Epidemiology of Mental Disorders (ESEMeD) findings into perspective. J Clin Psychiatr. 2007;2:10–19.
138. Goldberg DP, Wittchen HU, Zimmerman P, Pfifister H, Beesdo-Baum K. Anxious and non-anxious forms of major depression: familial, personality and symptom characteristics. Psychol Med. 2013;44:1223–1234. doi:10.1017/S0033291713001827
139. Glue P, Medlicott NJ, Harland S, et al. Ketamine’s dose-related effects on anxiety symptoms in patients with treatment refractory anxiety disorders. J Psychopharmacol. 2017;31(10):1302–1305. doi:10.1177/0269881117705089
140. Banov MD, Young JR, Dunn T, Szabo ST. Efficacy and safety of ketamine in the management of anxiety and anxiety spectrum disorders: a review of the literature. CNS Spectr. 2020;25(3):331–342. doi:10.1017/S1092852919001238
141. Fraga DB, Olescowicz G, Moretti M, et al. Anxiolytic effects of ascorbic acid and ketamine in mice. J Psychiatr Res. 2018;100:16–23. doi:10.1016/j.jpsychires.2018.02.006
142. Wu C, Wang Y, He Y, et al. Sub-anesthetic and anesthetic ketamine produce different long-lasting behavioral phenotypes (24 h posttreatment) via inducing different brain-derived neurotrophic factor (BDNF) expression level in the hippocampus. Neurobiol Learn Mem. 2019;167:107136. doi:10.1016/j.nlm.2019.107136
143. Pitsikas N, Boultadakis A. Pre-training administration of anesthetic ketamine differentially affects rats’ spatial and non-spatial recognition memory. Neuropharmacology. 2009;57(1):1–7. doi:10.1016/j.neuropharm.2009.03.015
144. Hayley A, Green M, Downey L, et al. Neurocognitive and behavioural performance of healthy volunteers receiving an increasing analgesic-range infusion of ketamine. Psychopharmacology. 2018;235:1273–1282. doi:10.1007/s00213-018-4842-7
145. Perry EB
146. de Souza I, Meurer Y, Tavares PM, et al. Episodic-like memory impairment induced by sub-anaesthetic doses of ketamine. Behav Brain Res. 2019;359:165–171. doi:10.1016/j.bbr.2018.10.031
147. Morrison RL, Fedgchin M, Singh J, et al. Effect of intranasal esketamine on cognitive functioning in healthy participants: a randomized, double-blind, placebo-controlled study. Psychopharmacology. 2018;235:1107–1119. doi:10.1007/s00213-018-4828-5
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.