")
Back to Journals » OncoTargets and Therapy » Volume 9
A meta-analysis of prognostic value of KIT mutation status in gastrointestinal stromal tumors
Authors Jiang Z, Zhang J, Li Z, Liu Y, Wang D, Han G
Received 4 December 2015
Accepted for publication 10 March 2016
Published 3 June 2016 Volume 2016:9 Pages 3387—3398
DOI https://doi.org/10.2147/OTT.S101858
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr William C. Cho
Zhiqiang Jiang, Jian Zhang, Zhi Li, Yingjun Liu, Daohai Wang, Guangsen Han
Department of General Surgery, Affiliated Tumor Hospital, Zhengzhou University, Zhengzhou, People’s Republic of China
Abstract: Numerous types of KIT mutations have been reported in gastrointestinal stromal tumors (GISTs); however, controversy still exists regarding their clinicopathological significance. In this study, we reviewed the publicly available literature to assess the data by a meta-analysis to characterize KIT mutations and different types of KIT mutations in prognostic prediction in patients with GISTs. Twenty-eight studies that included 4,449 patients were identified and analyzed. We found that KIT mutation status was closely correlated with size of tumors and different mitosis indexes, but not with tumor location. KIT mutation was also observed to be significantly correlated with tumor recurrence, metastasis, as well as the overall survival of patients. Interestingly, there was higher risk of progression in KIT exon 9-mutated patients than in exon 11-mutated patients. Five-year relapse-free survival (RFS) rate was significantly higher in KIT exon 11-deleted patients than in those with other types of KIT exon 11 mutations. In addition, RFS for 5 years was significantly worse in patients bearing KIT codon 557–558 deletions than in those bearing other KIT exon 11 deletions. Our results strongly support the hypothesis that KIT mutation status is another evaluable factor for prognosis prediction in GISTs.
Keywords: KIT, meta-analysis, prognosis, marker, therapy
Introduction
Gastrointestinal (GI) stromal tumors (GISTs), the most common mesenchymal neoplasms of the GI tract, are believed to originate from the interstitial cells of Cajal regulating GI motility. GISTs can be found anywhere within the GI tract; however, the stomach accounts for at least half of them and is the most common location.1 Up to 50% of patients developed tumor recurrence after initial resection for primary and localized GISTs, and median survival after recurrence was <2 years. The kinase mutational status has been accepted as the main pathogenic event, has been presented as the peculiar molecular hallmark of GISTs, and denoted as the best predictive biomarker of tumor response to tyrosine kinase inhibitor (TKI).1–4 The detection and analysis of somatic mutations from GIST tissue are the keys to understanding the genetic basis of tumor initiation, progression, therapy response, toxicity, and patient prognosis.
The KIT gene, the cellular homologue of the oncogene v-KIT, encodes a type III receptor tyrosine kinase, c-kit.5,6 KIT, a 145 kDa glycoprotein receptor of stem cell factor (SCF), is a member of the type III receptor tyrosine kinase family that contains the macrophage colony stimulating factor receptor, the Fl cytokine receptor, as well as the platelet-derived growth factor receptors-α and -β (PDGFRA and PDGFRB).7 Once interactions between c-kit and SCF occur, they lead to the activation of specific intracellular signaling pathways, such as PI3K, JAK/STAT, and Shc/Ras/MAPK cascades.8,9 Activation of the receptor tyrosine kinase c-kit is involved in numerous diseases, including mastocytosis,10 melanoma,11 multiple myeloma,12 and GISTs.13,14 The extracellular juxtamembrane domain of KIT is important for regulating receptor activation, and the differential activity of KIT splice forms is controlled by extracellular peptide insert length.15 The extended A-loop region also has a role in autoactivation of mutant KIT.16 A number of factors, such as interleukin-317 and the tyrosine kinase CSK,18 are able to regulate KIT and its downstream signaling.
The development of human lung cells, germ cells, erythrocytes, melanocytes, mast cells, and interstitial cells of Cajal occur through Kit–SCF interaction, while dysregulation of the complex KIT signaling network is known to be correlated with malignant transformation, tumor progression, such as lung cancer, gastric cancer, leukemias, mastocytosis, as well as GISTs.19–22 A number of studies have reported that c-kit dysregulation leads to tumor proliferation, development, heterogeneity, angiogenesis, survival, and resistance to anticancer therapy.22–27 The activation mechanism of the most commonly occurring mutation, D816V in exon 17 of KIT, has been well studied, while other mutations remain fairly uncharacterized in this respect. Recently, a lipid kinase-independent key role of PI3 kinase in KIT/V560D-mediated oncogenic signal transduction has been reported.28 Gain-of-function mutations of KIT or PDGFRA have been found in ~80%–85% of cases.3,29,30 Numerous types of KIT mutations, including point mutation, insertion, deletion, and duplication, involved in exons 9, 11, 13, and 17 have been reported in GISTs;31 however, controversy still exists regarding their prognostic value.32 After performing primary surgery and controlling unresectable tumors, treatment with TKIs is effective in reducing GIST recurrence.33 Thus, it is essential to assess the KIT mutation status to predict the mutation’s response to TKIs and prognosis. In this study, we review the publicly available literature to summarize the data by a meta-analysis of KIT mutations and analyze the clinicopathological significance and prognostic values of different types of KIT mutations in GISTs.
Methods
Search strategy
We searched PubMed, MEDLINE, and Web of Science from the earliest date up to May 2015 using the following search terms: “gastrointestinal stromal tumor” or “GIST”, “KIT”, and “c-KIT”. In this study, we did not include PDGFRA-mutant GIST patients. We also screened manually the reference lists of retrieved articles for additional articles. We screened the publications by titles first, then by the abstracts. After exclusion of duplicates and nonrelevant publications from the different databases, we then evaluated the full text version for inclusion and exclusion criteria. All clinical studies except case reports were chosen. All searched data were retrieved and evaluated. The references of selected studies and authors’ bibliographies were also searched for additional relevant studies.
Selection criteria
In this meta-analysis, we collected all eligible studies evaluating the relationship between KIT mutation and the clinicopathological significance of GISTs. We used the following inclusion criteria: 1) study design included KIT mutation status and the clinicopathological significance of GISTs; 2) studies that evaluated the correlation between KIT mutation status and prognosis in patients with GISTs. The following exclusion criteria were considered: 1) articles that showed insufficient data to calculate the odds ratio (OR); 2) case reports, letters, reviews, expert opinions, editorials, and conference abstracts; and 3) all articles involving cell lines, human xenografts, and in vitro/ex vivo studies.
Data extraction
The eligible studies were extracted by two investigators independently. Disagreements were resolved by discussions and consensus. We determined whether KIT mutations were detected in the primary tumor before treatment with imatinib and whether the report had sufficient available data (usually >15 cases). We recorded the following information for each study: year of publication, first author name, number of cases, sample source, KIT mutation status, and other clinicopathological parameters. Data for study characteristics and clinical information were summarized and converted into table format.
Statistics analysis
We used Review Manager 5.2 (Cochrane Collaboration, Oxford, UK) and the Stata 12.0 (Stata Corporation, College Station, TX, USA) for this analysis. Comparisons of dichotomous measures were determined by pooled estimates of ORs and their 95% confidence intervals (CIs). We used a random-effects model to pool the ORs when there was heterogeneity among studies; otherwise, a fixed-effect model was selected. The total variation among studies was estimated by I-square, with significance being set at I2>50%. Heterogeneity was determined by a chi-square test, with significance being set at P<0.10. P-value of <0.05 was considered to be statistically significant. A sensitivity analysis, in which one study was removed at a time, was conducted to assess the result stability. We used funnel plots for detection of publication bias.
Results
Identification of relevant studies
Six hundred and ninety-three publications were identified by the search method described. Six hundred and sixty-five of these were excluded because they were nonoriginal articles (reviews), laboratory studies, or studies irrelevant to the current analysis. There were 28 studies identified in the final meta-analysis (Figure 1).
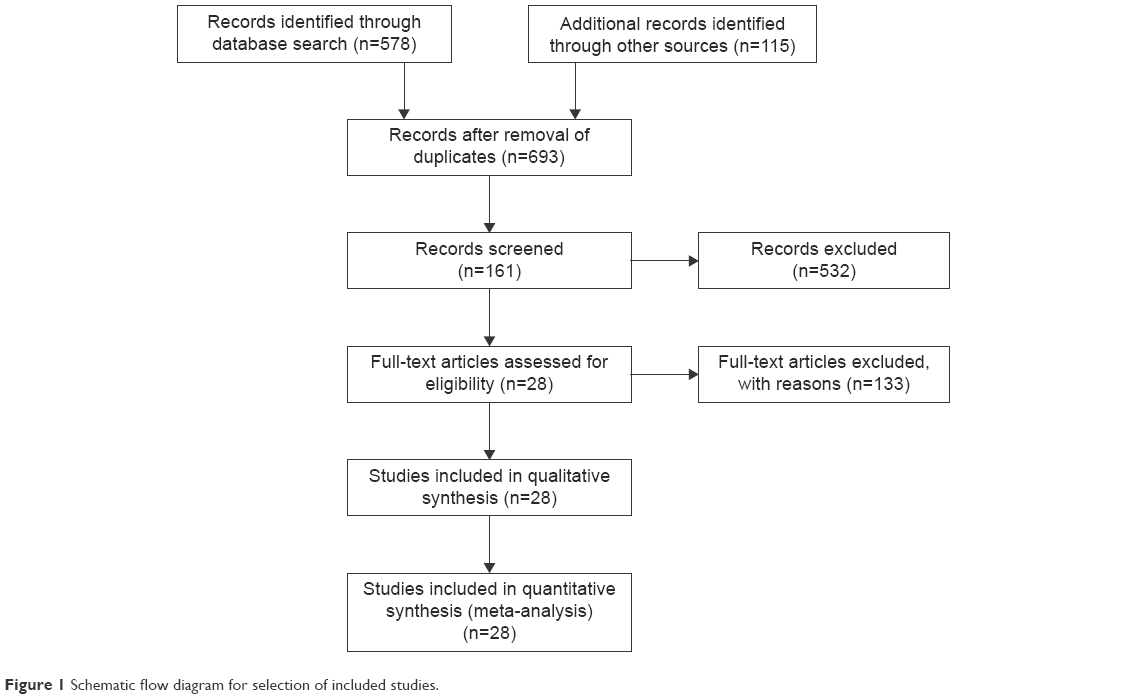 | Figure 1 Schematic flow diagram for selection of included studies. |
Study characteristics
Twenty-eight studies published from 1999 to 2014 were eligible for the analysis. A total of 4,449 GIST patients from the People’s Republic of China, Korea, Taiwan, Japan, Italy, Germany, Norway, Belgium, Spain, Greece, Sweden, and the USA were enrolled. As described earlier, the database search generated 693 articles from MEDLINE, PubMed, the Web of Science, Scopus, and Embase. The other 665 publications were excluded due to lack of full text or because they were in vitro/ex vivo studies, used cell lines and human xenografts, or were irrelevant studies. The following items were collected from each study: year of publication, first author’s name, countries, number of patients, tumor location, tumor size, the number of mitoses per 50 high-power fields (HPFs) in the GIST tumor section, KIT mutation status, treatment, and the time of follow-up. Their basic characteristics are summarized in Table 1.
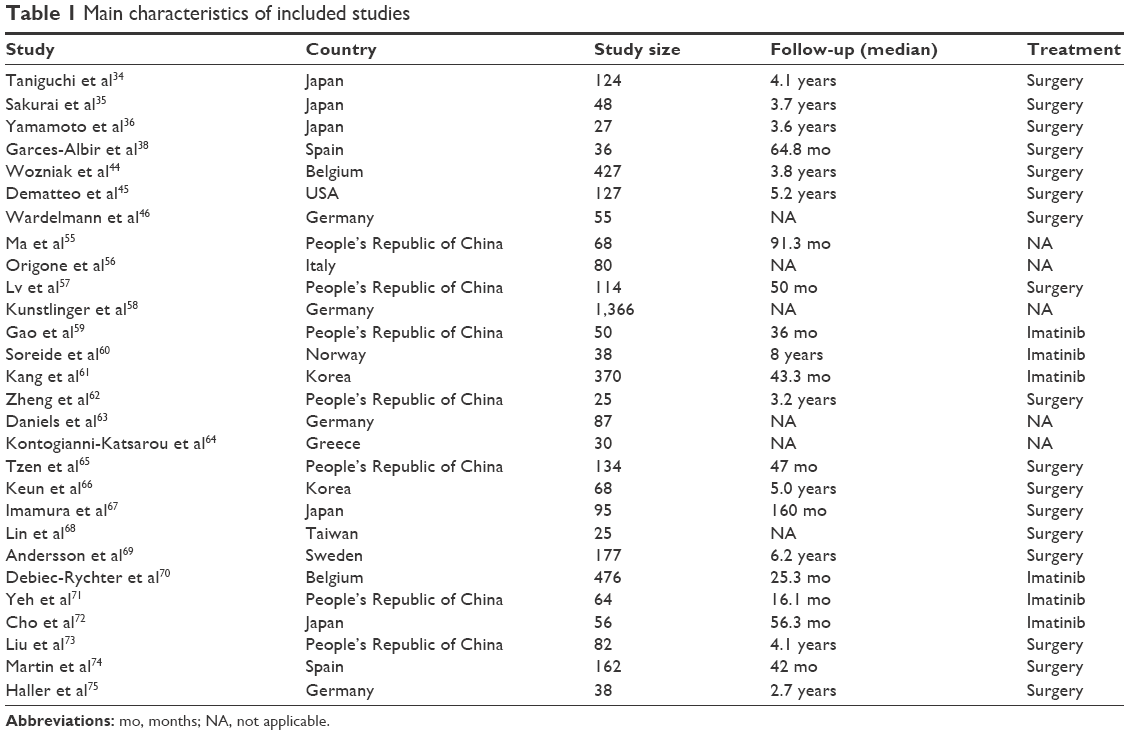 | Table 1 Main characteristics of included studies |
KIT mutation status and clinicopathological features
KIT mutation was not significantly associated with tumor location
To determine whether or not the KIT mutation could be linked to the location of tumor, we analyzed eight studies including 2,355 patients. OR was 1.00, 95% CI was in the range of 0.51–1.95, z=0.01, and P=0.99 (Figure 2), indicating that the rate of KIT mutation was not significantly changed between GISTs in stomachs and those in small intestines.
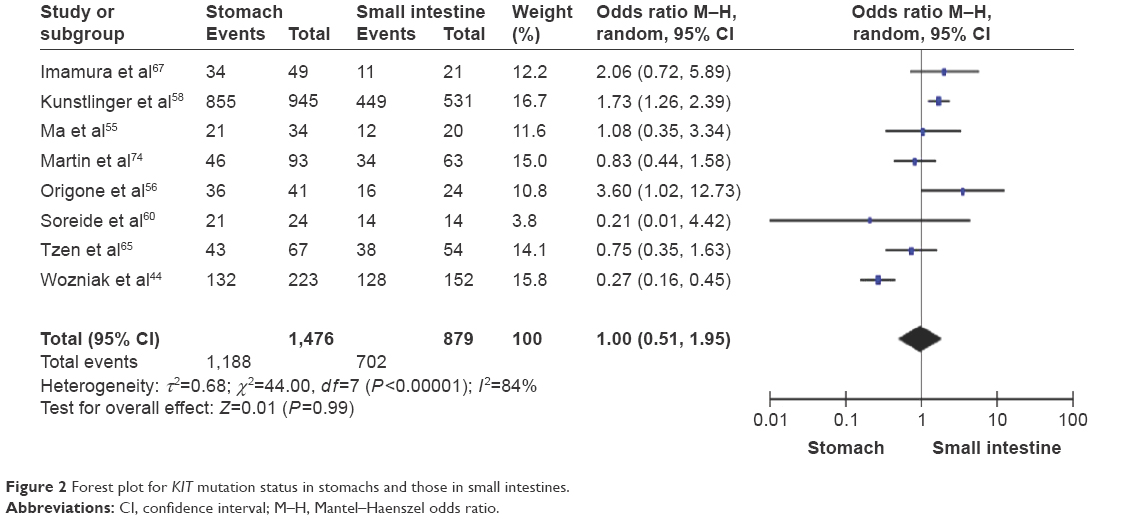 | Figure 2 Forest plot for KIT mutation status in stomachs and those in small intestines. |
KIT mutation was significantly associated with tumor size
Considering the tumor size, OR was 1.51, 95% CI: 1.05–2.17, z=2.22, and P=0.03 (Figure 3), indicating that KIT mutations were significantly more frequently observed in patients with larger size (>5 cm) of GISTs than those with smaller size (<5 cm) of GISTs.
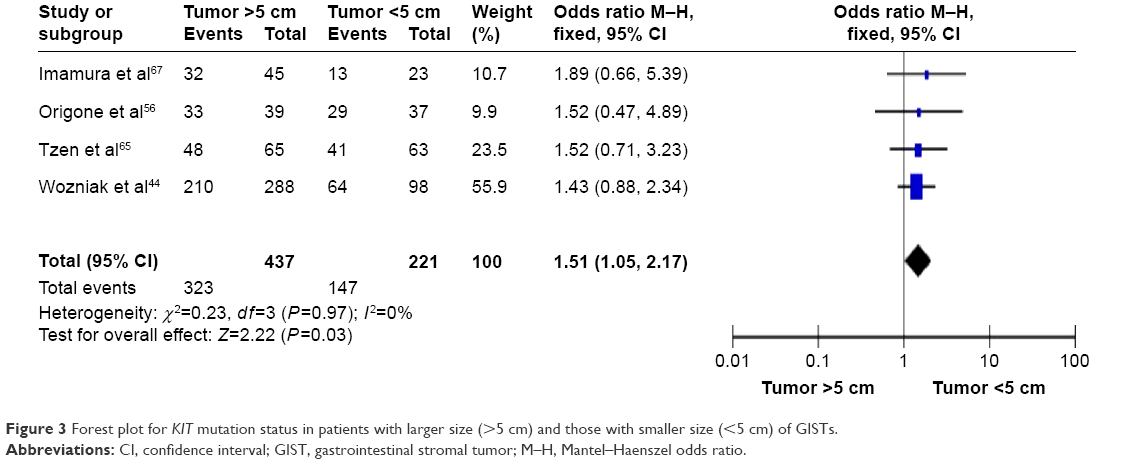 | Figure 3 Forest plot for KIT mutation status in patients with larger size (>5 cm) and those with smaller size (<5 cm) of GISTs. |
KIT mutation was significantly correlated with tumor mitosis index
To determine whether or not the KIT mutation could be linked to the tumor mitosis indexes (MIs), we analyzed seven studies including 899 patients. We found that the KIT mutation was significantly increased in patients with higher MIs (>5/50 HPFs) of GISTs compared to patients with lower MIs (<5/50 HPFs) of tumors. OR was 1.89, 95% CI ranged between 1.39 and 2.56, z=4.05, and P<0.0001 (Figure 4).
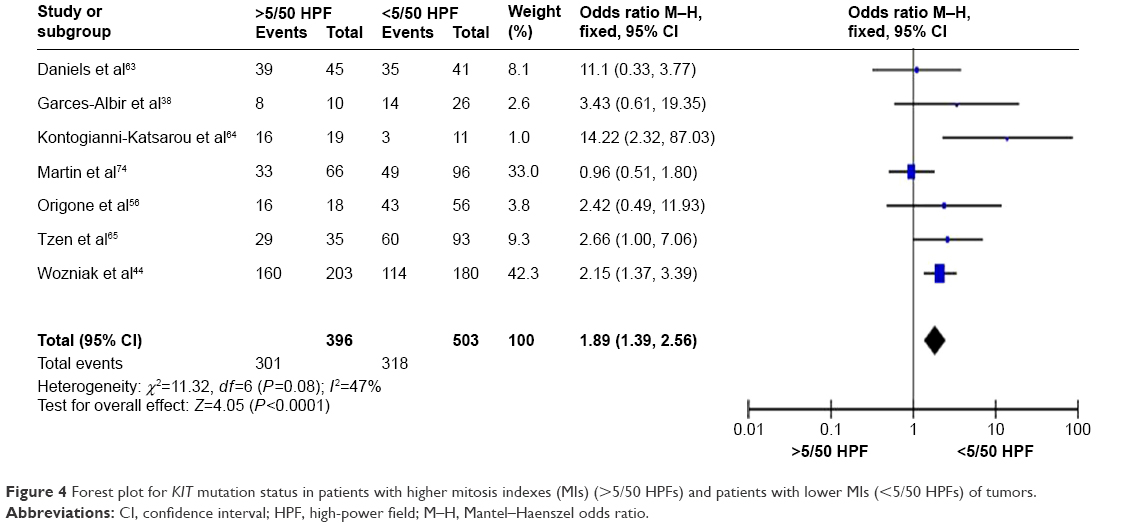 | Figure 4 Forest plot for KIT mutation status in patients with higher mitosis indexes (MIs) (>5/50 HPFs) and patients with lower MIs (<5/50 HPFs) of tumors. |
KIT mutation was significantly correlated with tumor recurrence
KIT mutation-positive patients showed a significantly higher rate of recurrence compared to KIT mutation-negative patients. OR was 2.06, 95% CI: 1.37–3.11, z=3.46, and P=0.0005 (Figure 5).
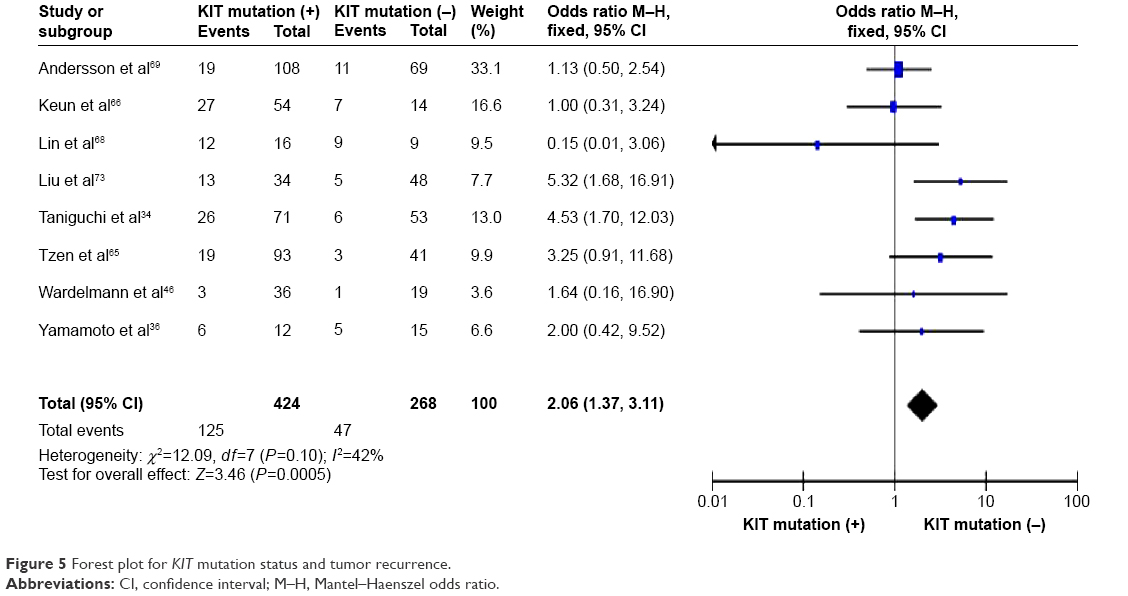 | Figure 5 Forest plot for KIT mutation status and tumor recurrence. |
KIT mutation was significantly correlated with tumor metastasis
KIT mutation-positive patients showed a significantly higher rate of tumor metastasis compared to KIT mutation-negative patients. OR was 2.77, 95% CI was 1.64–4.67, z=3.82, and P=0.0001 (Figure 6).
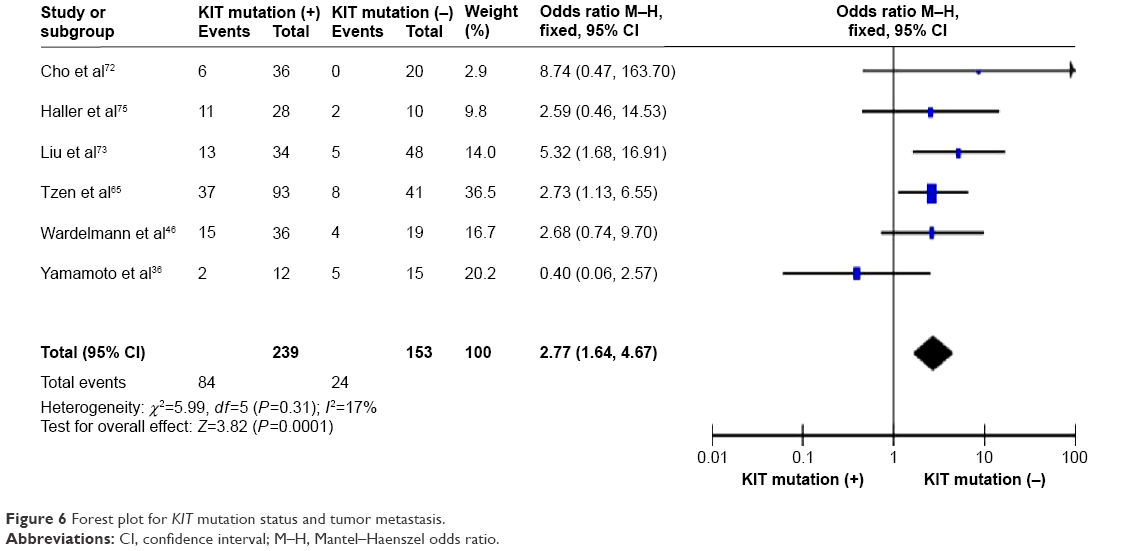 | Figure 6 Forest plot for KIT mutation status and tumor metastasis. |
KIT mutation was significantly correlated with the overall survival of patients
KIT mutation-positive patients showed a worse prognosis compared to KIT mutation-negative patients, which was supported by the 3-year overall survival analysis. OR was 0.47, 95% CI: 0.25–0.90, z=2.30, and P=0.02 (Figure 7).
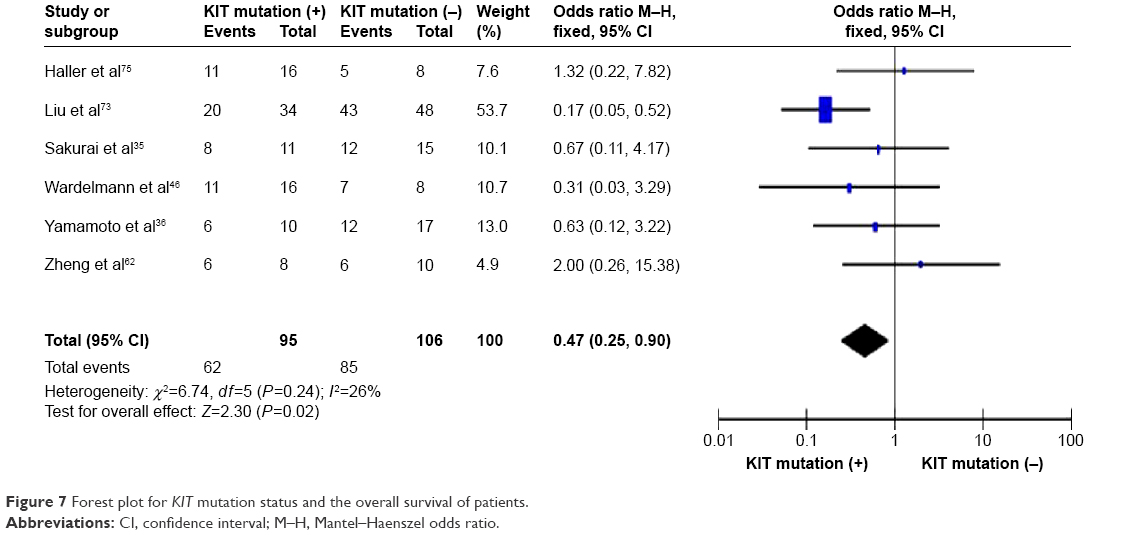 | Figure 7 Forest plot for KIT mutation status and the overall survival of patients. |
Further analysis of effects of different KIT mutations on patient overall survival
Finally, with respect to progression-free survival (PFS), OR was 3.60, 95% CI was 2.17–5.98, z=4.96, and P<0.00001 (Figure 8A), indicating that PFS was significantly worse in patients with KIT exon 9 mutations than in those with KIT exon 11 mutations. OR was 0.36, 95% CI 0.24–0.56, z=4.68, and P<0.00001 (Figure 8B), indicating that the 5-year PFS rate was significantly lower in patients with KIT exon 11 deletion than in those with other types of KIT exon 11 mutations. Moreover, OR was 0.19, 95% CI was 0.05–0.65, z=2.64, and P=0.008 (Figure 8C), indicating that 5-year PFS was significantly worse in patients with GISTs bearing deletions involving KIT codon 557–558 than in those bearing other deletions of KIT exon 11.
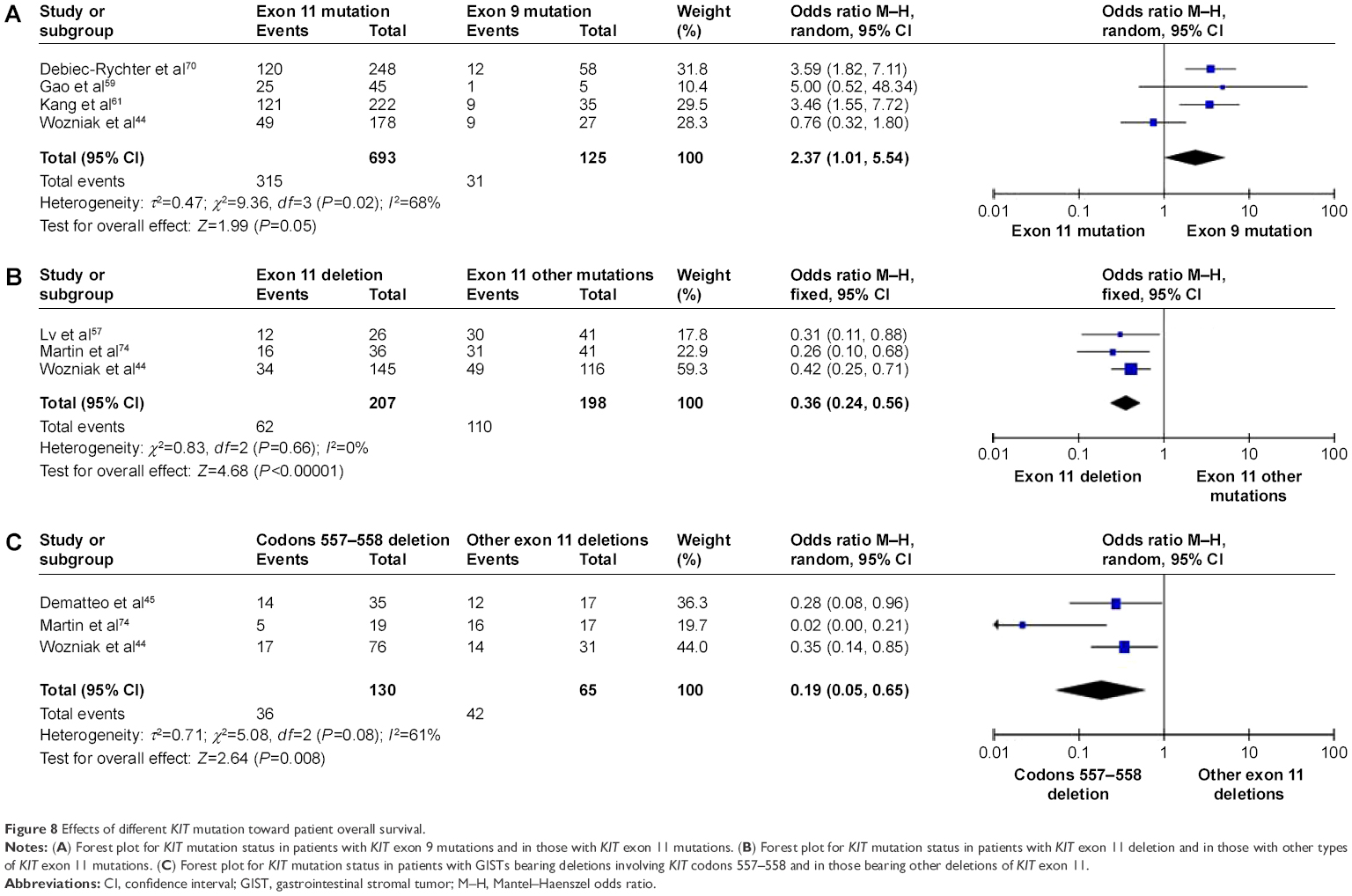 | Figure 8 Effects of different KIT mutation toward patient overall survival. |
Sensitivity analyses and publication bias
A sensitivity analysis was performed by testing the result stability by removing one study at a time. The pooled ORs were not significantly changed, which confirmed the stability of our analyses. The funnel plots were largely symmetric, suggesting that there were no publication biases in terms of KIT mutations and clinicopathological features (Figure 9).
 | Figure 9 Funnel plot for publication bias. |
Discussion
Previous studies have shown controversial results for the prognostic value of mutational status in GIST patients, in addition to tumor size, tumor site, and mitotic count, due to the relatively small number of tested samples in each study or the limited number of analyzed studies.13,34–37 The discrepancy between different studies could be explained by the variations in methods, varied interpretation of the results, heterogeneous patient populations, different clinical treatments, limited number of patients in studies, but most probably different types of KIT mutations. In this study, we first compared the frequency of KIT mutations in different locations, the sizes of tumors, and different MIs. Our results demonstrated that the rate of KIT mutation was not significantly changed between GISTs in stomachs and those in small intestines. However, KIT mutations were significantly more frequently observed in patients with larger sizes (>5 cm) of GISTs than in those with smaller sizes (<5 cm) of GISTs. In addition, KIT mutation was significantly increased in patients with higher MIs (>5/50 HPFs) of GISTs compared to patients with lower MIs (<5/50 HPFs) of tumors. Garces-Albir et al38 reported that GIST tumors >5 cm and the presence of >5 mitoses/50 HPFs were obviously associated with worse outcome. Tumor size and mitotic counts traditionally have been the two factors for estimation of prognosis.39 A previous study has also reported that there is a direct relationship between the presence of mutation in tumor, tumor size, and mitotic count,34 which is in agreement with our results. We further demonstrated by whole-gene sequencing that KIT mutation-positive patients showed a significantly higher rate of recurrence compared to KIT mutation-negative patients who did not have KIT gene mutations: OR was 2.06, 95% CI 1.37–3.11, z=3.46, and P=0.0005. KIT mutation-positive patients showed a significantly higher rate of tumor metastasis compared to KIT mutation-negative patients: OR was 2.77, 95% CI 1.64–4.67, z=3.82, and P=0.0001. In addition, the KIT mutation-positive patients showed a worse prognosis compared to the KIT mutation-negative patients, which was supported by the 3-year overall survival analysis: OR was 0.47, 95% CI 0.25–0.90, z=2.30, and P=0.02. Taken together, our results strongly support the hypothesis that KIT mutation status is another evaluable factor to estimate prognosis in GISTs, in addition to tumor size and mitotic counts. Therefore, determination of KIT mutations is a potential prognostic marker in GIST patients.
Mutations of the KIT gene in GISTs occur most frequently in KIT exon 11, followed by those in KIT exon 9; less frequently, mutations occur in exon 13 or exon 17.40 We determined that the PFS of GIST patients was significantly worse in patients with KIT exon 9 mutations than in those with KIT exon 11 mutations. A few studies have shown that tumors containing deletions in the KIT exon 11, which most frequently involved the 5′ portion between codons 550 and 560,41 are clinically more aggressive than tumors with other types of mutations. However, several studies have reported inconsistent results.42–45 Our result showed that 5-year RFS rate was significantly lower in patients with KIT exon 11 deletion than in those with other types of KIT exon 11 mutations. Deletions in KIT exon 11 were most frequently observed in the 5′ portion between codons 550 and 560 and occurred less frequently between codons 562 and 579.42,43,46 There was no significant difference in the rate of response to imatinib or the median PFS among patients with exon 11 deletion, point mutations, and mixed-type mutations.47,48 A few studies showed inconsistent results in terms of 5-year RFS in patients of GIST with codon 557–558 deletion and other deletions of KIT exon 11 due to the small number of patient samples.42,43,45,46 In this analysis, we showed that RFS for 5 years was significantly worse in patients with GISTs bearing deletions involving KIT codon 557–558 than in those bearing other deletions of KIT exon 11.
The GIST paradigm has been proven to be more complex than expected, due to a molecular heterogeneity within all GIST tumors and the identification of different subgroups characterized by a peculiar genotype–phenotype.49 With the application of high-throughput technologies of gene mutation analysis, a wide spectrum of other genomic alterations can be identified in GIST tumors. The biological role and clinical significance of most of these additional events, such as PDGFRA gain-of-function mutations, in GIST pathogenesis and development remain undefined. Besides the importance of KIT mutation status in predicting imatinib sensitivity and prognosis, the acquisition of secondary mutations in KIT represents the most frequent mechanism of imatinib resistance and worse prognosis in GIST patients. However, most of the studies till date have only reported one case or a few cases of secondary KIT mutations or have insufficient follow-up data;50–53 we are hence not able to perform a meta-analysis to compare the significance of primary and secondary KIT mutations in GIST patients. Acquired secondary KIT mutations are the major cause of secondary imatinib resistance and are important in the development of new therapeutic strategies in advanced GISTs.54 The predictive value of secondary KIT mutations in GIST patients needs further study. Therefore, additional research in the future, especially larger prospective studies, will be needed to evaluate the correlation between mutation status of KIT and/or other genes and their clinicopathological significance in GIST patients.
Conclusion
In summary, through the analysis of 4,449 patients from 28 eligible studies, we have shown that KIT mutation status is closely correlated with size of tumors and MIs, but not with tumor location. KIT mutation has also been observed to be significantly correlated with tumor recurrence, metastasis, and the overall survival of patients. GIST patients with KIT exon 9 mutations have higher risk of progression than those with exon 11 mutations, and 5-year RFS rate was significantly higher in patients with KIT exon 11 deletion than in those with other types of KIT exon 11 mutations. In addition, RFS for 5 years was significantly worse in patients with GISTs bearing deletions involving KIT codons 557–558 than in those bearing other deletions of KIT exon 11. Our results strongly support the hypothesis that KIT mutation status is another evaluable factor to estimate prognosis in GISTs, besides tumor size and mitotic counts. Therefore, determination of differential KIT mutation status is a potential prognostic marker for GIST patients.
Disclosure
The authors report no conflicts of interest in this work.
References
Liegl-Atzwanger B, Fletcher JA, Fletcher CD. Gastrointestinal stromal tumors. Virchows Arch. 2010;456(2):111–127. | ||
Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279(5350):577–580. | ||
Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299(5607):708–710. | ||
Heinrich MC, Maki RG, Corless CL, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol. 2008;26(33):5352–5359. | ||
Yarden Y, Kuang WJ, Yang-Feng T, et al. Human proto-oncogene c-kit: a new cell surface receptor tyrosine kinase for an unidentified ligand. EMBO J. 1987;6(11):3341–3351. | ||
Zsebo KM, Williams DA, Geissler EN, et al. Stem cell factor is encoded at the Sl locus of the mouse and is the ligand for the c-kit tyrosine kinase receptor. Cell. 1990;63(1):213–224. | ||
Hanks SK, Quinn AM, Hunter T. The protein kinase family: conserved features and deduced phylogeny of the catalytic domains. Science. 1988;241(4861):42–52. | ||
Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296(5573):1655–1657. | ||
Reith AD, Ellis C, Lyman SD, et al. Signal transduction by normal isoforms and W mutant variants of the Kit receptor tyrosine kinase. EMBO J. 1991;10(9):2451–2459. | ||
Arock M, Sotlar K, Akin C, et al. KIT mutation analysis in mast cell neoplasms: recommendations of the European Competence Network on Mastocytosis. Leukemia. 2015;29(6):1223–1232. | ||
Ronnstrand L, Phung B. Enhanced SOX10 and KIT expression in cutaneous melanoma. Med Oncol. 2013;30(3):648. | ||
Montero JC, Lopez-Perez R, San MJF, Pandiella A. Expression of c-Kit isoforms in multiple myeloma: differences in signaling and drug sensitivity. Haematologica. 2008;93(6):851–859. | ||
Yan L, Zou L, Zhao W, et al. Clinicopathological significance of c-KIT mutation in gastrointestinal stromal tumors: a systematic review and meta-analysis. Sci Rep. 2015;5:13718. | ||
Iorio N, Sawaya RA, Friedenberg FK. Review article: the biology, diagnosis and management of gastrointestinal stromal tumours. Aliment Pharmacol Ther. 2014;39(12):1376–1386. | ||
Phung B, Steingrimsson E, Ronnstrand L. Differential activity of c-KIT splice forms is controlled by extracellular peptide insert length. Cell Signal. 2013;25(11):2231–2238. | ||
Purohit R. Role of ELA region in auto-activation of mutant KIT receptor: a molecular dynamics simulation insight. J Biomol Struct Dyn. 2014;32(7):1033–1046. | ||
Kazi JU, Sun J, Ronnstrand L. The presence or absence of IL-3 during long-term culture of Flt3-ITD and c-Kit-D816V expressing Ba/F3 cells influences signaling outcome. Exp Hematol. 2013;41(7):585–587. | ||
Kazi JU, Vaapil M, Agarwal S, Bracco E, Pahlman S, Ronnstrand L. The tyrosine kinase CSK associates with FLT3 and c-Kit receptors and regulates downstream signaling. Cell Signal. 2013;25(9):1852–1860. | ||
Cohen PS, Chan JP, Lipkunskaya M, Biedler JL, Seeger RC. Expression of stem cell factor and c-kit in human neuroblastoma. The Children’s Cancer Group. Blood. 1994;84(10):3465–3472. | ||
Hassan S, Kinoshita Y, Kawanami C, et al. Expression of protooncogene c-kit and its ligand stem cell factor (SCF) in gastric carcinoma cell lines. Dig Dis Sci. 1998;43(1):8–14. | ||
Inoue M, Kyo S, Fujita M, Enomoto T, Kondoh G. Coexpression of the c-kit receptor and the stem cell factor in gynecological tumors. Cancer Res. 1994;54(11):3049–3053. | ||
Stankov K, Popovic S, Mikov M. C-KIT signaling in cancer treatment. Curr Pharm Des. 2014;20(17):2849–2880. | ||
Krystal GW, Hines SJ, Organ CP. Autocrine growth of small cell lung cancer mediated by coexpression of c-kit and stem cell factor. Cancer Res. 1996;56(2):370–376. | ||
Pietsch T, Kyas U, Steffens U, et al. Effects of human stem cell factor (c-kit ligand) on proliferation of myeloid leukemia cells: heterogeneity in response and synergy with other hematopoietic growth factors. Blood. 1992;80(5):1199–1206. | ||
Toyota M, Hinoda Y, Takaoka A, et al. Expression of c-kit and kit ligand in human colon carcinoma cells. Tumour Biol. 1993;14(5):295–302. | ||
Kumar A, Boyle EA, Tokita M, et al. Deep sequencing of multiple regions of glial tumors reveals spatial heterogeneity for mutations in clinically relevant genes. Genome Biol. 2014;15(12):530. | ||
Ammendola M, Sacco R, Sammarco G, et al. Mast cells positive to tryptase and c-kit receptor expressing cells correlates with angiogenesis in gastric cancer patients surgically treated. Gastroenterol Res Pract. 2013;2013:703163. | ||
Lindblad O, Kazi JU, Ronnstrand L, Sun J. PI3 kinase is indispensable for oncogenic transformation by the V560D mutant of c-Kit in a kinase-independent manner. Cell Mol Life Sci. 2015;72(22):4399–4407. | ||
Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279(5350):577–580. | ||
Miettinen M, Lasota J. Gastrointestinal stromal tumors: review on morphology, molecular pathology, prognosis, and differential diagnosis. Arch Pathol Lab Med. 2006;130(10):1466–1478. | ||
Corless CL. Gastrointestinal stromal tumors: what do we know now? Mod Pathol. 2014;27(suppl 1):S1–S16. | ||
Lasota J, Miettinen M. Clinical significance of oncogenic KIT and PDGFRA mutations in gastrointestinal stromal tumours. Histopathology. 2008;53(3):245–266. | ||
Rammohan A, Sathyanesan J, Rajendran K, et al. A gist of gastrointestinal stromal tumors: a review. World J Gastrointest Oncol. 2013;5(6):102–112. | ||
Taniguchi M, Nishida T, Hirota S, et al. Effect of c-kit mutation on prognosis of gastrointestinal stromal tumors. Cancer Res. 1999;59(17):4297–4300. | ||
Sakurai S, Fukasawa T, Chong JM, Tanaka A, Fukayama M. C-kit gene abnormalities in gastrointestinal stromal tumors (tumors of interstitial cells of Cajal). Jpn J Cancer Res. 1999;90(12):1321–1328. | ||
Yamamoto H, Oda Y, Kawaguchi K, et al. c-kit and PDGFRA mutations in extragastrointestinal stromal tumor (gastrointestinal stromal tumor of the soft tissue). Am J Surg Pathol. 2004;28(4):479–488. | ||
Lee JH, Kim Y, Choi JW, Kim YS. Correlation of imatinib resistance with the mutational status of KIT and PDGFRA genes in gastrointestinal stromal tumors: a meta-analysis. J Gastrointestin Liver Dis. 2013;22(4):413–418. | ||
Garces-Albir M, Marti-Obiol R, Lopez-Mozos F, Calabuig-Farinas S, Navarro-Ros S, Ortega-Serrano J. Results on prognostic value of mutations in localized gastrointestinal stromal tumors (GIST) in one single center. Rev Esp Enferm Dig. 2012;104(8):405–410. | ||
Casali PG, Jost L, Reichardt P, Schlemmer M, Blay JY. Gastrointestinal stromal tumours: ESMO clinical recommendations for diagnosis, treatment and follow-up. Ann Oncol. 2009;20(suppl 4):64–67. | ||
Corless CL, Barnett CM, Heinrich MC. Gastrointestinal stromal tumours: origin and molecular oncology. Nat Rev Cancer. 2011;11(12):865–878. | ||
Miettinen M, Lasota J. Gastrointestinal stromal tumors. Gastroenterol Clin North Am. 2013;42(2):399–415. | ||
Emile JF, Theou N, Tabone S, et al. Clinicopathologic, phenotypic, and genotypic characteristics of gastrointestinal mesenchymal tumors. Clin Gastroenterol Hepatol. 2004;2(7):597–605. | ||
Emile JF, Tabone-Eglinger S, Theou-Anton N, Lemoine A. Prognostic value of KIT exon 11 deletions in GISTs. Gastroenterology. 2006;131(3):976–977. author reply 7–8. | ||
Wozniak A, Rutkowski P, Piskorz A, et al. Prognostic value of KIT/PDGFRA mutations in gastrointestinal stromal tumours (GIST): polish clinical GIST registry experience. Ann Oncol. 2012;23(2):353–360. | ||
Dematteo RP, Gold JS, Saran L, et al. Tumor mitotic rate, size, and location independently predict recurrence after resection of primary gastrointestinal stromal tumor (GIST). Cancer. 2008;112(3):608–615. | ||
Wardelmann E, Losen I, Hans V, et al. Deletion of Trp-557 and Lys-558 in the juxtamembrane domain of the c-kit protooncogene is associated with metastatic behavior of gastrointestinal stromal tumors. Int J Cancer. 2003;106(6):887–895. | ||
Gao J, Dang Y, Sun N, Li J, Shen L. C-KIT mutations were closely associated with the response to Imatinib in Chinese advanced gastrointestinal stromal tumor patients. Med Oncol. 2012;29(5):3039–3045. | ||
Bachet JB, Hostein I, Le Cesne A, et al. Prognosis and predictive value of KIT exon 11 deletion in GISTs. Br J Cancer. 2009;101(1):7–11. | ||
Ricci R, Dei TAP, Rindi G. GISTogram: a graphic presentation of the growing GIST complexity. Virchows Arch. 2013;463(4):481–487. | ||
Wada N, Kurokawa Y, Takahashi T, et al. Detecting secondary C-KIT mutations in the peripheral blood of patients with imatinib-resistant gastrointestinal stromal tumor. Oncology. 2016;90(2):112–117. | ||
Spitaleri G, Biffi R, Barberis M, et al. Inactivity of imatinib in gastrointestinal stromal tumors (GISTs) harboring a KIT activation-loop domain mutation (exon 17 mutation pN822K). Onco Targets Ther. 2015;8:1997–2003. | ||
Kikuchi H, Miyazaki S, Setoguchi T, et al. Rapid relapse after resection of a sunitinib-resistant gastrointestinal stromal tumor harboring a secondary mutation in exon 13 of the c-KIT gene. Anticancer Res. 2012;32(9):4105–4109. | ||
Utsunomiya T, Okamoto M, Yano S, et al. Secondary c-kit mutation in a recurrent gastrointestinal stromal tumor under long-term treatment with imatinib mesylate: report of a case. Surg Today. 2008;38(1):65–67. | ||
Lim KH, Huang MJ, Chen LT, et al. Molecular analysis of secondary kinase mutations in imatinib-resistant gastrointestinal stromal tumors. Med Oncol. 2008;25(2):207–213. | ||
Ma YY, Yu S, He XJ, et al. Involvement of c-KIT mutation in the development of gastrointestinal stromal tumors through proliferation promotion and apoptosis inhibition. Onco Targets Ther. 2014;7:637–643. | ||
Origone P, Gargiulo S, Mastracci L, et al. Molecular characterization of an Italian series of sporadic GISTs. Gastric Cancer. 2013;16(4):596–601. | ||
Lv A, Li Z, Tian X, et al. SKP2 high expression, KIT exon 11 deletions, and gastrointestinal bleeding as predictors of poor prognosis in primary gastrointestinal stromal tumors. PLoS One. 2013;8(5):e62951. | ||
Kunstlinger H, Huss S, Merkelbach-Bruse S, et al. Gastrointestinal stromal tumors with KIT exon 9 mutations: update on genotype-phenotype correlation and validation of a high-resolution melting assay for mutational testing. Am J Surg Pathol. 2013;37(11):1648–1659. | ||
Gao J, Tian Y, Li J, Sun N, Yuan J, Shen L. Secondary mutations of c-KIT contribute to acquired resistance to imatinib and decrease efficacy of sunitinib in Chinese patients with gastrointestinal stromal tumors. Med Oncol. 2013;30(2):522. | ||
Soreide K, Sandvik OM, Soreide JA, Gudlaugsson E, Mangseth K, Haugland HK. Tyrosine-kinase mutations in c-KIT and PDGFR-alpha genes of imatinib naive adult patients with gastrointestinal stromal tumours (GISTs) of the stomach and small intestine: relation to tumour-biological risk-profile and long-term outcome. Clin Transl Oncol. 2012;14(8):619–629. | ||
Kang HJ, Ryu MH, Kim KM, et al. Imatinib efficacy by tumor genotype in Korean patients with advanced gastrointestinal stromal tumors (GIST): The Korean GIST Study Group (KGSG) study. Acta Oncol. 2012;51(4):528–536. | ||
Zheng S, Huang KE, Tao DY, Pan YL. Gene mutations and prognostic factors analysis in extragastrointestinal stromal tumor of a Chinese three-center study. J Gastrointest Surg. 2011;15(4):675–681. | ||
Daniels M, Lurkin I, Pauli R, et al. Spectrum of KIT/PDGFRA/BRAF mutations and phosphatidylinositol-3-kinase pathway gene alterations in gastrointestinal stromal tumors (GIST). Cancer Lett. 2011;312(1):43–54. | ||
Kontogianni-Katsarou K, Dimitriadis E, Lariou C, Kairi-Vassilatou E, Pandis N, Kondi-Paphiti A. KIT exon 11 codon 557/558 deletion/insertion mutations define a subset of gastrointestinal stromal tumors with malignant potential. World J Gastroenterol. 2008;14(12):1891–1897. | ||
Tzen CY, Wang MN, Mau BL. Spectrum and prognostication of KIT and PDGFRA mutation in gastrointestinal stromal tumors. Eur J Surg Oncol. 2008;34(5):563–568. | ||
Keun PC, Lee EJ, Kim M, et al. Prognostic stratification of high-risk gastrointestinal stromal tumors in the era of targeted therapy. Ann Surg. 2008;247(6):1011–1018. | ||
Imamura M, Yamamoto H, Nakamura N, et al. Prognostic significance of angiogenesis in gastrointestinal stromal tumor. Mod Pathol. 2007;20(5):529–537. | ||
Lin SC, Liu CL, Wang TI, Chang WS, Tzen CY, Huang MJ. Clinical implications of C-kit gene mutation in patients with large gastrointestinal stromal tumors. J Gastroenterol Hepatol. 2006;21(10):1604–1608. | ||
Andersson J, Bumming P, Meis-Kindblom JM, et al. Gastrointestinal stromal tumors with KIT exon 11 deletions are associated with poor prognosis. Gastroenterology. 2006;130(6):1573–1581. | ||
Debiec-Rychter M, Sciot R, Le Cesne A, et al. KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer. 2006;42(8):1093–1103. | ||
Yeh CN, Chen TW, Lee HL, et al. Kinase mutations and imatinib mesylate response for 64 Taiwanese with advanced GIST: preliminary experience from Chang Gung Memorial Hospital. Ann Surg Oncol. 2007;14(3):1123–1128. | ||
Cho S, Kitadai Y, Yoshida S, et al. Deletion of the KIT gene is associated with liver metastasis and poor prognosis in patients with gastrointestinal stromal tumor in the stomach. Int J Oncol. 2006;28(6):1361–1367. | ||
Liu XH, Bai CG, Xie Q, Feng F, Xu ZY, Ma DL. Prognostic value of KIT mutation in gastrointestinal stromal tumors. World J Gastroenterol. 2005;11(25):3948–3952. | ||
Martin J, Poveda A, Llombart-Bosch A, et al. Deletions affecting codons 557-558 of the c-KIT gene indicate a poor prognosis in patients with completely resected gastrointestinal stromal tumors: a study by the Spanish Group for Sarcoma Research (GEIS). J Clin Oncol. 2005;23(25):6190–6198. | ||
Haller F, Gunawan B, von HA, et al. Prognostic role of E2F1 and members of the CDKN2A network in gastrointestinal stromal tumors. Clin Cancer Res. 2005;11(18):6589–6597. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.