")
Back to Journals » International Journal of Nanomedicine » Volume 15
A Mansonone Derivative Coupled with Monoclonal Antibody 4D5-Modified Chitosan Inhibit AKR1C3 to Treat Castration-Resistant Prostate Cancer
Authors Zhou M, Wang X, Xia J , Cheng Y, Xiao L, Bei Y, Tang J, Huang Y , Xiang Q , Huang S
Received 6 December 2019
Accepted for publication 8 April 2020
Published 1 May 2020 Volume 2020:15 Pages 3087—3098
DOI https://doi.org/10.2147/IJN.S241324
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Meng Zhou,1,* Xiaoyu Wang,1,* Jie Xia,2 Yating Cheng,1 Lichun Xiao,1 Yu Bei,3 Jianzhong Tang,3 Yadong Huang,1,3 Qi Xiang,1,3 Shiliang Huang2
1Institute of Biomedicine and Guangdong Provincial Key Laboratory of Bioengineering Medicine, Jinan University, Guangzhou 510632, People’s Republic of China; 2School of Pharmaceutical Sciences, Sun Yat-Sen University, Guangzhou 510006, People’s Republic of China; 3Biopharmaceutical R&D Center of Jinan University, Guangzhou 510630, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Qi Xiang; Shiliang Huang Tel +86 20-85563234
; +86 20- 39943052
Fax +86 20-85565109
; +86 20-39943056
Email [email protected]; [email protected]
Purpose: Aldo-ketoreductase (AKR) 1C3 is crucial for testosterone synthesis. Abnormally high expression/activity of AKR1C3 can promote castration-resistant prostate cancer (CRPC). A mansonone derivative and AKR1C3 inhibitor, 6e, was combined with 4D5 (extracellular fragment of the monoclonal antibody of human epidermal growth factor receptor-2)-modified chitosan to achieve a nanodrug-delivery system (CS-4D5/6e) to treat CRPC.
Materials and Methods: Morphologies/properties of CS-4D5/6e were characterized by atomic force microscopy, zeta-potential analysis, and Fourier transform-infrared spectroscopy. CS-4D5/6e uptake was measured by immunofluorescence under confocal laser scanning microscopy. Testosterone in LNCaP cells overexpressing human AKR1C3 (LNCaP-AKR1C3) and cell lysates was measured to reflect AKR1C3 activity. Androgen receptor (AR) and prostate-specific antigen (PSA) expression was measured by Western blotting. CS-4D5/6e-based inhibition of AKR1C3 was evaluated in tumor-xenografted mice.
Results: CS-4D5/6e was oblate, with a particle size of 200– 300 nm and thickness of 1– 5 nm. Zeta potential was 1.39± 0.248 mV. 6e content in CS-4D5/6e was 7.3± 1.4% and was 18± 3.6% for 4D5. 6e and CS-4D5/6e inhibited testosterone production significantly in a concentration-dependent manner in LNCaP-AKR1C3 cells, and a decrease in expression of AKR1C3, PSA, and AR was noted. Half-maximal inhibitory concentration of CS-4D5/6e on LNCaP-AKR1C3 cells was significantly lower than that in LNCaP cells (P< 0.05). CS-4D5/6e significantly reduced growth of 22Rv1 tumor xenografts by 57.00% compared with that in the vehicle group (P< 0.01).
Conclusion: We demonstrated the antineoplastic activity of a potent AKR1C3 inhibitor (6e) and its nanodrug-delivery system (CS-4D5/6e). First, CS-4D5/6e targeted HER2-positive CRPC cells. Second, it transferred 6e (an AKR1C3 inhibitor) to achieve a reduction in intratumoral testosterone production. Compared with 6e, CS-4D5/6e showed lower systemic toxicity. CS-4D5/6e inhibited tumor growth effectively in mice implanted with tumor xenografts by downregulating testosterone production mediated by intratumoral AKR1C3. These results showed a promising strategy for treatment of the CRPC that develops invariably in prostate-cancer patients.
Keywords: mansonone derivative, conjugate of HER2 monoclonal antibody extracellular fragment 4D5, castration-resistant prostate cancer
Introduction
Cancer of the prostate gland (also termed “prostate cancer” (PC)) is one of the most commonly diagnosed malignancies and second leading cause of cancer-related death in the USA.1 First-line treatment for PC is androgen-deprivation therapy (ADT). The latter includes orchiectomy, 5-α reductase inhibitors, and agonists of luteinizing hormone-releasing hormone, which can delay PC development.2 However, after a long period of ADT, nearly all patients showed high levels of prostate-specific antigen (PSA) and growth of tumor cells. Re-growth of PC cells has been found to be due to reactivation of the androgen-signaling system in tumor cells and, thus, termed “castration-resistant prostate cancer” (CRPC). If PC develops into CRPC, patient survival is, in general, <2 years.3 Therefore, preventing the transition from PC to CRPC and treating CRPC are important problems that must be solved.
Typically, after ADT, two main mechanisms aggravate PC developing into CRPC: (i) activation of the androgen receptor (AR) signaling pathway and changes in activation of AR domains and AR ligand-independent domains;4–7 (ii) PC cells synthesize androgens using their own enzyme systems. PC cells can express all the enzymes needed for androgen synthesis, so long-term ADT can induce upregulation of expression of these enzymes. PC cells are not dependent upon androgen levels in the blood circulation after castration, but generate androgens within themselves and continue to activate ARs to promote the transformation of PC into CRPC.8 Therefore, reducing androgen synthesis in tumor cells is an important goal of therapy against CRPC, which can be achieved by inhibiting expression of the key enzymes in the testosterone-synthesis pathway.
Aldo-ketoreductase (AKR)1C3 is a crucial androgenic enzyme that shows high expression in metastatic tumors, recurrent tumors, or prostate xenograft tumors.9 Abnormally high expression/activity of AKR1C3 can promote CRPC development. Hence, AKR1C3 is a potential therapeutic target after treatment failure of CRPC or ADT failure. AKR1C3 has pivotal roles in the pathogenesis and progression of CRPC because it catalyses the conversion of weak androgen precursors to potent AR ligands: testosterone and 5α-dihydrotestosterone (5α-DHT).10
There are several subtypes of AKR1C, but AKR1C3 is found mainly in tissues of the human prostate gland. In addition, the AKR1C3 level in the PC tissues of patients with CRPC is significantly higher than that in cases with benign PC or early PC.11–13 AKR1C3 “moonlights” by acting as a co-activator of the AR and stabilizes ubiquitin ligases that are independent of its enzyme activity.14 Moreover, AKR1C3 has an important role in abiraterone (an inhibitor of cytochrome P450 (CYP)17A1) resistance in metastatic CRPC by increasing androgen synthesis within tumors.15 The ability of AKR1C3 to regulate ligand access to the AR and estrogen receptor (ER) in a tumor-specific fashion makes it a superior drug target than AR antagonists or ER antagonists.
Indomethacin is an AKR1C3 inhibitor. It can overcome abiraterone resistance and enhance abiraterone therapy in vitro and in vivo by reducing the levels of intracrine androgens and diminishing the transcriptional activity of ARs.16 Potent and selective inhibitors of AKR1C3 have been designed and synthesized based on nonsteroidal, steroidal and natural-product scaffolds. AKR1C3 inhibitors have progressed to clinical trials for CRPC but with mixed success. However, AKR1C3 inhibitors require optimization for testing in animals and humans, especially in terms of potency, selectivity and lower toxicity.17 Therefore, development of AKR1C3 inhibitors with better selectivity for preventing and treating CRPC is very important.
Mansonone F is a tricyclic sesquiterpenoid. It occurs naturally as o-naphthoquinone and has an unusual oxaphelane skeleton. It was isolated first from the heartwood of the west African tree Mansonia altissima A. Chev. (Sterculiaceae).18,19 A derivative of mansonone F, 6e, has been optimized for inhibiting testosterone production in LNCaP cells overexpressing human AKR1C3 (LNCaP-AKR1C3).
Tumor-targeted delivery of cytotoxins presents considerable advantages over that of passive transport. Previously, we noted that intervention against human epidermal growth factor receptor 2 (HER2) to deplete tumor-initiating cells can optimize chemotherapy management and prevent CRPC progression.20 HER2 (ErbB-2/Neu) is important for mediating the ligand-dependent and -independent activation of ARs in androgen-sensitive (AS) and androgen-independent (AI)/castration-resistant (CR) PC cells, respectively, for the progression and survival of PC cells.21
scFv 4D5 is a fragment of the humanized anti-HER2 monoclonal antibody. As a “mini-antibody”, scFv 4D5 is an example of a high-efficiency HER2/neu-targeting vehicle that represents a single-chain variable fragment of immunoglobulin molecules.22,23 scFv 4D5 exhibits lower cross-reactivity and immunogenicity and faster penetration in tissue in comparison with the corresponding full-size antibody. There have been several inspiring success stories of scFv 4D5 coupled with other therapeutic drugs representing a new class of antibody-targeted immunotoxin therapy.24 Covalent bioconjugation of scFv 4D5 to the polymeric surface of nanomedicines can enable recognition by HER2 protein and uptake into HER2 cancer cells. Simultaneously, 4D5 has a low molecular weight, low immunogenicity, and good thermal stability, which enable 4D5 to infiltrate the HER2 receptor.25
In targeted cancer therapy using nanodrug-delivery systems, chitosan has attracted considerable attention as a carrier material for drug-loaded nanoparticles.26 Modified chitosan-based nanoparticles can deliver various anti-tumor agents to specific tumor tissues efficiently.
To improve the prostate gland-targeting effects of 6e (see above), scFv 4D5-modified chitosan (CS) was used as a drug carrier to prepare a new nanodrug-delivery system. Physical and chemical characterization and pharmacodynamics investigation in vitro and in vivo were conducted to evaluate whether this new nanodrug-delivery system can be used to treat CRPC.
In summary, CRPC tumors that have escaped systemic androgen deprivation have measurable intratumoral levels of testosterone, suggesting that a resistance mechanism is dependent upon androgen-simulated growth.27 We have found that AKR1C3 is expressed in the tumor microenvironment of CRPC metastases in addition to epithelial cells.28 Also, the relative abundance of AKR1C3 in the epithelium compared with that in stroma varies substantially between metastatic sites. AKR1C3 inhibitors may have distinct advantages over existing therapeutics for CRPC treatment. Here, we designed a nanomedicine, CS-4D5/6e, that could inhibit AKR1C3 (using 6e) and target HER2-positive CRPC (using a fragment of the monoclonal antibody 4D5). Experiments (in vivo and in vitro) verified our hypothesis. CS-4D5/6e, as a nanodrug carrier, suppressed intratumoral levels of testosterone effectively, demonstrated the capabilities of 6e as an AKR1C3 inhibitor, and could improve tumor targeting significantly. Hence, CS-4D5/6e could be a promising therapeutic strategy for CRPC.
Materials and Methods
Ethical Approval of the Study Protocol
The experimental protocols used in this study were approved by the Animal Care and Use Committees of Jinan University (approval number: 2019228) in Tianhe, China, and the Chinese Academy of Medical Science (Beijing, China). Experiments were conducted in accordance with the guidelines for animal care and use set by the Chinese government.
Cell Culture
22Rv1 (ATCC® CRL-2505™) and LNCaP (ATCC® CRL-1740™) cells were purchased from the Chinese Academy of Sciences (Shanghai, China). They were cultured in RPMI-1640 supplemented with 10% fetal bovine serum (FBS). LNCaP-AKR1C3 cells overexpressing AKR1C3 were generated by Cyagen China (Guangzhou, China). Cells at passage nine or lower were used. Where indicated, cells were also cultured in charcoal-stripped serum (CSS) medium prepared by supplementing RPMI-1640 without phenol red with charcoal-stripped FBS (Biological Industries, Beit HaEmek, Israel). All cells were maintained at 37°C in a humidified incubator in an atmosphere of 5% carbon dioxide. Indomethacin (CAS: 53-86-1) was purchased from MedChemExpress (Monmouth Junction, NJ, USA). EDC·HCl, NHS, and related other chemical reagents were purchased from Maklin (Shanghai, China).
Mice
Balb/c athymic nude mice were purchased from the Animal Centre of Guangdong Province (number 11401500058336). They were caged under controlled room temperature, humidity, and light (12-h light-dark cycle) conditions with access to water and food ad libitum.
Synthesis and Characterization of CS-4D5/6e
Briefly, 10 mg/mL of chitosan (5000 Da; deacetylation = 95%; Golden-Shell Pharmaceuticals, Zhejiang, China) solution was added to a solution of 4D5 (0.8 mg/mL; 50 mL; Biomedical Center of Jinan University), EDC·HCl (1.5 mg/mL; 50 mL) and NHS (0.9 mg/mL; 50 mL) and stirring carried out overnight at 4°C. This solution was dialyzed (7000 Da; catalog number, T34-70-001 MD34; Viskase, Lombard, IL, USA) against ultrapure water for 24 h at 4°C to remove excess EDC·HCl, NHS, and free-CS molecules. The resulting solution was designated “CS-4D5” and centrifuged at 15,000 rpm for 10 min at 4°C. Then, 6e was mixed with CS-4D5 solution for 2 h at 4°C. This mixed solution was centrifuged at 9500 rpm for 30 min at 4°C to remove unlabelled 6e. This nanomaterial was named “CS-4D5/6e”. Finally, high-performance liquid chromatography (HPLC) was undertaken to determine the 6e concentration in CS-4D5/6e. 4D5 was detected by Western blotting, and quantitative analysis was done using ImageJ (National Institutes of Health, Bethesda, MD, USA).
Purified CS-4D5/6e was characterized by atomic force microscopy (AFM), analysis of the zeta potential, size analyses, and Fourier transform-infrared (FT-IR) spectroscopy.
4D5-Mediated Targeting of HER2 Receptors on Cells in vitro
Targeting of HER2 receptors mediated by 4D5 was evaluated using cellular-uptake assays. LNCaP cells and 22Rv1 cells were cultured in 12-well plates at 5×104 cells/2 mL/well for 24 h, respectively. Next, 4D5, CS-4D5, or CS-4D5-6e (normalized with 2 mg/mL of 4D5) were added and co-incubated for 24 h. The drug-containing medium was removed, and cells were washed thrice with phosphate-buffered saline (PBS). The intracellular location of 4D5, CS-4D5, and CS-4D5/6e was observed by confocal laser scanning microscopy using a LSM700 system (Zeiss, Oberkochen, Germany). Briefly, cells were fixed with 4% paraformaldehyde, washed thrice with PBS, incubated with anti-4D5 antibody (rabbit polyclonal antibody (pAb), 1:75 dilution; Nanjing Zoonbio Biotechnology, Nanjing, China) and anti-HER2 antibody (100 μL; 1:100 dilution; bsm-33051M; Bioss, Woburn, MA, USA) overnight at 4°C, and then incubated with different fluorescent secondary antibodies. Nuclei were stained blue with Hoechst 33258. Red fluorescence (Alexa Fluor 555) represented the location of the HER2 receptor. Green fluorescence (Alexa Fluor 488) represented the location of CS-4D5 and CS-4D5-6e.
Cell Viability Assays
Cells were seeded at 8000 cells/well in 96-well plates and then incubated in normal medium (24 h) or CSS medium (24 h). Treatments with CS, CS-4D5, 4D5, 6e, or CS-4D5/6e (concentrations were measured in terms of 6e, diluted from 15 μM to 0.47 μM) were carried out with or without 10 nM AD (AKR1C3 substrate) and incubated up to 24 h. Cell viability was determined by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, as described previously.29,30
Inhibition of Testosterone Production by AKR1C3 Inhibitors of 6e and CS-4D5/6e
Testosterone Production by AKR1C3 in LNCaP-AKR1C3 Cell Lysates
LNCaP cells or LNCaP-AKR1C3 cells were seeded into six-well plates at 3×105 cells/2-mL well in RPMI-1640 supplemented with 10% heat-inactivated FBS (Gibco, Grand Island, NY, USA). After overnight incubation at 37°C in an atmosphere of 5% CO2, AD (final concentration, 300 nmol/L) was added to each well. At 24 h after incubation, cell supernatants were collected to measure the testosterone concentration using a radioimmunoassay kit (North Bioengineering Institute, Beijing, China) according to manufacturer instructions.
Inhibition of Testosterone Production by AKR1C3 Inhibitors
Systems (1 mL) containing potassium phosphate (pH 7.0), 300 nM AD, the reduced form of nicotinamide adenine dinucleotide phosphate (NADPH; 2 mM), LNCaP-AKR1C3 cell lysates (10 μg/mL) and different concentrations of 6e and CS-4D5/6e (0, 10, 20, 40, 80 μM, concentration of CS-4D5/6e was measured in terms of 6e) were incubated for 2 h at 37°C. Then, 1 mL of ether (pre-cooled in a 4°C refrigerator) was added and mixed evenly to terminate the reaction. Finally, the ether was dried under nitrogen gas, and the remaining liquid was stored at 4°C until use. The concentration of testosterone was measured with a radioimmunoassay kit.
Inhibition of Testosterone Production in LNCaP-AKR1C3 Cells
LNCaP-AKR1C3 cells digested with 0.1% protease from Streptomyces griseus (Sigma-Aldrich, Saint Louis, MO, USA) were seeded into six-well plates at 2.5×105 cells/2-mL well in RPMI-1640 supplemented with 5% CD-FBS. After overnight incubation in an atmosphere of 5% CO2 at 37°C, 300 nM AD and different concentrations of 6e, CS-4D5, and CS-4D5/6e (0, 0.94, 1.88, 3.75, 7.5, 15 μM, the concentration was measured in terms of 6e) were added to each well. After culturing for 4 h, the medium was collected and the testosterone content in each group measured with a radioimmunoassay kit.
PSA-Expression Assay in LNCaP-AKR1C3 Cells in vitro
LNCaP-AKR1C3 cells were seeded into six-well plates at 1.5×105 cells/2-mL well in CSS medium containing 10% FBS for 24 h. Cells were treated with CS-4D5/6e and cultured for ~48 h. The CS-4D5/6e concentration (in 6e) was 0.16, 0.32, 0.64, 1.25, and 2.5 μM; the blank group was treated with 2 mL of 0.4% CSS medium (n = 3). Cell pellets were lysed in RIPA-1640 (Cell Signaling Technology, Danvers, MA, USA) on ice for 30 min. Then, Western blotting was done to measure expression of PSA and ARs.
In vivo Model for Intracrine Testosterone Synthesis in LNCaP-AKR1C3 Cell Xenografts
Five-week-old male Balb/c athymic nude mice were used to prepare an LNCaP-AKR1C3 xenograft model. LNCaP-AKR1C3 cells in the logarithmic growth phase were collected, resuspended in PBS, and mixed with Matrigel® (356,234; BD Biosciences, Franklin Lakes, NJ, USA) (1:9 v/v) to a concentration of 7.5×107 cells/mL. Next, 200 μL of the cell suspension was inoculated (s.c.) into nude mice.
Approximately 2 weeks after implantation, the tumor volume in nude mice was observed to be 50–100 mm3 using the formula V = 1/2 ab2 (where a is the length and b is the width). Mice carrying LNCaP-AKR1C3 tumors were divided into five groups of six mice. Group A was a control group for background used to determine the intratumoral testosterone concentration (no AD). Group B was a placebo group (AD) treated with vehicle (0.5% methylcellulose). Group C was treated with 6e (10 mg/kg) dissolved in 5% dimethyl sulfoxide (DMSO) + propanediol:ethanol (97:3) (v/v). Group D was the indomethacin group (indomethacin with an intraperitoneal injection dose of 10 mg/kg, dissolved in 5% DMSO + propylene glycol:ethanol at 97:3). Group E was CS-4D5/6e with an intraperitoneal injection of 6e (10 mg/kg) dissolved in 10% DMSO + PBS. Each injection volume was 150 μL, and the injection was once every other day.
Four hours after administration, nude mice were sacrificed. AD was injected intratumorally 1 h before sacrifice at 1 ng/100 mm3 tumor. Tumor tissues were removed and weighed. Next, 200 μL of 200 mM phosphate buffer (pH 7.4) was added to the homogenate, which was centrifuged at 13,000 rpm for 10 min at 4°C; the supernatant was stored at 4°C. Testosterone was extracted using tert-butyl methyl ether. The testosterone concentrations in the reconstituted extracts were determined with a radioimmunoassay kit according to manufacturer instructions.
Plasma samples were obtained from the eye sockets of nude mice before sacrifice and, after 30 min, centrifuged at 3000 rpm for 10 min at 4°C. The plasma supernatants were collected, and the testosterone content in serum in each group was measured with a radioimmunoassay kit.
Antitumor Effect in a 22Rv1 Xenograft Mouse Model
Five-week-old male Balb/c athymic nude mice were used as the 22Rv1 xenograft model. Digested cells were suspended in PBS and then mixed with Matrigel (1:1, v/v) solution to 2.5×107 cells/mL. Approximately 200 μL of the cell suspension was injected (s.c.) into the flank of each mouse. Approximately 2 weeks after implantation, mice carrying 22Rv1 tumors of similar sizes were divided into eight groups of six mice. Group A was the solvent group (5% DMSO + propanediol:ethanol at 97:3). Group B was the physiologic (0.9%) saline group. In group C, mice were administered 6e (2.5 mg/kg) dissolved in 5% DMSO + propanediol:ethanol at 97:3 (v/v). The indomethacin group was group D (indomethacin with an intraperitoneal injection dose of 3 mg/kg, dissolved in 5% DMSO + propylene glycol:ethanol at 97:3). Group E was CS-4D5/6e with an intraperitoneal injection dose of 6e (1 mg/kg) dissolved in 10% DMSO + PBS. Group F was an intraperitoneal injection dose of 6e (2.5 mg/kg) of CS-4D5/6e, dissolved in 10% DMSO + PBS. Group G was an intraperitoneal injection dose of 6e (5 mg/kg) of CS-4D5/6e, dissolved in 10% DMSO + PBS. Group H was the CS-4D5 group (intraperitoneal injection dose of CS-4D5 (50 mg/kg) in PBS). Each injection volume was 100 μL, and injection was carried out once every other day. Mice were observed 14 days after administration. The bodyweight of mice was measured on the next day, and the tumor volume was measured with Vernier calipers at this time.
Surviving mice were sacrificed 24 h after the final treatment. Tumors were collected to carry out histology and Western blotting. The orbital blood of nude mice in each group was collected to determine the testosterone content.
Western Blotting
Cells and tumor tissues were lysed in RIPA-1640 (Cell Signaling Technology) on ice for 30 min. After centrifugation at 12,000 rpm for 20 min at 4°C, proteins were quantified and electrophoresed, as described previously.31 Cell membranes were probed with primary antibodies overnight against AR (rabbit monoclonal antibody (mAb); 5153S; Cell Signaling Technology), PSA (rabbit mAb; 5877S; Cell Signaling Technology), AKR1C3 (rabbit mAb; ab203834; Abcam, Cambridge, UK), HER2 (mouse mAb; bsm-33051M; Bioss) or α-tubulin (rabbit mAb; A01080; Abbkine, Wuhan, China) followed by incubation with anti-mouse (AA75181; Bioworld Technology, Saint Louis Park, MN, USA) or anti-rabbit (AA01191; Bioworld Technology) horseradish peroxidase-conjugated secondary antibody (1:5000 dilution) for 1 h, followed by chemiluminescence detection. All antibodies were diluted to 1:2000. Bands were quantified by densitometry using ImageJ.
Statistical Analyses
Experiments were repeated at least thrice. Data are the mean ± SD. Groups were compared using Prism 6 (GraphPad, La Jolla, CA, USA) by one-way analysis of variance followed by Tukey’s test. P<0.05 was considered significant. The half-maximal inhibitory concentration (IC50) was calculated using Prism 6.
Results
Synthesis and Characterization of CS-4D5/6e
FT-IR spectroscopy was undertaken to confirm combination of CS-4D5 (Figure 1). The spectrum showed that the -NH2 peak of chitosan at 1639 cm−1 shifted to 1649 cm−1 for CS-4C5. This occurred because the unshared electron pair on the nitrogen atoms of chitosan can be p-π-conjugated with the carbonyl group of 4D5, causing a characteristic peak shift. In addition, an in-plane bending vibration peak of CS-4D5 was observed at 1540 cm−1. The series of changes in peak position shown in Figure 1A indicate formation of new amide bonds in CS-4D5, which combined with CS to form CS-4D5. The 4D5 content in CS-4D5 and CS-4D5/6e was 24.9±6.4% and 18.0±3.6%, respectively. The 6e content in CS-4D5/6e was determined quantitatively by HPLC, with drug-loading reaching ≤10%. The mean particle size of CS-4D5/6e was 330.4±22.59 nm, and the zeta potential was −1.39 ± 0.248 mV (Figure 1B). AFM showed that CS-4D5/6e was an oblate spheroid with a particle size of 200–300 nm and thickness of 1–5 nm.
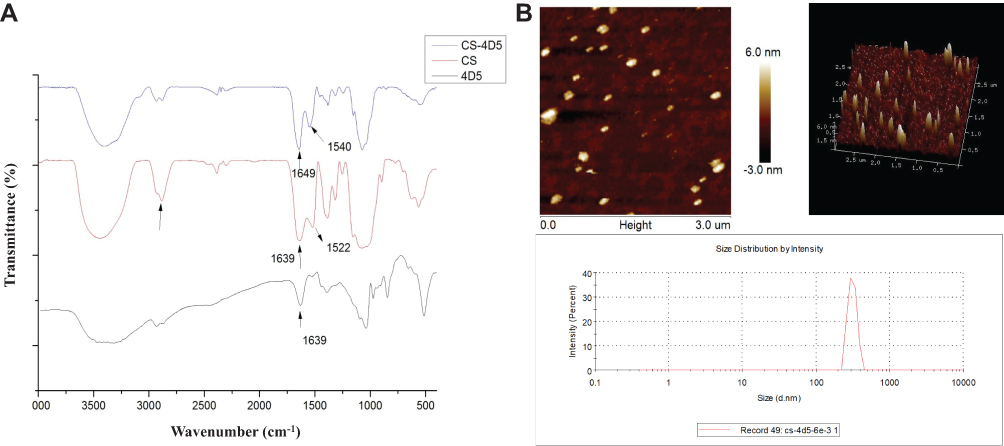 |
Figure 1 Structural characterization of CS-4D5 and CS-4D5/6e. (A) Infrared spectra of CS, 4D5, and CS-4D5. (B) AFM images and Marvin particle size distribution chart of CS-4D5/6e. Abbreviations: CS, chemically chitosan; 4D5, single chain antibody fragment 4D5; 6e, a derivative of mansonone F; AFM, atomic force microscopy. |
HER2 Receptor-Dependent Accumulation of CS-4D5/6e in PC Cells
HER2 shows high expression in CRPC cell lines.32 LNCaP cells with a relatively high HER2 receptor content (Figure 2A) could take in more CS-4D5/6e compared with 22Rv1 cells. According to the location and intensity of green fluorescence (4D5) and red fluorescence (HER2 receptor), 4D5 and CS-4D5/6e showed significant HER2 receptor-dependent accumulation in LNCaP cells, with higher expression of the HER2 receptor than that in 22Rv1 cells because of 4D5 immunoaffinity to HER2 (Figure 2B).
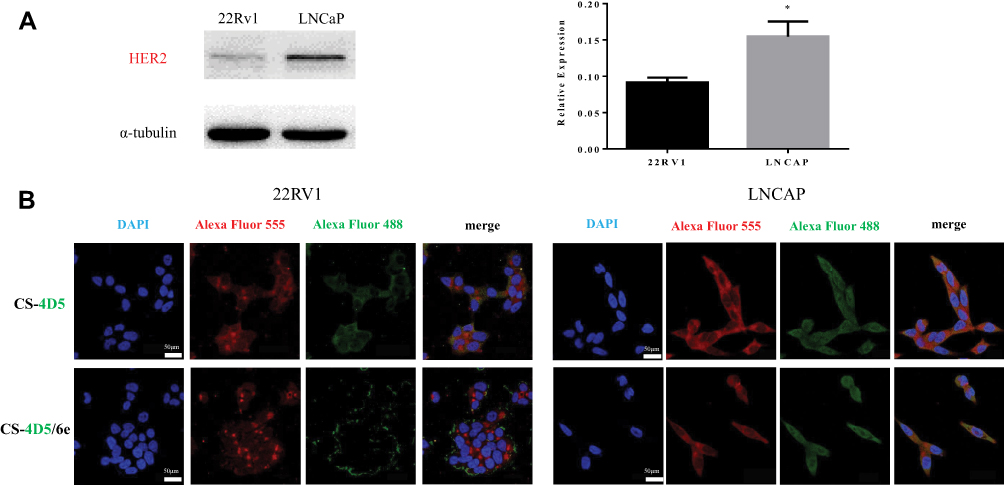 |
Figure 2 Affinity of CS-4D5 and CS-4D5/6e for 22Rv1 and LNCaP cells. (A) Western blot analysis relative expression of HER2 receptor in 22RV1 and LNCaP cells. Data are expressed as the mean SEM (n = 3). *P < 0.05 vs α-tubulin. (B) Immunofluorescence double co-localization assay was used to compare the affinity of CS-4D5 and CS-4D5/6e in 22RV1 and LNCAP cells. Red fluorescence (Alexa Fluor 555) represents the location of the HER2 receptor, whereas green fluorescence (Alexa Fluor 488) represents the location of CS-4D5 and CS-4D5/6e. Abbreviations: HER2, human epidermal growth factor receptor 2; CS, chemically chitosan; 4D5, single chain antibody fragment 4D5; 6e, a derivative of mansonone F; DAPI, 4ʹ,6-diamidino-2-phenylindole; SEM, standard error of mean. |
AKR1C3 Is Required for Conversion of AD to Testosterone
Different concentrations of AD were applied to LNCaP cells and LNCaP-AKR1C3 cells, and the testosterone content in the culture medium was determined after 24 h. LNCaP-AKR1C3 cells could produce more testosterone at the same substrate concentration (Figure 3A and B), further demonstrating the key role of AKR1C3 in testosterone synthesis.
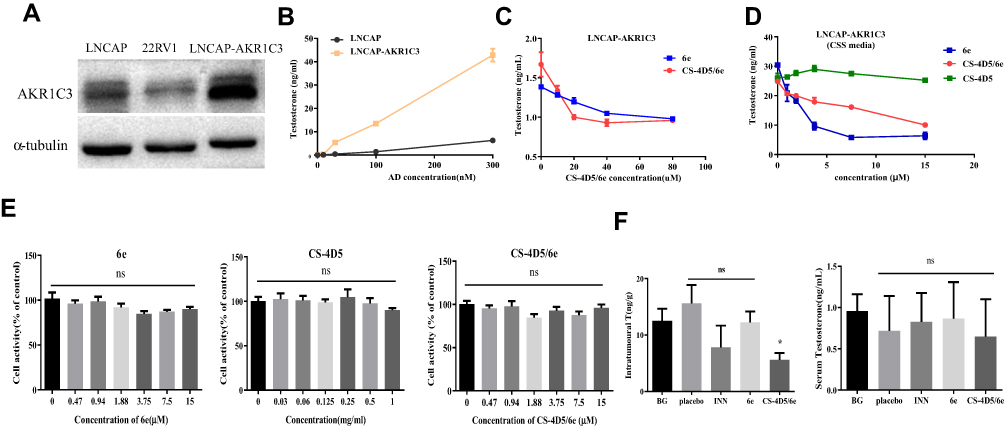 |
Figure 3 CS-4D5/6e inhibits AKR1C3 activity by inhibiting the conversion of AD to testosterone. (A) Western blot analysis relative expression of AKR1C3 receptor in LNCaP, 22RV1, and LNCAP-AKR1C3 cells. (B) Comparison of testosterone formation between LNCaP cells and LNCaP cells stably expressing AKR1C3 (LNCaP-AKR1C3). Cells were treated with AD at various concentrations (range: 10–300 nmol/L). Testosterone concentration in the medium were measured 24 h after administration of AD, respectively. (C) Testosterone production were detected after different concentrations of 6e and CS-4D5/6e were applied to LNCAP-AKR1C3 cells for 4 h. (D) The content of testosterone in the system after different concentrations of 6e (0, 0.47, 0.94, 1.88, 3.75, 7.5, 15 μM) concentration of CS-4D5/6e was measured in terms of 6e, CS-4D5, and CS-4D5/6e were incubated with LNCaP-AKR1C3 cell lysates in vitro for 2 h (concentration of CS-4D5 and CS-4D5/6e was measured in terms of 6e). (E) MTT assay for cytotoxicity of different concentrations of 6e, CS-4D5, and CS-4D5/6e. Cell viability was measured by MTT at 4 h after adding different concentrations of 6e (0, 0.47, 0.94, 1.88, 3.75, 7.5, 15 μM), CS-4D5, and CS-4D5/6e. Concentration of CS-4D5 and CS-4D5/6e was measured in terms of 6e. Data are expressed as the mean ± SD (n = 6, ns means no significant difference vs control, P > 0.05). (F) Mice were sacrificed at 2 h after administration of a single oral dose of INN, 6e, or CS-4D5/6e (10 mg/kg), left shows serum testosterone levels and right shows prostate tumour testosterone levels (n = 6, means ± SD, *P < 0.05, ns means no significant difference vs BG, P > 0.05). Abbreviations: AKR1C3, aldo-ketoreductase1C3; AD, androgen; CS, chemically chitosan; 4D5, single chain antibody fragment 4D5; 6e, a derivative of mansonone F; CSS, charcoal-stripped serum; MTT, methyl thiazolyl tetrazolium; BG, background group; INN, indomethacin; T, testosterone; SD, standard deviation. |
Antitumor Activity of CS-4D5/6e in vitro
The MTT assay was used to investigate the effects of CS-4D5/6e on the proliferation of LNCaP cells and LNCaP-AKR1C3 cells under different culture conditions. The IC50 of CS-4D5/6e was 2.34±0.48 μM in CSS medium for LNCaP-AKR1C3 cells (Figure 4A). The IC50 in normal medium was 7.29±0.31 μM and, when 10 nM AD was added to the CSS medium, the IC50 was 4.45±0.53 μM (Figure 4B). This result was because AKR1C3 induced AD to produce testosterone and attenuated the inhibition of tumor-cell growth by CS-4D5/6e. The IC50 of 6e for LNCaP-AKR1C3 cells was also lower than that of LNCaP, confirming the selectivity of 6e for AKR1C3 (Figure 4C-D, Table 1). These results demonstrated that CS-4D5/6e was AKR1C3-dependent and that AKR1C3 was a pharmacodynamic target of CS-4D5/6e.
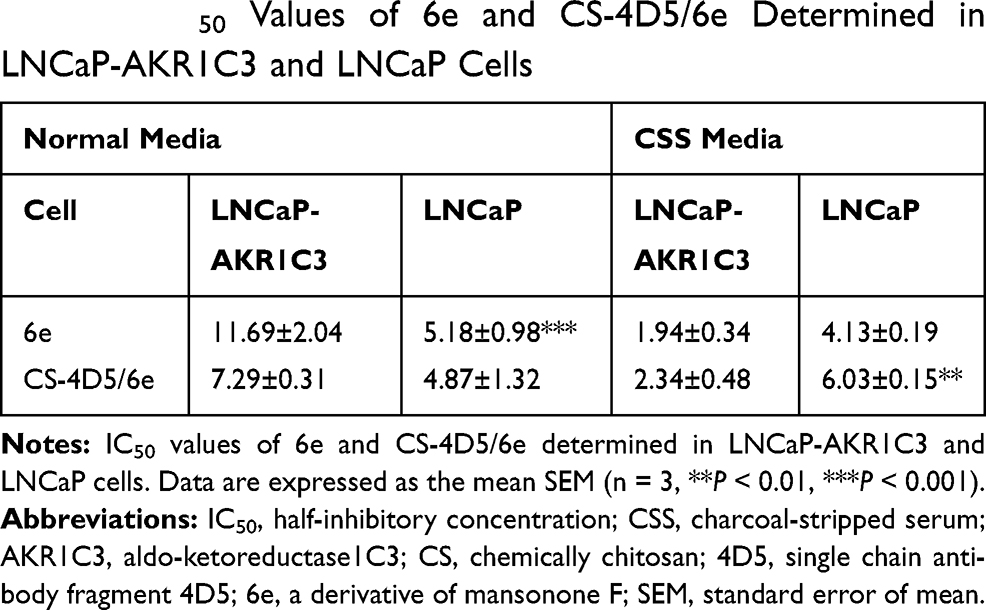 |
Table 1 IC50 Values of 6e and CS-4D5/6e Determined in LNCaP-AKR1C3 and LNCaP Cells |
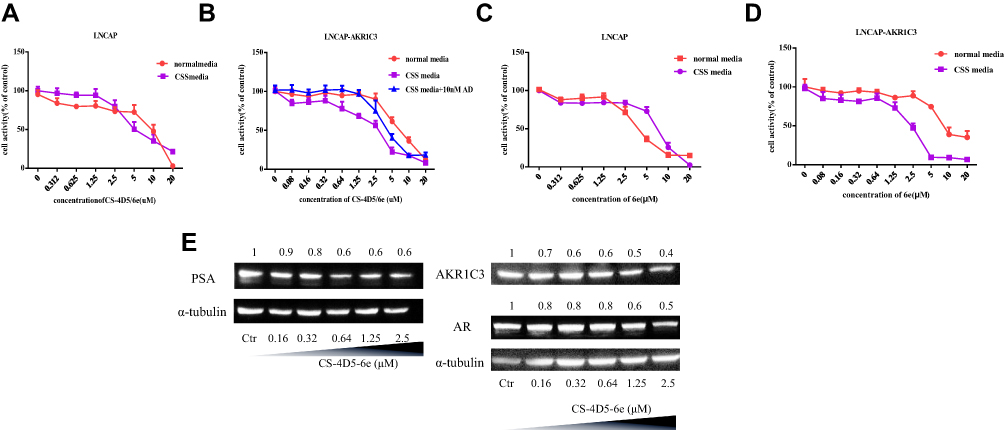 |
Figure 4 CS-4D5/6e targets AKR1C3, and overexpression of AKR1C3 enhances the cytotoxic effect of CS-4D5/6e on cells. Percentage cell viability of (A) LNCAP cells cultured in normal and CSS media, (B) LNCaP-AKR1C3 cells cultured in normal, CSS, and CSS media supplemented with 10 nM Δ4-androstenedione after CS-4D5/6e treatment for 24 h. Percentage cell viability of (C) LNCAP cells cultured in normal and CSS media, (D) LNCaP-AKR1C3 cells cultured in normal, CSS, and CSS media supplemented with 10 nM Δ4-androstenedione after 6e treatment for 24 h. (E) Western blot analysis the expression of AKR1C3, PSA, and AR after LNCAP-AKR1C3 cells treated with CS-4D5/6e. Treatment with CS-4D5/6e at indicated concentration for 24 h in LNCaP-AKR1C3 cells cultured in CSS media. Abbreviations: CS, chemically chitosan; 4D5, single chain antibody fragment 4D5; 6e, a derivative of mansonone F; AD, androgen; CSS, charcoal-stripped serum; PSA, prostate specific antigen; AKR1C3, aldo-ketoreductase1C3; AR, androgen receptor; Ctr, control. |
CS-4D5/6e Inhibits AKR1C3 Activity in PC Cell Lines by Inhibiting the Conversion of AD to Testosterone
In the enzymatic reaction system composed of cell lysates of LNCaP-AKR1C3 in vitro, 6e and CS-4D5/6e at different concentrations inhibited testosterone formation in a concentration-dependent manner and finally stabilized (Figure 3B). This observation indicated that CS-4D5/6e blocked testosterone production by inhibiting AKR1C3 activity.
Further information was obtained by treating LNCaP-AKR1C3 cells with 6e, CS-4D5, or CS-4D5/6e for 4 h. CS-4D5/6e inhibited testosterone production in LNCaP-AKR1C3 cells significantly (Figure 3C), whereas CS-4D5 had no effect (Figure 3D). To avoid the reduction in testosterone content because of drug cytotoxicity, we conducted a 4-h cytotoxicity test at the corresponding concentration (Figure 3E). The activity of LNCaP-AKR1C3 cells was not affected by CS-4D5 or CS-4D5/6e within the time period and concentration range tested, indicating that the decrease in testosterone concentration was caused by inhibition of AKR1C3 expression.
Molecular Changes Following CS-4D5/6e Treatment in PC Cells
PSA and AR are key factors in androgen signaling. A low concentration of CS-4D5/6e without toxicity was applied to LNCaP-AKR1C3 cells for 24 h, followed by Western blotting to measure protein expression. Expression of AKR1C3, PSA, and ARs in LNCaP-AKR1C3 cells showed a concentration-dependent decrease (Figure 4E).
CS-4D5/6e Inhibits Intratumoral Testosterone Production in Mice Bearing LNCaP-AKR1C3 Xenografts
To ascertain if CS-4D5/6e inhibits intratumoral testosterone production, mice bearing LNCaP-AKR1C3 xenografts were used. Four hours after single administration, the serum testosterone content in the five groups was measured (Figure 3F). There was no significant difference in serum testosterone content between groups (P>0.05 for all). Compared with the blank group, CS-4D5/6e inhibited testosterone production in LNCaP-AKR1C3 xenograft tumors significantly (P < 0.05). This finding suggested that CS-4D5/6e did not reduce the testosterone concentration in the general blood circulation but instead acted on AKR1C3 in tumors to inhibit testosterone production.
Antitumor Effect of CS-4D5/6e in a 22Rv1 Xenograft Model
The rate of tumor growth in the vehicle group and physiologic-saline group was relatively fast, and that in the CS-4D5/6e group was significantly slower, than that in the vehicle group and PBS group (Figure 5A). CS-4D5/6e reduced the growth of 22Rv1 tumor xenografts by 57.00% (dose: 2.5 mg of 6e per kg) and 64.36% (dose: 5 mg of 6e per kg) significantly compared with that in the vehicle group (P<0.01). On day-14 of treatment, the 2.5 and 5 mg/kg groups of CS-4D5/6e exhibited significant inhibition of tumor growth when compared with the physiologic-saline and solvent groups (P<0.01). After mice had been sacrificed, the mean tumor-bearing volume (in mm3) of each group was 486.33±23.87 for indomethacin, 419.11±44.02 for 6e, 514.60±36.54 for 1 mg/kg CS-4D5/6e, 344.56±45.34 for 2.5 mg/kg CS-4D5/6e, 318.33±35.34 for 5 mg/kg CS-4D5/6e, 433.00±44.68 for CS-4D5, 638.33±34.11 for physiologic saline, and 632.5±96.84 for vehicle, respectively. Figure S1 shows that there was no significant difference in the mean body weight between the eight groups (P>0.05). Figure S2 shows no significant morphological changes in the vehicle and drug-treated nude mice.
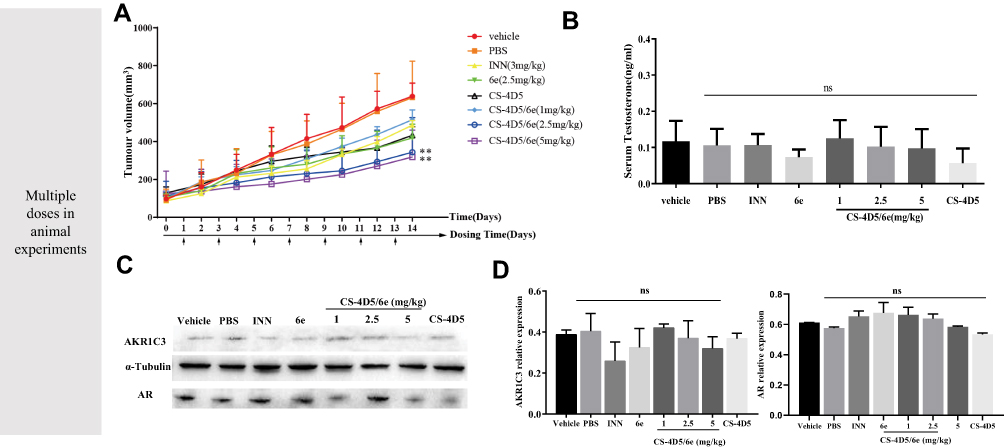 |
Figure 5 Notes: Antitumor effect of CS-4D5/6e in a 22Rv1 xenograft model. (A) Reduction in prostate tumour volume in each group for 14 days after administration. (n = 6, means ± SD, **P < 0.01). (B) Serum testosterone levels of nude mice in each group for 14 days after administration. (C) Comparison of AKR1C3 expression levels in 22Rv1 xenografted nude mice by Western blot analysis, using rabbit monoclonal anti-AKR1C3 antibody and α-tubulin as a loading control. The dose of INN was 3 mg/kg and that of 6e was 2.5 mg/kg. (D) Western blot and analysed by greyscale scanning AKR1C3 (n = 6, mean ± SD, ns: no significant difference vs vehicle, P > 0.05). Abbreviations: PBS, phosphate buffered saline; INN, indomethacin; CS, chemically chitosan; 4D5, single chain antibody fragment 4D5; 6e, a derivative of mansonone F; AR, androgen receptor; AKR1C3, aldo-ketoreductase1C3; SD, standard deviation. |
Fourteen days after drug administration, the serum testosterone content in the eight groups was measured (Figure 5B). There was no significant difference in serum testosterone content between groups (P>0.05 for all). Hence, CS-4D5/6e did not reduce the testosterone content in the blood circulation but instead acted on AKR1C3 in tumor tissues to inhibit testosterone production.
Figure 5C and D shows that expression of AKR1C3 and ARs in the CS-4D5/6e group was decreased slightly with increasing doses, but there was no significant difference between the eight groups (P>0.05 for all).
Discussion
Biocompatible multifunctional nanomedicines have emerged as attractive candidates for targeted theranostics, and have advantageous compared with molecular conjugates.24 The most important advantage is that the nanomedicine surface can be functionalized with one or more biological ligands having specific affinity to receptors in tumors.
To target CRPC cells efficiently and accumulate in tumors, we synthesized CS-4D5 through an amidation reaction. 4D5 showed good retention during preparation of CS-4D5 or CS-4D5/6e, and the latter was an oblate spheroid of size 200–300 nm. For nanomedicines, tumoral uptake is tightly correlated to particle size. Nanoparticles of diameter 100–200 nm have been shown to possess a fourfold higher rate of uptake in tumors compared with particles >300 nm or <50 nm in diameter.33 The size of CS-4D5/6e was slightly larger than expected, so we needed to improve the particle size and zeta potential to create our nanodrug-delivery system.
CS-4D5/6e uptake by LNCaP cells and 22Rv1 cells was investigated by immunofluorescence co-localization. Compared with 22Rv1 cells, the red fluorescence of HER2 antibody in LNCaP cells was stronger, which was consistent with the results of Western blotting. When both cell types were treated with CS-4D5 or CS-4D5/6e, we observed stronger green fluorescence labeling of 4D5 in LNCaP cells than that in 22Rv1 cells, indicating that 4D5 combined with HER2 and mediated 6e uptake into cells.
AKR1C3 plays a key part in CRPC development. To investigate the effect of CS-4D5/6e on AKR1C3, LNCaP cells overexpressing AKR1C3 (LNCaP-AKR1C3) were constructed. The AKR1C3 content in LNCaP-AKR1C3 cells was significantly higher than that in LNCaP cells at gene and protein levels (Figure 3A), which indicated construction of LNCaP-AKR1C3 cells. To investigate the effect of CS-4D5/6e on testosterone production, we constructed an enzymatic reaction system in vitro. Total protein in LNCaP-AKR1C3 cell lysates was used as the enzyme source to investigate the effect of different concentrations of CS-4D5/6e on testosterone synthesis. CS-4D5/6e and 6e inhibited testosterone formation in the in vitro enzymatic reaction system (Figure 3D). To further illustrate the inhibitory effect of CS-4D5/6e on testosterone synthesis, testosterone content was measured after different concentrations of CS-4D5/6e had been applied to LNCaP-AKR1C3 cells for 4 h. Treatment with CS-4D5 or CS-4D5/6e in LNCaP-AKR1C3 cells did not alter cell survival significantly.
Studies have shown that hormones in the culture medium can affect the inhibitory effect of a drug on AKR1C3, thereby impacting the IC50.34 Therefore, we investigated the toxicity of CS-4D5/6e on LNCaP cells and LNCaP-AKR1C3 cells in different media. As the substrate, AD (10 nM) was used to mimic the serum level of androgens in patients with CRPC.35–39 The IC50 of LNCaP-AKR1C3 increased after AD addition. This finding suggested that the decreased cytotoxicity of CS-4D5/6e was due to the formation of DHT and testosterone, and confirmed that AKR1C3 was the target of the inhibitor. In addition, we measured expression of androgen-dependent genes after CS-4D5/6e treatment and observed that levels of AKR1C3, ARs, and PSA were reduced in a concentration-dependent manner. These results suggest that CS-4D5/6 downregulated AKR1C3 expression and reduced expression of ARs and PSA. At the molecular level, the inhibitory effect of CS-4D5/6e on PC was demonstrated. To further investigate testosterone production in plasma and tumor tissue, a nude mouse model bearing LNCaP-AKR1C3 tumors were created. Single administration of CS-4D5/6e caused no significant change in the plasma testosterone concentration between groups (P>0.05) but there was a significant change in the testosterone concentration in the tumor tissues of the CS-4D5/6e group compared with that in the placebo group (P<0.05).
Numerous studies have shown that HER2 can activate ARs during androgen deficiency.40 In addition, epidermal growth factor receptor/HER2 expression is increased with the progression of PC to CRPC. Thus, we prepared a mouse model of CRPC with high expression of HER2. However, few mice carrying LNCaP-AKR1C3 cells were prepared because of the weak tumorigenicity of this model. Therefore, we used castration-resistant PC cells (22Rv1) for tumor-bearing experiments in nude mice with continuous multiple dosing for 14 days. 22Rv1 cells also express the HER2 receptor. Mice bearing LNCaP-AKR1C3 xenografts were used to demonstrate that CS-4D5/6e inhibited intratumoral testosterone production after single administration. CS-4D5/6e inhibited tumor growth effectively compared with that in the solvent group (Figure 5A). Interestingly, CS-4D5/6e exhibited stronger inhibitory effects compared with 6e, which may have been due to 4D5 targeting HER2 and, therefore, increasing 6e accumulation in the tumor. Compared with indomethacin (a selective inhibitor of AKR1C3), CS-4D5/6e inhibited tumor growth more strongly. At the end of experimentation, detailed dissection of mice was undertaken. The liver, spleen, kidneys, heart and lungs were examined using H&E staining. We did not observe significant morphologic changes in vehicle- and drug-treated nude mice. During experimentation, the fluctuation in body weight was normal. CS-4D5/6e was relatively safe within the experimental doses tested (Figure S2).
Conclusions
We demonstrated the antineoplastic activity of a potent AKR1C3 inhibitor (6e) and its nanodrug-delivery system (CS-4D5/6e). First, CS-4D5/6e targeted HER2-positive CRPC cells. Second, it transferred 6e (a AKR1C3 inhibitor) to achieve a reduction in testosterone production within the tumor. Compared with 6e, CS-4D5/6e showed lower systemic toxicity. CS-4D5/6e inhibited tumor growth effectively in nude mice implanted with tumor xenografts by downregulating testosterone production mediated by AKR1C3 in the tumor. These results showed a promising strategy for treatment of the CRPC that develops invariably in PC patients.
Abbreviations
PCa, prostate cancer; ADT, androgen-deprivation therapy; AR, androgen receptor; AKR1C3, Aldo-ketoreductase1C3; T, testosterone; 5α-DHT, 5α-dihydrotestosterone HER2, human epidermal growth factor receptor 2; CS, chemically chitosan; AFM, atomic force microscopy; FBS, foetal bovine serum; CSS, charcoal-stripped serum; FT-IR, Furier transform infrared spectroscopy; CD, charcoal-dextran; MTT, methyl thiazolyl tetrazolium; AD, androgen; NADPH, nicotinamide adenine dinucleotide phosphate; PSA, prostate-specific antigen; INN, Indomethacin; PBS, phosphate-buffered saline; DMSO, dimethyl sulfoxide; EDC·HCL, 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride; NHS, N-Hydroxy succinimide; DAPI, 4ʹ,6-diamidino-2-phenylindole.
Acknowledgment
The authors thank Dr. Qihao Zhang, Dr. Zhijian Su, and Dr. Yan Yang for their support.
Funding
This work was supported by the Major Scientific and Technological Special Project of the Administration of Ocean and Fisheries of Guangdong Province [grant number GDME-2018C013, Yuecainong, 2017, no. 17], Guangzhou Science and Technology Program Key Projects [grant number 201803010044], Science and Technology Program of Tianhe District, Guangzhou City [grant number 201704YG066], and the National Natural Science Project [grant number 21272288].
Disclosure
The authors declare no conflicts of interest.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7–34. doi:10.3322/caac.21551
2. Crawford ED, Heidenreich A, Lawrentschuk N, et al. Androgen-targeted therapy in men with prostate cancer: evolving practice and future considerations. Prostate Cancer Prostatic Dis. 2019;22(1):24–38. doi:10.1038/s41391-018-0079-0
3. Lowrance WT, Murad MH, Oh WK, Jarrard DF, Resnick MJ, Cookson MS. Castration-resistant prostate cancer: AUA guideline amendment 2018. J Urol. 2018;200(6):1264–1272. doi:10.1016/j.juro.2018.07.090
4. Yuan X, Cai C, Chen S, Chen S, Yu Z, Balk SP. Androgen receptor functions in castration-resistant prostate cancer and mechanisms of resistance to new agents targeting the androgen axis. Oncogene. 2014;33(22):2815–2825. doi:10.1038/onc.2013.235
5. Massard C, Fizazi K. Targeting continued androgen receptor signaling in prostate cancer. Clin Cancer Res. 2011;17(12):3876–3883. doi:10.1158/1078-0432.CCR-10-2815
6. Buchanan G. Collocation of androgen receptor gene mutations in prostate cancer. Clin Cancer Res. 2001;7(5):1273–1281.
7. Lapouge G, Marcias G, Erdmann E, et al. Specific properties of a C-terminal truncated androgen receptor detected in hormone refractory prostate cancer. Advan Exp Med Biol. 2008;617(01):529–534.
8. Chandrasekar T, Yang JC, Gao AC, Evans CP. Mechanisms of resistance in castration-resistant prostate cancer (CRPC). Transl Androl Urol. 2015;4(3):365–380. doi:10.3978/j.issn.2223-4683.2015.05.02
9. Miyazaki Y, Teramoto Y, Shibuya S, et al. Consecutive prostate cancer specimens revealed increased Aldo(-) keto reductase family 1 member C3 expression with progression to castration-resistant prostate cancer. J Clin Med. 2019;8:5. doi:10.3390/jcm8050601
10. Adeniji AO, Chen M, Penning TM. AKR1C3 as a target in castrate resistant prostate cancer. J Steroid Biochem Mol Biol. 2013;137(18):136–149. doi:10.1016/j.jsbmb.2013.05.012
11. Hofland J, van Weerden WM, Dits NF, et al. Evidence of limited contributions for intratumoral steroidogenesis in prostate cancer. Cancer Res. 2010;70(3):1256–1264. doi:10.1158/0008-5472.CAN-09-2092
12. Jernberg E, Thysell E, Bovinder Ylitalo E, et al. Characterization of prostate cancer bone metastases according to expression levels of steroidogenic enzymes and androgen receptor splice variants. PLoS One. 2013;8(11):e77407. doi:10.1371/journal.pone.0077407
13. Mitsiades N, Sung CC, Schultz N, et al. Distinct patterns of dysregulated expression of enzymes involved in androgen synthesis and metabolism in metastatic prostate cancer tumors. Cancer Res. 2012;72(23):6142–6152. doi:10.1158/0008-5472.CAN-12-1335
14. Penning TM. AKR1C3 (type 5 17beta-hydroxysteroid dehydrogenase/prostaglandin F synthase): roles in malignancy and endocrine disorders. Mol Cell Endocrinol. 2019;489:82–91. doi:10.1016/j.mce.2018.07.002
15. Zhao J, Zhang M, Liu J, et al. AKR1C3 expression in primary lesion rebiopsy at the time of metastatic castration-resistant prostate cancer is strongly associated with poor efficacy of abiraterone as a first-line therapy. Prostate. 2019;79(13):1553–1562. doi:10.1002/pros.23875
16. Liu C, Armstrong CM, Lou W, Lombard A, Evans CP, Gao AC. Inhibition of AKR1C3 activation overcomes resistance to abiraterone in advanced prostate cancer. Mol Cancer Ther. 2017;16(1):35–44. doi:10.1158/1535-7163.MCT-16-0186
17. Kshitij V, Nehal G, Tianzhu Z, et al. AKR1C3 inhibitor KV-37 exhibits antineoplastic effects and potentiates enzalutamide in combination therapy in prostate adenocarcinoma cells. mol Cancer Ther. 2017;17(9):1833–45.
18. Wu WB, Ou J-B, Huang Z-H, et al. Synthesis and evaluation of mansonone F derivatives as topoisomerase inhibitors. Eur J Med Chem. 2011;46(8):3339–3347. doi:10.1016/j.ejmech.2011.04.059
19. Wang D, Xia M, Cui Z, Tashiro S-I, Onodera S, Ikejima T. Cytotoxic effects of mansonone E and F isolated from ulmus pumila. Biol Pharm Bull. 2004;27(7):1025–1030. doi:10.1248/bpb.27.1025
20. Jathal MK, Steele TM, Siddiqui S, et al. Dacomitinib, but not lapatinib, suppressed progression in castration-resistant prostate cancer models by preventing HER2 increase. Br J Cancer. 2019;121(3):237–248. doi:10.1038/s41416-019-0496-4
21. Muniyan S, Chen SJ, Lin FF, et al. ErbB-2 signaling plays a critical role in regulating androgen-sensitive and castration-resistant androgen receptor-positive prostate cancer cells. Cell Signal. 2015;27(11):2261–2271. doi:10.1016/j.cellsig.2015.08.002
22. Ivanova JL, Edelweiss EF, Leonova OG, Balandin TG, Popenko VI, Biochimie SMDJ. Application of fusion protein 4D5 scFv-dibarnase:barstar-gold complex for studying P185HER2 receptor distribution in human cancer cells. Biochimie. 2012;94(8):1833–1836.
23. Lv X, Zhang J, Xu R, et al. Gigantoxin-4-4D5 scFv is a novel recombinant immunotoxin with specific toxicity against HER2/neu-positive ovarian carcinoma cells. Appl Microbiol Biotechnol 100(14):6403–6413.
24. Herve-Aubert K, Allard-Vannier E, Joubert N, et al. Impact of site-specific conjugation of ScFv to multifunctional nanomedicines using second generation maleimide. Bioconjug Chem. 2018;29(5):1553–1559. doi:10.1021/acs.bioconjchem.8b00091
25. Silvia S, Alessio L, Agnese S, et al. Highly efficient production of anti-HER2 scFv antibody variant for targeting breast cancer cells. Appl Microbiol Biotechnol. 2011;91(3):613–621. doi:10.1007/s00253-011-3306-3
26. Zhang E, Xing R, Liu S, Qin Y, Li K, Li P. Advances in chitosan-based nanoparticles for oncotherapy. Carbohydr Polym. 2019;222:115004. doi:10.1016/j.carbpol.2019.115004
27. Kaipainen A, Zhang A, Gil da Costa RM, et al. Testosterone accumulation in prostate cancer cells is enhanced by facilitated diffusion. Prostate. 2019;79(13):1530–1542. doi:10.1002/pros.23874
28. Wang B, Gu Y, Hui K, et al. AKR1C3, a crucial androgenic enzyme in prostate cancer, promotes epithelial-mesenchymal transition and metastasis through activating ERK signaling. Urol Oncol. 2018;36(10):472e411–472 e420. doi:10.1016/j.urolonc.2018.07.005
29. Kumar P, Nagarajan A, Uchil PD. Analysis of cell viability by the MTT assay. Cold Spring Harb Protoc. 2018;2018:6.
30. Wako K, Kawasaki T, Yamana K, et al. Expression of androgen receptor through androgen-converting enzymes is associated with biological aggressiveness in prostate cancer. J Clin Pathol. 2008;61(4):448–454. doi:10.1136/jcp.2007.050906
31. Tobrain E. Penfluridol: an antipsychotic agent suppresses metastatic tumor growth in triple-negative breast cancer by inhibiting integrin signaling axis. Cancer Res. 2016;76(4):877–890. doi:10.1158/0008-5472.CAN-15-1233
32. Tai W, Mahato R, Cheng K. The role of HER2 in cancer therapy and targeted drug delivery. J Controlled Release off J Controlled Release Soc. 2010;146(3):264–275. doi:10.1016/j.jconrel.2010.04.009
33. Li SD, Huang L. Pharmacokinetics and biodistribution of nanoparticles. Mol Pharm. 2008;5(4):496–504. doi:10.1021/mp800049w
34. Verma K, Gupta N, Zang T, et al. AKR1C3 inhibitor KV-37 exhibits antineoplastic effects and potentiates enzalutamide in combination therapy in prostate adenocarcinoma cells. Mol Cancer Ther. 2018;17(9):
35. Byrns MC, Mindnich R, Duan L, Penning TM. Overexpression of aldo-keto reductase 1C3 (AKR1C3) in LNCaP cells diverts androgen metabolism towards testosterone resulting in resistance to the 5alpha-reductase inhibitor finasteride. J Steroid Biochem Mol Biol. 2012;130(1–2):7–15. doi:10.1016/j.jsbmb.2011.12.012
36. Mowday AM, Ashoorzadeh A, Williams EM, et al. Rational design of an AKR1C3-resistant analog of PR-104 for enzyme-prodrug therapy. Biochem Pharmacol. 2016;116:176–187. doi:10.1016/j.bcp.2016.07.015
37. Adeniji A, Uddin MJ, Zang T, et al. Discovery of (R)-2-(6-Methoxynaphthalen-2-yl) butanoic acid as a potent and selective aldo-keto reductase 1C3 inhibitor. J Med Chem. 2016;59(16):7431–7444. doi:10.1021/acs.jmedchem.6b00160
38. Gregory CW, Fei X, Ponguta LA, et al. Epidermal growth factor increases coactivation of the androgen receptor in recurrent prostate cancer. J Biol Chem. 2004;279(8):7119–7130. doi:10.1074/jbc.M307649200
39. Gregory CW, Whang YE, McCall W, et al. Heregulin-induced activation of HER2 and HER3 increases androgen receptor transactivation and CWR-R1 human recurrent prostate cancer cell growth. Clin Cancer Res. 2005;11(5):1704–1712. doi:10.1158/1078-0432.CCR-04-1158
40. He L, Du Z, Xiong X, et al. Targeting androgen receptor in treating HER2 positive breast cancer. Sci Rep. 2017;7(1):14584. doi:10.1038/s41598-017-14607-2
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.