")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 12
A New Compound Heterozygous Mutation Of BSCL2 In A Chinese Zhuang Ethnic Family With Congenital Generalized Lipodystrophy
Authors Qin Y, Zhang X, Xiang L, Shan Q, Li S, Yan J, Lin F
Received 1 March 2019
Accepted for publication 14 October 2019
Published 5 December 2019 Volume 2019:12 Pages 2583—2587
DOI https://doi.org/10.2147/DMSO.S207293
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Antonio Brunetti
Yuan-yuan Qin,1 Xuan Zhang,1 Li-qun Xiang,1 Qing-wen Shan,2 Shou-dong Li,3 Jie Yan,1 Fa-quan Lin1
1Department of Clinical Laboratory, The First Affiliated Hospital of Guangxi Medical University, Nanning, Guangxi Zhuang Autonomous Region, People’s Republic of China; 2Department of Pediatric, The First Affiliated Hospital of Guangxi Medical University, Nanning, Guangxi Zhuang Autonomous Region, People’s Republic of China; 3Guangxi Anren Xin Bio Technology Co., Ltd, Nanning, Guangxi Zhuang Autonomous Region, People’s Republic of China
Correspondence: Fa-quan Lin
The First Affiliated Hospital of Guangxi Medical University, Nanning, Guangxi Zhuang Autonomous Region, People’s Republic of China
Tel +86-771-5329287
Email [email protected]
Purpose: This study aims to report the clinical features of an infant with CGL in a Chinese Zhuang ethnic family, whose family members were discovered to carry new pathogenic mutations in the BSCL2.
Patients and methods: In this study, we report clinical and molecular investigations of CGL disease in a family of 4 members (parents and two sons). We used whole exome sequencing (WES) in the family to examine the genetic cause of the disease.
Results: The proband presented with skin pigmentation, hypertriglyceridemia and diabetes. WES identified a previously unreported compound heterozygous mutation in the BSCL2 (c.545_546insCCG heterozygous mutation and exon 3 heterozygous deletion) in the proband. His mother is a heterozygous carrier of the c.545_546insCCG mutation and his father and brother are carriers of the exon 3 heterozygous deletion.
Conclusion: Compound heterozygous mutation of the BSCL2 (new c.545_546insCCG heterozygous mutation and new exon 3 heterozygous deletion) was detected in the proband with characteristic clinical manifestations of CGL2.
Keywords: congenital generalized lipodystrophy, pigmentation, hypertriglyceridemia, diabetes, BSCL2 mutation, whole exome sequencing
Introduction
Congenital generalized lipodystrophy (CGL), also known as Berardinelli-Seip congenital lipodystrophy (BSCL), is an autosomal recessive disorder characterized by a near-total loss of adipose tissue, severe hypertriglyceridemia, insulin resistance, and fatty liver.1–3 CGL was first reported by Berardinelli from Brazil and Seip from Scandinavia in the 1950s.4,5 Patients have since been reported worldwide, including patients of African, European, Middle Eastern, Native American, and Latino descent.6 Currently, the disease is classified into four subtypes (CGL1-4) based on the clinical characteristics and genetic mutations in four genes: AGPAT2 (CGL1),7 BSCL2 (CGL2),2 CAV1 (CGL3),8 and PTRF (CGL4)9 CGL2 have been reported in patients of various ethnicities, including patients from Europe, Mediterranean and Middle Eastern Arabs, and Japanese.10–14 The worldwide prevalence of CGL is estimated to be 1 in 10 million population and 95% of cases are CGL1 or CGL2.15
Here, we report the clinical features of an infant with CGL in a Chinese Zhuang ethnic family. Whole exome sequencing (WES) analysis of the patient and family members discovered new pathogenic mutations in the BSCL2.
Materials And Methods
The study was approved by the Ethics Committee of the First Affiliated Hospital of Guangxi Medical University in People’s Republic of China. We have received written consent from the patient’s parents for the case details to be published. The proband of the study was a 3-month-old boy of Zhuang ethnicity from Nanning, Guangxi Zhuang Autonomous Region, People’s Republic of China. He was hospitalized for skin pigmentation and diarrhea. The mother stated that the skin pigmentation was found at 1 month after birth and the diarrhea occurred 1 month prior to the hospitalization. There was a prominent white spot on the forehead and both sides of the mandible, separately, and then the spots spontaneously receded, but the body skin pigmentation gradually increased, accompanied by feeding difficulties. At the time of admission in the hospital, the proband presented with malnutrition, emaciation,pigmentation of the entire body, abdomen distension, and umbilical protrusion. Laboratory tests were performed and the results are as in the following:1 routine blood analysis: red blood cells (RBC): 3.80×1012/L, hemoglobin (Hb): 129.00g/L, white blood cells (WBC): 12.49×109/L, platelets: 567.00×109/L, absolute neutrophil count: 2.20×109/L.2 Biochemical tests showed no functional abnormalities in the liver and kidney, the fasting blood glucose (FBG) level was 9.06 mmol/L, sodium (Na) was 133.5mmol/L, ferrum (Fe) was 17.9μmol/L, triglyceride (TG) was 26.63mmol/L.3 Immunological examination did not show significant abnormality in five items of thyroid function, but estradiol was 5.00 Pg/mL (20–75), testosterone was 0.85ng/mL (1.75–7.81).4 Electrocardiogram showed sinus tachycardia.5 Abdominal B ultrasound showed a heterogeneous echo mass, suggesting an inguinal hernia, in the right inguinal region and hepatomegaly.6 Cardiac B ultrasound showed small atrial septal defect.7 Intelligence measurement showed mild mental retardation. Discharge after symptomatic treatment pays attention to diet during the period. He was hospitalized again at 6 months. Laboratory tests were performed and the results are as in the following:1 TG was 4.22mmol/L;2 FBG level was 7.4 mmol/L.
Whole Exome Sequencing (WES)
Peripheral blood samples from the proband and his family members were collected in EDTA tubes and then genomic DNA was extracted from the peripheral blood lymphocytes with a DNA extraction kit (TIANGEN, Beijing, People’s Republic of China) according to the manufacturer’s instructions. DNA samples were sent to Guangxi Anrenxin Biotechnology Ltd. (Nanning, People’s Republic of China) for WES analysis. All exons of protein-coding genes as well as some important other genomic regions were enriched and amplified utilizing the SureSelect Human All Exon +UTR V5 kit (Agilent, USA). Enriched DNA libraries were pooled and sequenced on Illumina HiSeq X Ten system. In general, test platform examined >95% of the targeted regions with sensitivity of above 99%. The average coverage of the target bases was >150x. Bioinformatics analysis of the sequencing results was performed using BWA aligner, GATK, and ANNOVAR open access software as well as public databases and standard bioinformatics software such as CADD-Phred, SIFT, and M-CAP. Genome build GRCh37 was used for mapping and variant calling.
Results
Clinical Evaluation
The laboratory test results of the proband and his family are shown in Table 1. The proband’s parents were not consanguineous, no family history of disease and no history of intrauterine infection. The patient appeared lean since 1 month of age and had poor weight in early infancy. He presented with skin pigmentation at 2 months old, especially in the face.
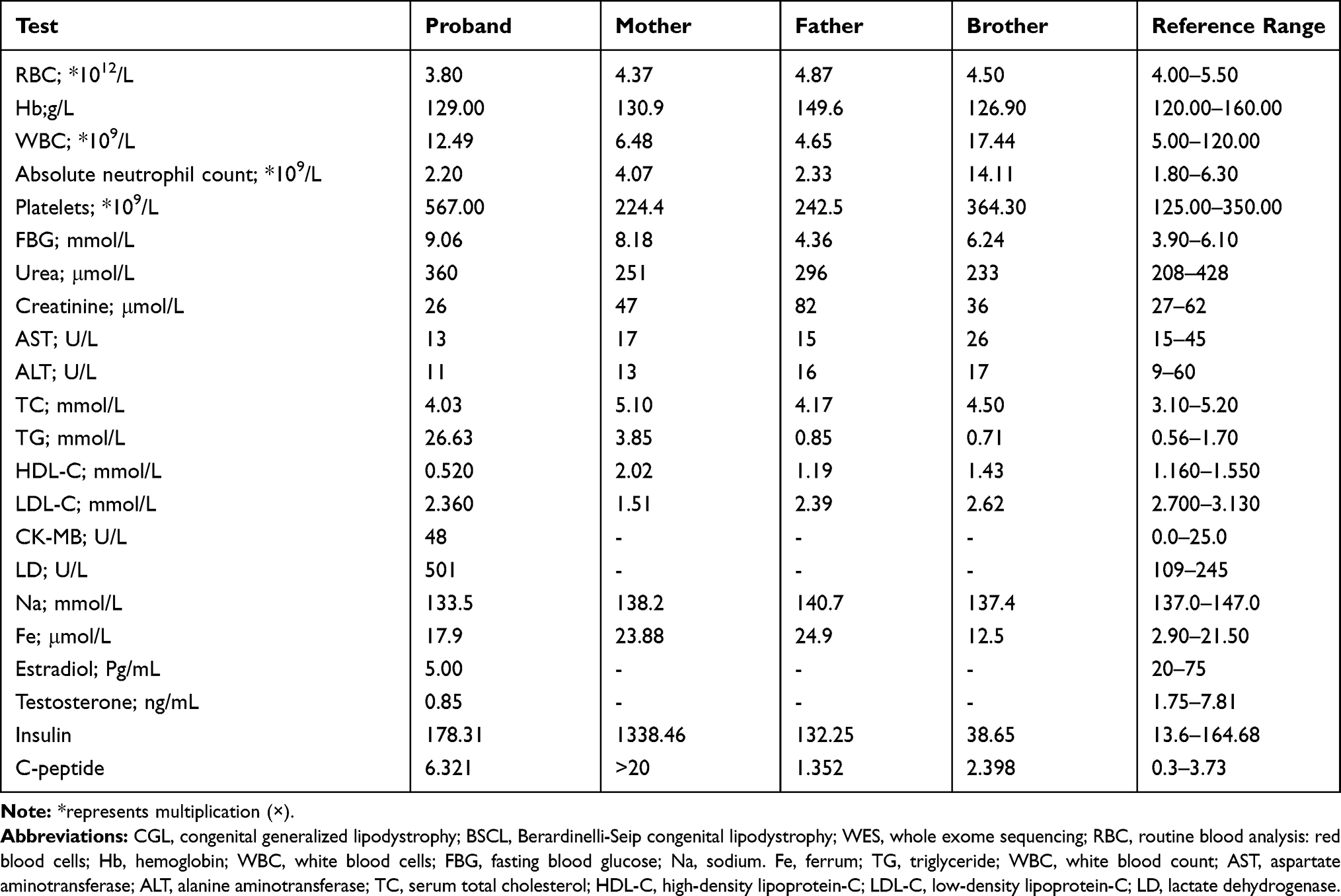 |
Table 1 The Laboratory Test Results Of The Proband And His Family |
Mutational Analysis
WES analysis revealed a new compound heterozygous mutation in the BSCL2 of the proband (c.545_546insCCG heterozygous mutation and exon 3 heterozygous deletion). The c.545_546insCCG mutation was predicted to cause deletion of Glu and insertion of AspArg residues at position 182 of the BSCL2 protein. His mother was a heterozygous carrier of the c.545_546insCCG mutation and his father and brother were carriers of the exon 3 heterozygous deletion, as shown in Figure 1. Both mutations were confirmed absent from the NCBI SNP database (http://www.ncbi.nlm.nih.gov/SNP).
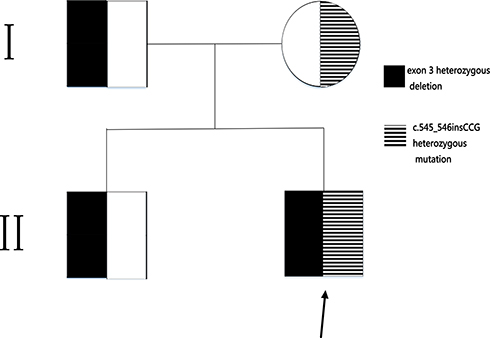 |
Figure 1 Pedigree chart of the congenital generalized lipodystrophy family. Arrowhead indicates the proband. |
Discussion
CGL2 is caused by recessive mutations in Berardinelli-Seip congenital lipodystrophy 2 (BSCL2) gene. The BSCL2, located on chromosome 11q13, encodes a 398 amino acid transmembrane protein, also called seipin.16 Seipin is an important regulator of fat formation and plays an important role in lipid droplet formation and adipocyte differentiation.17,18 Seipin regulates lipid balance by limiting adipogenesis and lipid droplet accumulation in non-adipocytes, while promoting adipogenesis during periods of excess energy storage.17 Seipin deficiency leads to abnormal differentiation and development of adipose tissue.19 Seipin deficiency results in lipodystrophy in human and mouse.19,20
Patients with homozygous or compound heterozygous loss of function mutations in BSCL2 showed the most severe CGL2 type. At present, nearly three-quarters of the reported mutations in BSCL2 of patients with CGL2 are null, remainders are missense mutations.2,21 Individuals with CGL2 are usually recognized at birth or shortly after birth because they have almost no fat at all. Late features include insulin resistance, hypertriglyceridemia, fatty liver, and early onset of diabetes. The prevalence of mild to moderate mental retardation is high. Other features include acanthosis nigricans, hepatomegaly, hyperandrogenism, and skeletal muscle hypertrophy.2 The proband reported here has characteristic phenotypes of CGL 2, including a near-total lack of body fat, hypertriglyceridemia, early onset of diabetes, mild intellectual impairment, acanthosis nigricans, and hepatomegaly. Schuster et al22 identified a Pakistani family with autosomal recessive phenotype, which was characterized by muscle hypertrophy, mild mental retardation, and skeletal abnormalities, using WES to bypass the missing clinical data. WES analysis of the family revealed a mutation in the receptor splicing site of intron 5 in the BSCL2 (c.574-2A > G) and gene expression analysis revealed that the mutation was associated with skipping of exon 6. Gomes et al23 found that 669insA mutation in exon 4 of BSCL2 was considered to be the main genetic change leading to BSCL in 22 patients from Greater Pradesh in northeastern Brazil. In this study, two new mutations in the BSCL2 (c.545_546insCCG and exon 3 heterozygous deletion) were found in a Chinese Zhuang ethnic family with compound heterozygous mutations in the proband. We speculate that this compound heterozygous mutation may significantly reduce seipin protein activity and induce lipodystrophy, hypertriglyceridemia, and early onset of diabetes. The biochemical mechanism of different lipid droplets caused by which BSCL2 mutation remains unclear.
Recent studies have used the BSCL2 knockout mouse models to summarize the phenotype of human CGL2 and to observe changes in adipose tissue metabolism in mice.24 Chen et al25 found that knockdown of BSCL2 mice develops severe lipodystrophy of white adipose tissue, dyslipidemia, insulin resistance, and hepatic steatosis, and underscores the fundamental role of regulated cAMP/PKA-mediated lipolysis in adipose differentiation and identifies BSCL2 as a novel cell-autonomous determinant of activated lipolysis essential for terminal adipocyte differentiation. Seipin knockout mice resulted in adipocyte hypertrophy, lipid droplets increase, lipolysis decrease, adipose tissue inflammation, white and brown adipose tissue gradually decrease, insulin resistance, hepatic steatosis.26 Magre et al2 propose that BSCL2 may maintain a certain role in the formation of lipid droplets (intracellular storage of triglyceride organelles). Using BSCL2 knockout (Ad-mKO) mouse model, Zhou et al27 found that deletion of BSCL2 could reduce obesity, prevent high fat diet-induced obesity, and reduce food intake and down-regulated expression of adipose β3-adrenergic receptor. These results suggest that BSCL2 regulates lipolysis of adipocytes and the production ofβ-adrenergic signals, which have complex effects on adipose tissue and energy balance in the whole body. In addition, Liu et al26 showed that adipose-specific seipin knockout (ASKO) mice had hypertrophic adipocytes, increased lipid droplets, decreased lipolysis, inflammation of adipose tissue, progressive loss of white and brown adipose tissue, insulin resistance, and hepatic steatosis. Wang et al28 found that seipin null mice were unable to respond appropriately to long-term fasting and developed postprandial hypertriglyceridemia. Impaired very low-density lipoprotein (VLDL) secretion and enhanced triglyceride-rich lipoproteins (TRL) clearance were also observed in the seipin null mice.
CGL treatment should focus on improving insulin resistance and metabolic disturbance. The treatment of congenital total body lipid dystrophy includes drug therapy, diet control, prevention of complications, and related rehabilitation therapy. Current drug treatment for CGL includes leptin, thiazolidinediones (TZDs), metformin, and fibroblast growth factor 21 (FGF21). At present, the treatment of CGL in People’s Republic of China is mostly based on insulin hypoglycemic therapy and blood lipid control. Early diagnosis of pathophysiology and early identification of genetic mutations can help us better treat the disease, reduce complications, and improve the quality of life of the patients.29
Conclusion
We present here the identification of two new CGL-associated mutations in BSCL2 using WES analysis. The clinical features of our patients are characteristics of CGL2 phenotype. Our finding extends the evidence for the role of BSCL2 in severe lipodystrophy phenotype. Understanding the molecular defects of BSCL2 in CGL2 patients can be helpful in the genetic counseling and prenatal diagnosis of affected families and may help to improve specific therapeutic interventions.
Acknowledgments
We would like to thank the Department of Clinical Laboratory, The First Affiliated Hospital of Guangxi Medical University, People’s Republic of China.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Agarwal AK, Garg A. Genetic basis of lipodystrophies and management of metabolic complications. Annu Rev Med. 2006;57:297–311. PubMed PMID: 16409151. doi:10.1146/annurev.med.57.022605.114424
2. Magre J, Delepine M, Khallouf E, et al. Identification of the gene altered in Berardinelli-Seip congenital lipodystrophy on chromosome 11q13. Nat Genet. 2001;28(4):365–370. PubMed PMID: 11479539. doi:10.1038/ng585
3. Pagac M, Cooper DE, Qi Y, et al. SEIPIN Regulates Lipid Droplet Expansion and Adipocyte Development by Modulating the Activity of Glycerol-3-phosphate Acyltransferase. Cell Rep. 2016;17(6):1546–1559. PubMed PMID: 27806294. Pubmed Central PMCID: 5647143. doi:10.1016/j.celrep.2016.10.037
4. Berardinelli W. An undiagnosed endocrinometabolic syndrome: report of 2 cases. J Clin Endocrinol Metab. 1954;14(2):193–204. PubMed PMID: 13130666. doi:10.1210/jcem-14-2-193
5. Seip M. Lipodystrophy and gigantism with associated endocrine manifestations. A new diencephalic syndrome? Acta Paediatr. 1959;48:555–574. PubMed PMID: 14444642. doi:10.1111/j.1651-2227.1959.tb17558.x
6. Agarwal AK, Simha V, Oral EA, et al. Phenotypic and genetic heterogeneity in congenital generalized lipodystrophy. J Clin Endocrinol Metab. 2003;88(10):4840–4847. PubMed PMID: 14557463. doi:10.1210/jc.2003-030855
7. Agarwal AK, Arioglu E, De Almeida S, et al. AGPAT2 is mutated in congenital generalized lipodystrophy linked to chromosome 9q34. Nat Genet. 2002;31(1):21–23. PubMed PMID: 11967537. doi:10.1038/ng880
8. Kim CA, Delepine M, Boutet E, et al. Association of a homozygous nonsense caveolin-1 mutation with Berardinelli-Seip congenital lipodystrophy. J Clin Endocrinol Metab. 2008;93(4):1129–1134. PubMed PMID: 18211975. doi:10.1210/jc.2007-1328
9. Hayashi YK, Matsuda C, Ogawa M, et al. Human PTRF mutations cause secondary deficiency of caveolins resulting in muscular dystrophy with generalized lipodystrophy. J Clin Invest. 2009;119(9):2623–2633. PubMed PMID: 19726876. Pubmed Central PMCID: 2735915. doi:10.1172/JCI38660
10. Van Maldergem L, Magre J, Khallouf TE, et al. Genotype-phenotype relationships in Berardinelli-Seip congenital lipodystrophy. J Med Genet. 2002;39(10):722–733. PubMed PMID: 12362029. Pubmed Central PMCID: 1734991. doi:10.1136/jmg.39.10.722
11. Magre J, Delepine M, Van Maldergem L, et al. Prevalence of mutations in AGPAT2 among human lipodystrophies. Diabetes. 2003;52(6):1573–1578. PubMed PMID: 12765973. doi:10.2337/diabetes.52.6.1573
12. Cristancho AG, Lazar MA. Forming functional fat: a growing understanding of adipocyte differentiation. Nat Rev Mol Cell Biol. 2011;12(11):722–734. PubMed PMID: 21952300. doi:10.1038/nrm3198
13. Miranda DM, Wajchenberg BL, Calsolari MR, et al. Novel mutations of the BSCL2 and AGPAT2 genes in 10 families with Berardinelli-Seip congenital generalized lipodystrophy syndrome. Clin Endocrinol (Oxf). 2009;71(4):512–517. PubMed PMID: 19226263. doi:10.1111/j.1365-2265.2009.03532.x
14. Ebihara K, Kusakabe T, Masuzaki H, et al. Gene and phenotype analysis of congenital generalized lipodystrophy in Japanese: a novel homozygous nonsense mutation in seipin gene. J Clin Endocrinol Metab. 2004;89(5):2360–2364. PubMed PMID: 15126564. doi:10.1210/jc.2003-031211
15. Wee K, Yang W, Sugii S, Han W. Towards a mechanistic understanding of lipodystrophy and seipin functions. Biosci Rep. 2014;34(5):583–591. PubMed PMID: 25195639. Pubmed Central PMCID: 4182903. doi:10.1042/BSR20140114
16. Szymanski KM, Binns D, Bartz R, et al. The lipodystrophy protein seipin is found at endoplasmic reticulum lipid droplet junctions and is important for droplet morphology. Proc Natl Acad Sci U S A. 2007;104(52):20890–20895. PubMed PMID: 18093937. Pubmed Central PMCID: 2409237. doi:10.1073/pnas.0704154104
17. Payne VA, Grimsey N, Tuthill A, et al. The human lipodystrophy gene BSCL2/seipin may be essential for normal adipocyte differentiation. Diabetes. 2008;57(8):2055–2060. PubMed PMID: 18458148. Pubmed Central PMCID: 2494687. doi:10.2337/db08-0184
18. Yang W, Thein S, Guo X, et al. Seipin differentially regulates lipogenesis and adipogenesis through a conserved core sequence and an evolutionarily acquired C-terminus. Biochem J. 2013;452(1):37–44. PubMed PMID: 23458123. doi:10.1042/BJ20121870
19. Cui X, Wang Y, Tang Y, et al. Seipin ablation in mice results in severe generalized lipodystrophy. Hum Mol Genet. 2011;20(15):3022–3030. PubMed PMID: 21551454. Epub 2011/05/10. eng. doi:10.1093/hmg/ddr205
20. Cartwright BR, Goodman JM. Seipin: from human disease to molecular mechanism. J Lipid Res. 2012;53(6):1042–1055. PubMed PMID: 22474068. Pubmed Central PMCID: 3351812. doi:10.1194/jlr.R023754
21. Patni N, Garg A. Congenital generalized lipodystrophies–new insights into metabolic dysfunction. Nat Rev Endocrinol. 2015;11(9):522–534. PubMed PMID: 26239609. doi:10.1038/nrendo.2015.123
22. Schuster J, Khan TN, Tariq M, et al. Exome sequencing circumvents missing clinical data and identifies a BSCL2 mutation in congenital lipodystrophy. BMC Med Genet. 2014;24(15):71. PubMed PMID: 24961962. Pubmed Central PMCID: 4076434. doi:10.1186/1471-2350-15-71
23. Gomes KB, Pardini VC, Ferreira AC, Fonseca CG, Fernandes AP. Founder effect of the 669insA mutation in BSCL2 gene causing Berardinelli-Seip congenital lipodystrophy in a cluster from Brazil. Ann Hum Genet. 2007;71(Pt 6):729–734. PubMed PMID: 17535271. doi:10.1111/j.1469-1809.2007.00369.x
24. Dollet L, Magre J, Cariou B, Prieur X. Function of seipin: new insights from Bscl2/seipin knockout mouse models. Biochimie. 2014;96:166–172. PubMed PMID: 23831461. doi:10.1016/j.biochi.2013.06.022
25. Chen W, Chang B, Saha P, et al. Berardinelli-seip congenital lipodystrophy 2/seipin is a cell-autonomous regulator of lipolysis essential for adipocyte differentiation. Mol Cell Biol. 2012;32(6):1099–1111. PubMed PMID: 22269949. Pubmed Central PMCID: 3295006. doi:10.1128/MCB.06465-11
26. Liu L, Jiang Q, Wang X, et al. Adipose-specific knockout of SEIPIN/BSCL2 results in progressive lipodystrophy. Diabetes. 2014;63(7):2320–2331. PubMed PMID: 24622797. doi:10.2337/db13-0729
27. Zhou H, Lei X, Benson T, et al. Berardinelli-Seip congenital lipodystrophy 2 regulates adipocyte lipolysis, browning, and energy balance in adult animals. J Lipid Res. 2015;56(10):1912–1925. PubMed PMID: 26269358. Pubmed Central PMCID: PMC4583079. Epub 2015/08/14.eng. doi:10.1194/jlr.M060244
28. Wang M, Gao M, Liao J, Han Y, Wang Y, Liu G. Dysfunction of lipid metabolism in lipodystrophic Seipin-deficient mice. Biochem Biophys Res Commun. 2015;461(2):206–210. PubMed PMID: 25866184. doi:10.1016/j.bbrc.2015.03.117
29. Lima JG, Nobrega LH, de Lima NN, Do Nascimento Santos MG, Baracho MF, Jeronimo SM. Clinical and laboratory data of a large series of patients with congenital generalized lipodystrophy. Diabetol Metab Syndr. 2016;8:23. PubMed PMID: 26985241. Pubmed Central PMCID: PMC4793761. Epub 2016/03/18.eng. doi:10.1186/s13098-016-0140-x
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.