")
Back to Journals » Cancer Management and Research » Volume 11
A modified method for isolating human quiescent pancreatic stellate cells
Authors Meng FT, Huang M, Fan FF, Shao F, Wang C, Huang Q
Received 26 October 2018
Accepted for publication 17 January 2019
Published 14 February 2019 Volume 2019:11 Pages 1533—1539
DOI https://doi.org/10.2147/CMAR.S192354
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Antonella D'Anneo
Fu-Tao Meng,1,2,* Mei Huang,1,2,* Fang-Fang Fan,3,* Feng Shao,1,2 Chao Wang,1,2 Qiang Huang1,2
1Department of General Surgery, Anhui Provincial Hospital Affiliated to Anhui Medical University, Hefei, Anhui Province, People’s Republic of China; 2Anhui Province Key Laboratory of Hepatopancreatobiliary Surgery, Anhui Provincial Hospital, Hefei, Anhui Province, People’s Republic of China; 3Perelman School of Medicine, University of Pennsylvania, Pennsylvania, PA, USA
*These authors contributed equally to this work
Background: This study explored a simple, high-yield method for isolating quiescent human pancreatic stellate cells (PSCs) to provide sufficient and reliable raw materials for PSC-related studies.
Materials and methods: Single-cell suspensions were prepared from normal human pancreatic tissue specimens using the gentleMACS™ tissue processor, which enhanced the yield and viability of the suspensions. Percoll density gradient centrifugation was then performed to isolate quiescent normal PSCs (NPSCs). Cell viability was determined by trypan blue staining, and the states of the NPSCs were determined by autofluorescence and oil red O staining. The purity of human activated PSCs (APSCs) was determined by immunofluorescence assays.
Results: The yield of NPSCs was ~(2.75±0.65)×106 cells/g. The maximum cell viability was 92%, whereas the maximum cell purity was 95%.
Conclusion: The method employed in this study to isolate PSCs is a simple, high-yield and stable method that is worth popularizing.
Keywords: pancreatic stellate cells, gentleMACS™ Dissociator, Percoll, primary cell culture
Background
Pancreatic ductal adenocarcinoma (PDAC) is a malignancy that ranks fourth among the causes of cancer-related death in Western countries. It is estimated that PDAC will become the second leading cause of cancer-related death by the year 2030.1,2 Both pancreatic cancer and chronic pancreatitis exhibit the pathological features of pancreatic fibrosis. The main cause of extensive hyperplasia of fibrous connective tissue in the pancreatic interstitium is the secretion of large amounts of extracellular matrix components by activated pancreatic stellate cells (APSCs).3–6 In recent years, Apte et al3,5,7 have continuously explored and improved primary pancreatic stellate cell (PSC) culture techniques, and their efforts have significantly enhanced the in vitro culture efficiency of primary PSCs and accelerated the study of the mechanisms of action of PSCs in benign and malignant pancreatic diseases. Primary culture technology is essential for PSCs’ research. However, current studies are mostly limited to mouse-derived PSCs. Many technical difficulties are encountered in obtaining human PSCs, and the efficiency is relatively low. Therefore, efficient and convenient acquisition of high-purity human PSCs is crucial for conducting further successful research.
Traditional methods of culturing and isolating PSCs include tissue explant culture and enzymatic perfusion with digestion-density gradient centrifugation.3,5,7 The former is easy to perform but cannot be used to establish a pure cell culture. The latter involves density gradient centrifugation, which solves the problem of cell purification. However, that method requires specific perfusion equipment and is difficult to perform. Moreover, the experimental results obtained using that method are greatly affected by the extent of enzymatic digestion. Therefore, the above two methods have their advantages and disadvantages but require further improvements. Based on the methods commonly used to isolate PSCs, the present study modified and improved the procedure for isolating and culturing quiescent normal pancreatic stellate cells (NPSCs). In this study, the classical, enzymatic digestion-based isolation method was subjected to technical improvement and process optimization and was combined with the gentle mechanical dissociation function of the gentleMACS™ tissue processor,8,9 which greatly increased the yield and significantly enhanced the viability of NPSCs. The proposed method is more stable and effective than the traditional methods. Application of the modified method significantly enhanced the efficiency of human PSC isolation and provided reliable support for the culture of primary human PSCs.
Materials and methods
Materials
The tissue specimens used in the present study were obtained from patients with pancreatic diseases who were treated in the Department of Biliary-Pancreatic Surgery, Anhui Provincial Hospital, People’s Republic of China. All patients included in the study provided informed consent. The study was approved by the Ethics Committee of the Anhui Provincial Hospital, and this study was conducted in accordance with the Declaration of Helsinki. Normal pancreatic tissues were obtained from surgically treated patients without tumors (such as pancreatic neuroendocrine tumors and duodenal adenomas). A total of 28 patients at Anhui Provincial Hospital between September 2017 and November 2018 were enrolled in this study.
A Tumor Dissociation Kit (human) was purchased from Miltenyi Biotec (Germany). Enzyme H solution was prepared by mixing lyophilized enzyme H powder thoroughly with 3 mL of Roswell Park Memorial Institute (RPMI) 1640 medium or DMEM and was then aliquoted and stored at –20°C. Repeated freeze-thaw cycles were avoided. Enzyme R solution was prepared by mixing lyophilized enzyme R powder thoroughly with 2.7 mL of RPMI 1640 medium or DMEM and then stored at –20°C. Prior to storage, enzyme R solution was aliquoted to avoid repeated freeze-thaw cycles. Enzyme A solution was prepared by mixing lyophilized enzyme A powder with 1 mL of buffer A without vigorous shaking. Enzyme A solution was then aliquoted and stored at –20°C. All enzyme solutions were sterilized by filtration before aliquoting. Percoll solution (density: 1.131 g/mL) was purchased from Beijing Solarbio Science & Technology Co. Ltd. (Beijing, People’s Republic of China). RPMI 1640 and DMEM/F12 media were purchased from HyClone Laboratories Inc. (Logan City, UT, USA). FBS was obtained from Biological Industries (Trasff, Israel). Oil red O (AR) was purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, People’s Republic of China). A Trypan Blue Staining Kit was purchased from Beyotime Biotechnology Co., (Shanghai, People’s Republic of China). The primary antibodies used in immunofluorescence assays included rabbit monoclonal anti-vimentin antibody (Abcam, Cambridge, UK), rabbit monoclonal anti-alpha-smooth muscle actin (α-SMA) antibody (Cell Signaling Technology, Inc., Danvers, MA, USA), rabbit monoclonal anti-glial fibrillary acidic protein (GFAP) antibody (Abcam) and rabbit monoclonal anti-desmin antibody (Abcam). The secondary antibody used in the immunofluorescence assays, goat anti-rabbit IgG, was purchased from Abcam.
Separation steps
Preparation of specimens and single-cell suspension
Surgically resected fresh normal pancreatic tissues were collected using aseptic techniques. The tissue specimens were immersed in serum-containing F12 medium, stored at 4°C on ice and quickly transported to the tissue culture room. The tissue specimens were placed into Petri dishes and rinsed three times with sterile PBS. The adipose tissue, connective tissue and blood vessels were removed from the specimens using ophthalmic scissors and hemostatic forceps, leaving tissue blocks with a diameter of ~1 cm. The tissue blocks were weighed, cut into small pieces ~1–2 mm in size and placed in glass dishes.
Preparation of enzyme digestion solution
The mixed enzyme solution (containing a sufficient amount of enzyme for digestion of 1–2 g of tissues) was prepared by thoroughly mixing 5 mL of RPMI 1640 medium with 200 µL of enzyme H, 100 µL of enzyme R and 25 µL of enzyme A working solutions.
Preparation of single-cell suspension
After processing, the tissues were mechanically dissociated using the gentleMACS™ tissue processor and digested with the mixed enzymes at 37°C for 40 minutes with constant shaking. Single-cell suspensions were then prepared according to a predetermined procedure. Subsequently, the cells were washed with F12 medium, filtered through SmartStrainers (70 µm) and collected in 50 mL centrifuge tubes. The SmartStrainers (70 µm) were washed with 20 mL of F12 medium. The resulting cell suspensions were centrifuged at 300× g for 7 minutes. After removal of the supernatants, the cells were resuspended (final volume, 4 mL) and subjected to density gradient centrifugation.
Percoll density gradient centrifugation
Two 15-mL centrifuge tubes were filled consecutively with 3 mL of 30% Percoll, 4 mL of 10% Percoll and 2 mL of cell suspension using pipettes. Subsequently, the tubes were centrifuged at 900× g for 20 minutes.
Primary culture
After centrifugation, the cell layer between the 10% and 30% Percoll gradients was carefully collected and transferred to another centrifuge tube (Figure 1A). The cells were then mixed thoroughly with three volumes of 1× PBS and centrifuged at 1,500 rpm for 5 minutes. After removal of the supernatant, the cell pellet was resuspended in culture medium and stained with trypan blue, and cells were counted (Figure 1B). Subsequently, the cells were cultured in F12 medium containing 10% FBS in a humidified 37°C, 5% CO2 incubator (Figure 1C). After 12 hours of cultivation, NPSCs gradually attached to the wall (Figure 1D). Because the NPSCs did not attach to the wall firmly, the culture medium was aspirated with caution and replaced with fresh medium once. Afterward, culture medium was changed every 24 hours. All steps described above were completed under aseptic conditions.
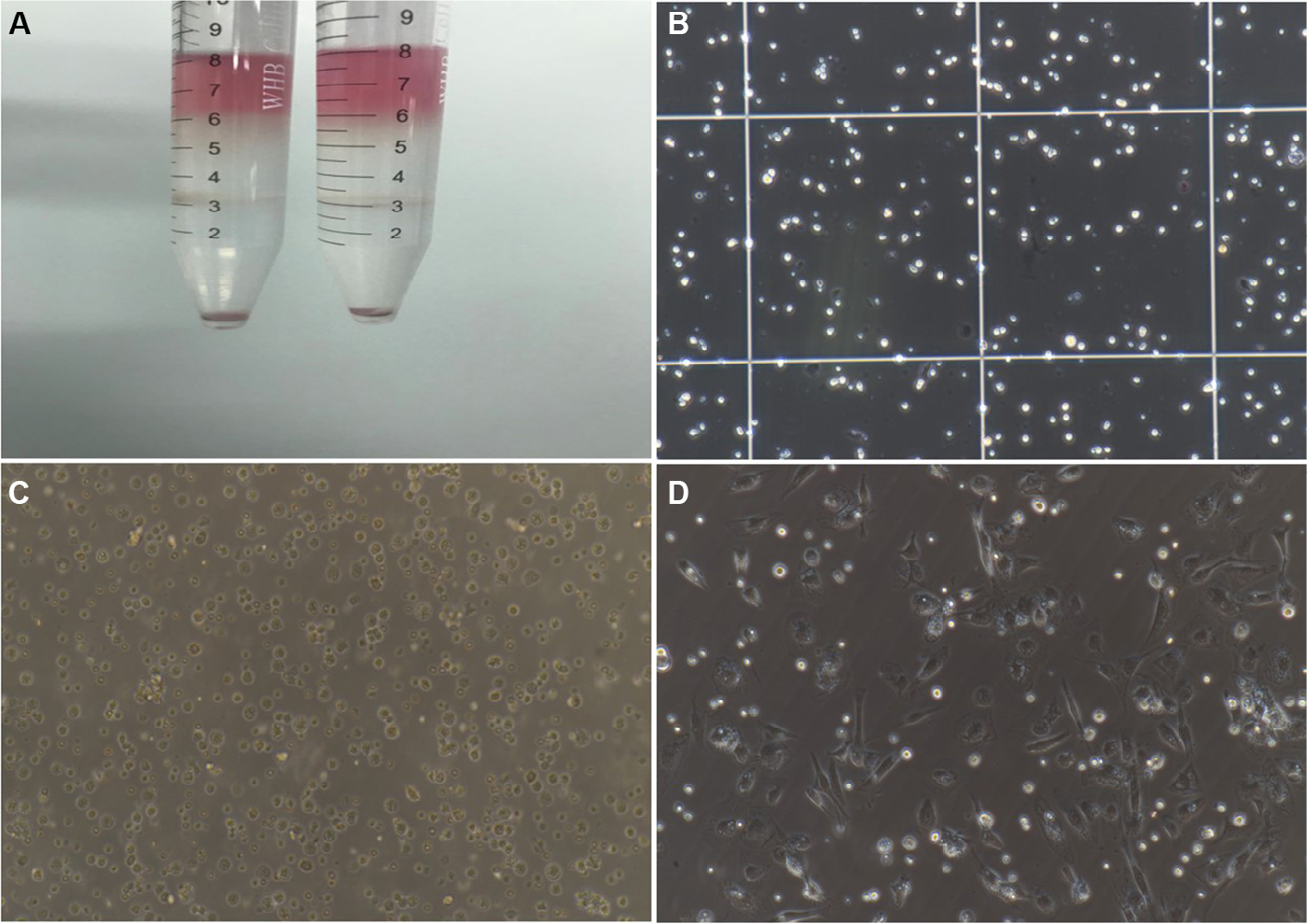 | Figure 1 Isolation and culture of NPSCs. Note: (A) Percoll density gradient centrifugation layering; (B) cell count after isolation (200×); (C) the cell morphology of freshly isolated NPSCs (200×); (D) cell morphology of NPSCs gradually adhering after 12 hours of culture (200×). Abbreviation: NPSC, normal pancreatic stellate cell. |
Oil red O staining
Two milliliters of PBS (prewarmed at 37°C) was added into six-well plates inoculated with primary PSCs using a Papillon dropper. The plates were then gently shaken in a crosswise manner. After the adherent cells were gently rinsed twice with PBS, residual buffer was completely removed using filter paper. The cells were then fixed at room temperature (RT) in 1 mL of formaldehyde solution (100 g/L) for 15 minutes. After removal of the formaldehyde solution, the fixed cells were rinsed twice with PBS. Subsequently, 3 mL of freshly prepared oil red O working solution was added slowly along the wall of the wells. The plate was covered with a lid and sealed with Parafilm. The cells were then dynamically examined under an inverted biological microscope, which showed gradual deepening of the color of the fat droplets. Once the fat droplets appeared as strings of bright red beads of varying sizes, the staining reaction was terminated by completely aspirating the oil red O solution. Subsequently, the cells were carefully rinsed two to three times with PBS and incubated with the nuclear counterstain hematoxylin for 30 seconds. The cells were washed again with PBS and subjected to differentiation/decolorization. The cells were then imaged under a microscope.
Immunofluorescence assay
Activated PSCs were seeded into six-well plates containing coverslips and cultured for 48 hours under standard conditions (37°C and 5% CO2). The coverslips were removed from the plates and washed twice with PBS. The cells grown on the coverslips were fixed with 4% paraformaldehyde for 15 minutes, washed with PBS and incubated with blocking buffer for 60 minutes. The blocking solution was then discarded. The cells were incubated with the corresponding primary antibodies (rabbit anti-vimentin antibody, 1:100 dilution; rabbit anti-α-SMA antibody, 1:100 dilution; rabbit anti-GFAP antibody, 1:100 dilution; and rabbit anti-desmin antibody, 1:100 dilution) at 4°C overnight. After washing with PBS, the cells were incubated with fluorescent dye-conjugated secondary antibodies for 1–2 hours at RT in the dark. The cells were washed again with PBS and then incubated with the nuclear counterstain DAPI. After the cells were labeled, the coverslips were mounted onto microscope slides. Finally, the cells were examined and imaged using a fluorescence microscope, and the purity of the PCSs was analyzed.
Cell purity (%) = the number of positive cells/total number of DAPI-stained cells × 100%
Western blot
Total proteins were extracted from the 3-day NPSC and 10-day APSC cultures, and the protein concentration was measured using a BCA assay kit (Sigma-Aldrich Co., St Louis, MO, USA) according to the manufacturer’s instructions. Thirty micrograms of total protein was separated by SDS/PAGE and transferred electrophoretically to polyvinylidene fluoride membranes (Thermo Fisher Scientific, Waltham, MA, USA). The membranes were incubated with specific primary antibodies diluted with TBS (0.1% Tween 20) (α-SMA: 1:500 dilution, vimentin: 1:500 dilution, GFAP: 1:500 dilution, desmin: 1:500 dilution, β-actin: 1:1,000 dilution). A Vectastain ABC kit (Vector Laboratories, Burlingame, CA, USA) was used for 3,3′-diamino benzidine chromogenic detection (Sigma-Aldrich Co.) according to the manufacturer’s instructions.
Ethics statement
Ethical approval was obtained from the ethics committee of the Anhui Provincial Hospital Affiliated to Anhui Medical University, and written informed consent was obtained from the patients whose tissue and medical data were used in this study.
Results
Isolation and autofluorescence-based identification of NPSCs
Approximately (2.75±0.65)×106 NPSCs were obtained from 1 g of normal pancreatic tissue. Trypan blue staining showed that the mean cell viability was ~90%. The obtained cells attached to the wall at 8–24 hours after seeding. Moreover, the cells displayed typical characteristics of quiescent cells, including small volume, slow proliferation, round or polygonal shape and richness in lipid droplets (Figure 2A). Fluorescence microscopy revealed transient spontaneous blue–green fluorescence in NPSCs at an excitation wavelength of 328 nm (Figure 2B). The cells were gradually activated after several passages and exhibited the phenotypic characteristics of activation-state cells, such as pseudopod extension, disappearance of intracellular lipid droplets and an increase in cell volume.
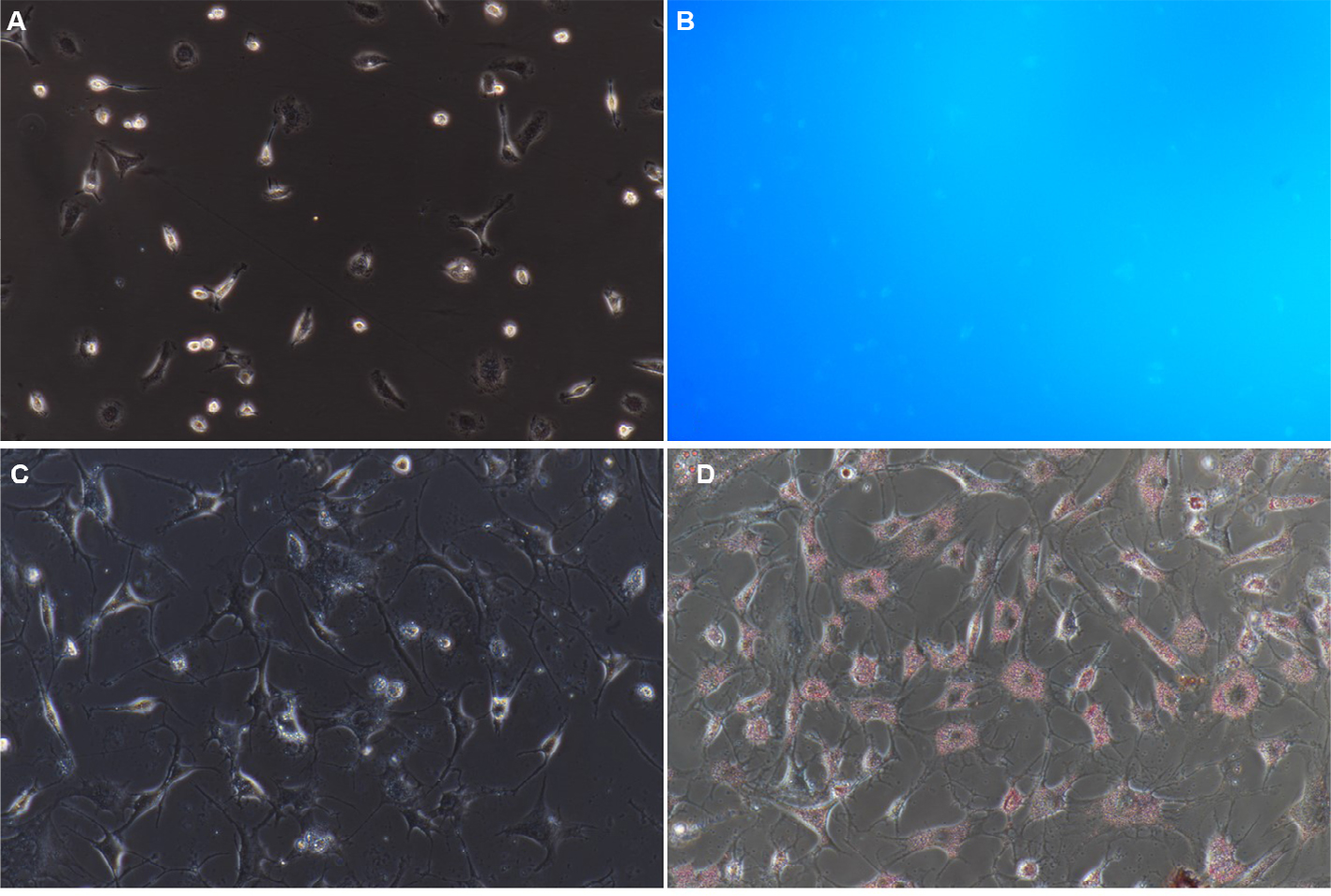 | Figure 2 Identification of NPSCs by blue–green autofluorescence at 328 nm and oil red O staining. Notes: (A) NPSC autofluorescence bright field control (200×); (B) NPSC blue–green autofluorescence at 328 nm (200×); (C) NPSC oil red O staining bright field control (200×); (D) positive oil red staining of NPSCs (200×). Abbreviation: NPSC, normal pancreatic stellate cell. |
Identification of NPSCs by oil red O staining
In NPSCs, the fat droplets were stained bright red by oil red O (Figure 2C, D). The bead-like “oil droplets” of varying sizes were distributed throughout the cytoplasm. These droplets connected onto ring-like structures around the nucleus.
Immunofluorescence-based phenotypic analysis
NPSCs were activated after 1 week of culture (Figure 3A). Immunofluorescence staining demonstrated that NPSCs expressed the activation state-specific markers vimentin, α-SMA, GFAP and Desmin (Figure 3B1–B3, C1–C3, D1–D3, E1–E3).
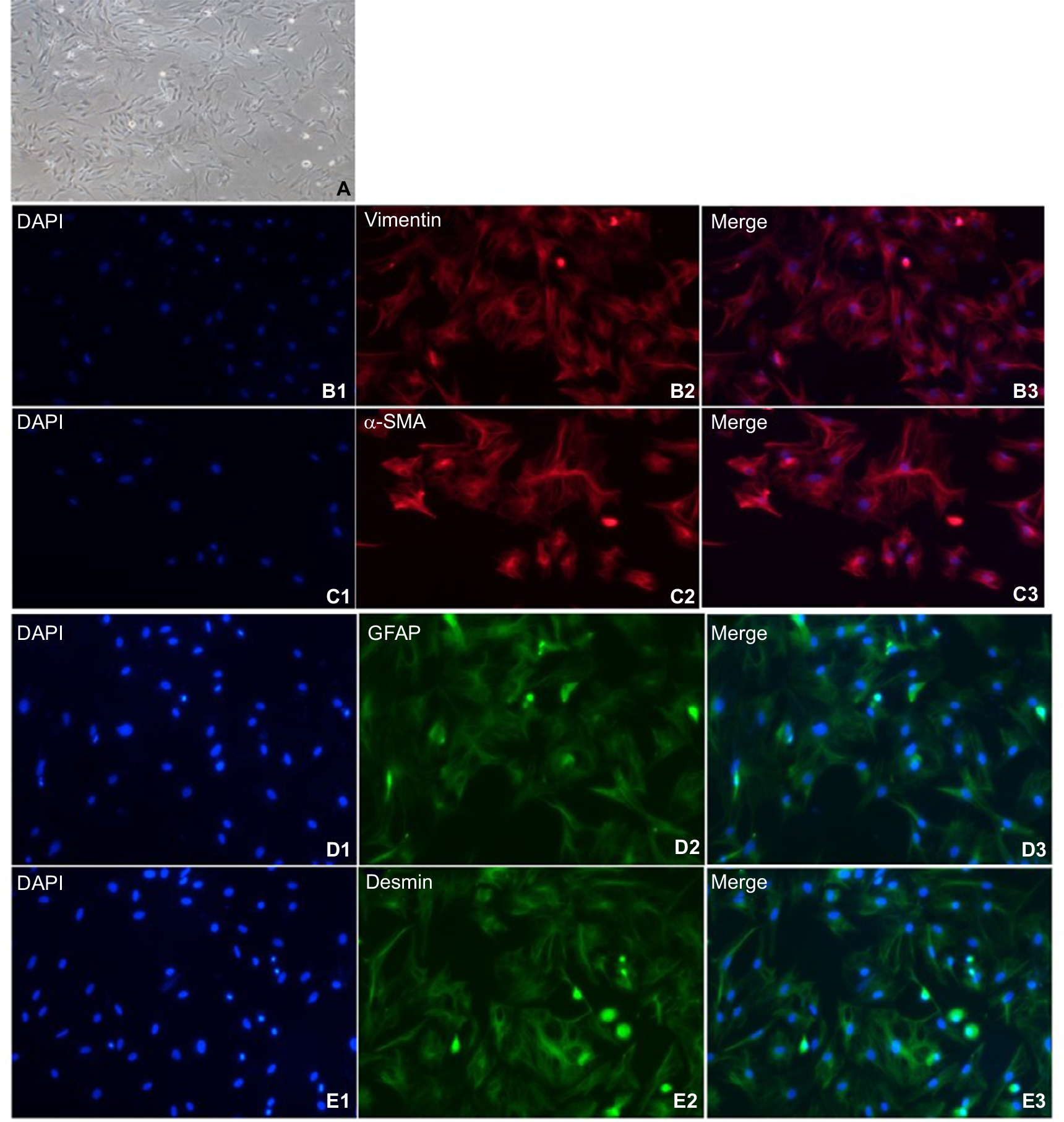 | Figure 3 Expression of cellular markers in primary isolated APSCs. Notes: (A) After about 1 week of culture, NPSCs gradually activate. APSCs exhibit typical activation characteristics, with large cell volume, no lipid droplets in the cells, and a multi-angled shape. (B1–B3) APSCs’ positive expression marker protein vimentin. Blue is the nuclear marker DAPI; red is the vimentin-positive expression localization (200×); (C1–C3) APSCs’ positive expression marker protein α-SMA. Blue is the nuclear marker DAPI; red is the α-SMA-positive expression localization (200×); (D1–D3) APSCs’ positive expression marker protein GFAP. Blue is the nuclear marker DAPI; green is the GFAP-positive expression localization (200×); (E1–E3) APSCs’ positive expression marker protein desmin. Blue is the nuclear marker DAPI; green is the desmin-positive expression localization (200×). Abbreviations: APSC, activated pancreatic stellate cell; GFAP, glial fibrillary acidic protein; NPSC, normal pancreatic stellate cell; α-SMA, alpha-smooth muscle actin. |
Western blot measurement expression of α-SMA, vimentin, GFAP and Desmin in NPSCs and APSCs
As shown in Figure 4, the expression of vimentin, α-SMA, GFAP and Desmin were significantly increased in APSCs cultured for 10 days compared to freshly isolated NPSCs for 3 cultured days.
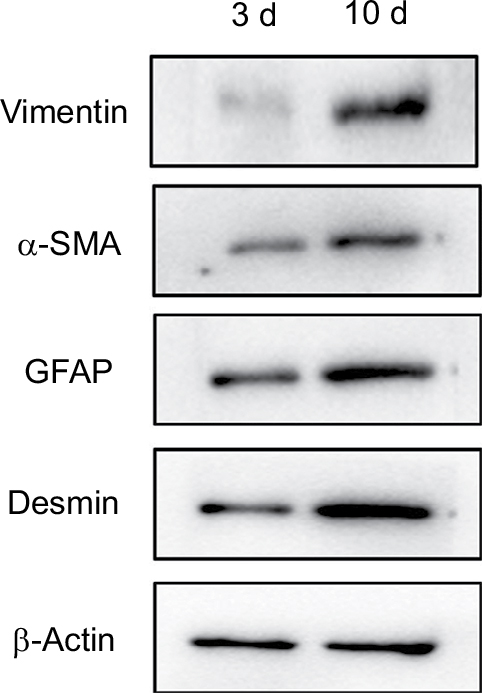 | Figure 4 PSC activation-related indicator expression by Western blot detection. Abbreviations: APSC, activated pancreatic stellate cell; NPSC; normal pancreatic stellate cell; PSC, pancreatic stellate cell; 3 d, freshly isolated NPSCs cultured for 3 days; 10 d, APSCs cultured for 10 days. |
Discussion
The role of hepatic stellate cells (HSCs) in the liver fibrosis process has been widely recognized.10,11 Currently, the HSC mechanisms of action are being studied in depth at the molecular level. Activated PSCs are similar to HSCs and may play an initiating role in the process of pancreatic fibrosis. Therefore, PSCs may serve as a main target in pancreatic fibrosis.3,5,12,13 The key role of PSCs in pancreatic fibrosis has been constantly demonstrated and confirmed in recent years.6,12–15 Effective isolation and culture of PSCs allows in vitro and in vivo studies of PSCs and provides a technical guarantee for further exploration of the pathogenic role of PSCs in chronic pancreatitis and pancreatic cancer-associated fibrosis. At present, immortalized PSCs have been established by only a few laboratories.16,17 PSCs are nontumoros cells. Currently, it remains controversial18 whether immortalized PSCs will fully differentiate into fibroblasts as the passage number increases. Therefore, primary cell culture is still the most important means of obtaining PSCs.
The purification and yield of PSCs are the main challenges encountered during primary PSC isolation and culture. At present, only a few laboratories have established immortalized PSC lines. Moreover, significant heterogeneity exists between the PSCs isolated from different patients. Therefore, studies of PSCs are conducted mainly on the basis of primary PSC isolation and cultivation. In the present study, innovations and improvements were made based on the existing research. Specifically, the Tumor Dissociation Kit was used, digestive enzyme solution of an appropriate concentration was prepared, and normal pancreatic tissues were subjected to gentle and standardized mechanical disruption using the gentleMACS™ Tissue Dissociator. As a result, massive cell lysis caused by excessive mechanical disruption was avoided, and a single-cell suspension was obtained quickly and efficiently. Because of the digestive activity of pancreatic enzymes in excised normal pancreatic tissue specimens, the specimens were kept in culture medium containing 20% FBS to neutralize the enzymes. In addition, the appropriate concentration of the digestive enzyme solution was determined in a preliminary experiment, which allowed replacement of the traditional enzyme perfusion method with the gentleMACS-based mechanical disruption method. Because normal human pancreatic tissue specimens often account for a small portion of the surgical margin and have been removed from living body, it is difficult to identify the pancreatic ducts and corresponding large blood vessels for digestive enzyme perfusion. Therefore, the improved human NPSC isolation method is simpler and easier to perform. After the single-cell suspension was obtained, density gradient centrifugation was performed. At present, the commonly used density gradient media include Nycodenz, Percoll, Ficoll and Dextran, among which Ficoll19 and Dextran20 gradients are often used to isolate islet cells. Percoll medium was used in this study. Compared to Nycodenz,3,7 Percoll medium possesses several advantages such as low cytotoxicity and simple creation of a density gradient. Attached cells can be readily eluted if Percoll medium is used. Moreover, Percoll medium is rather inexpensive. Water-based Percoll stock solutions are commercially available and are convenient to use. After the above improvements were made, the yield of PSCs and success rate of PSC isolation were enhanced significantly.
Conclusion
The existing PSC culture methods have their limitations.3,5,7 In contrast, the enzyme digestion-gentleMACS™ Dissociator-density gradient centrifugation method developed in the present study displays a number of advantages over the traditional enzyme perfusion-density gradient centrifugation method. The method developed in the present study is easy to implement, exhibits good repeatability and allows high-purity isolation of PSCs. Therefore, the method is worth popularizing.
Acknowledgment
This study was supported by the National Natural Science Foundation of China (No. 81501354), the Natural Science
Foundation of Anhui Province (No. 1608085QH197), and
the Programs for Science and Technology Development of
Anhui Province in 2016 (No. 1606c08234).
Disclosure
The authors report no conflicts of interest in this work.
References
Rahib L, Smith BD, Aizenberg R, et al. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74(11):2913–2921. | ||
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30. | ||
Apte MV, Haber PS, Applegate TL, et al. Periacinar stellate shaped cells in rat pancreas: identification, isolation, and culture. Gut. 1998;43(1):128–133. | ||
Apte MV, Xu Z, Pothula S, et al. Pancreatic cancer: the microenvironment needs attention too! Pancreatology. 2015;15(4 Suppl):S32–S38. | ||
Bachem MG, Schneider E, Groß H, et al. Identification, culture, and characterization of pancreatic stellate cells in rats and humans. Gastroenterology. 1998;115(2):421–432. | ||
Wilson JS, Pirola RC, Apte MV. STARS and stripes in pancreatic cancer: role of stellate cells and stroma in cancer progression. Front Physiol. 2014;5:52. | ||
Vonlaufena A, Phillipsa PA, Yanga L, et al. Isolation of quiescent human pancreatic stellate cells: a promising in vitro tool for studies of human pancreatic stellate cell biology. Pancreatology. 2010;10(4):434–443. | ||
Messier EM, Mason RJ, Kosmider B. Efficient and rapid isolation and purification of mouse alveolar type II epithelial cells. Exp Lung Res. 2012;38(7):363–373. | ||
Pennartz S, Reiss S, Biloune R, Hasselmann D, Bosio A. Generation of single-cell suspensions from mouse neural tissue. JoVE. 2009;29(29):1267. | ||
Friedman SL. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem. 2000;275(4):2247–2250. | ||
Cui W, Wang M, Maegawa H, Teranishi Y, Kawada N. Inhibition of the activation of hepatic stellate cells by arundic acid via the induction of cytoglobin. Biochem Biophys Res Commun. 2012;425(3):642–648. | ||
Cabrera MC, Tilahun E, Nakles R, et al. Human pancreatic cancer-associated stellate cells remain activated after in vivo chemoradiation. Front Oncol. 2014;4:102. | ||
McCarroll JA, Naim S, Sharbeen G, et al. Role of pancreatic stellate cells in chemoresistance in pancreatic cancer. Front Physiol. 2014;5(270):141. | ||
Tang D, Yuan Z, Xue X, et al. High expression of galectin-1 in pancreatic stellate cells plays a role in the development and maintenance of an immunosuppressive microenvironment in pancreatic cancer. Int J Cancer. 2012;130(10):2337–2348. | ||
Tang D, Zhang J, Yuan Z, et al. Pancreatic satellite cells derived galectin-1 increase the progression and less survival of pancreatic ductal adenocarcinoma. PLoS One. 2014;9(3):e90476. | ||
Ohuchida K, Mizumoto K, Murakami M, et al. Radiation to stromal fibroblasts increases invasiveness of pancreatic cancer cells through tumor-stromal interactions. Cancer Res. 2004;64(9):3215–3222. | ||
Endo S, Nakata K, Ohuchida K, et al. Autophagy is required for activation of pancreatic stellate cells, associated with pancreatic cancer progression and promotes growth of pancreatic tumors in mice. Gastroenterology. 2017;152(6):1424:1492–1506.e24. | ||
Erkan M, Adler G, Apte MV, et al. StellaTUM: current consensus and discussion on pancreatic stellate cell research. Gut. 2012;61(2):172–178. | ||
Zongyi Y, Funian Z, Hao L, et al. A rapid, efficient, and economic device and method for the isolation and purification of mouse islet cells. PLoS One. 2017;12(2):e0171618. | ||
Andersen PL, Vermette P. Biomimetic surfaces supporting dissociated pancreatic islet cultures. Colloids Surf B Biointerfaces. 2017;159:166–173. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.