")
Back to Journals » International Journal of General Medicine » Volume 14
A KRT6A and a Novel KRT16 Gene Mutations in Chinese Patients with Pachyonychia Congenita
Authors Gong L, Guo S, Wang D, Wang T, Ren X, Yuan Y , Cui H
Received 9 September 2020
Accepted for publication 16 February 2021
Published 17 March 2021 Volume 2021:14 Pages 903—907
DOI https://doi.org/10.2147/IJGM.S280160
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Li Gong,1,2 Shuping Guo,1 Detong Wang,3 Ting Wang,4 Xiaoli Ren,1 Yuting Yuan,1,2 Hongzhou Cui1
1Department of Dermatology, First Hospital of Shanxi Medical University, Taiyuan, People’s Republic of China; 2The First Clinical Medical College of Shanxi Medical University, Taiyuan, People’s Republic of China; 3Tonghua Hospital of Traditional Chinese Medicine, Tonghua, People’s Republic of China; 4Department of Dermatology, Shanxi Hospital of Integrated Traditional and Western Integrated Medicine, Taiyuan, People’s Republic of China
Correspondence: Hongzhou Cui
Department of Dermatology, First Hospital of Shanxi Medical University, No. 85 Jiefang South Road, Taiyuan, Shanxi, 030001, People’s Republic of China
Email [email protected]
Background: Pachyonychia congenita (PC) is a rare, autosomal dominant genodermatosis characterized by palmoplantar keratoderma, nail dystrophy, cystic lesions, follicular hyperkeratosis, mucosal leukokeratoses, hyperhidrosis, hoarseness, and, rarely, natal teeth. Five keratin genes, KRT6A, KRT6B, KRT6C, KRT16 and KRT17, have been found to be associated with PC.
Methods: Using polymerase chain reaction and Sanger sequencing techniques, the purpose of the present study was to investigate the clinical features associated with PC and discover disease-associated variants. The KRT6A, KRT16, KRT17, and KRT6B exonic and flanking region sequences were amplified and directly sequenced to detect mutations.
Results: Across two independent instances of PC, we identified a previously reported c.1393T>C (p.Tyr465His) mutation in exon 7 of KRT6A, and a novel c.1237G>C (p.Glu413Gln) heterozygous missense mutation in exon 6 of the KRT16 gene.
Conclusion: Through phenotype-genotype analysis among PC pedigrees, confirmed diagnoses of PC-K6a and PC-K16 were made in the two patients who presented with symptoms of PC. A new pathogenic mutation site in PC-K16 was potentially discovered.
Keywords: KRT6A gene, KRT16 gene, pachyonychia congenital, phenotype-genotype
Introduction
Characterized by palmoplantar keratoderma, nail dystrophy, cystic lesions, follicular hyperkeratosis, mucosal leukokeratoses, hyperhidrosis, hoarseness, and, rarely, natal teeth, pachyonychia congenita (PC) is a rare, autosomal dominant genodermatosis.1,2 PC has historically been classified into two types, namely, Jadassohn-Lewandowski syndrome (type 1 PC) and Jackson-Lawler syndrome (type 2 PC).4,5 While it is estimated that there are 5000–10000 PC patients worldwide,8 no correlative evidence supportive of any association with ethnicity has been observed. At present, 115 distinct mutations have been identified in different keratin genes and are reflected in the International Pachyonychia Congenita Research Registry (IPCRR; http://registry.pachyonychia org/s3/IPCR). Most of these variants are missense mutations or small in-frame insertion or deletions.9 Among these, KRT6A, KRT16, KRT17, KRT6B, and KRT6C mutations have been reported in 39%, 33%, 16%, 9%, and 3% of cases, respectively.
Case Series
Subjects
The cases of two unrelated northern Chinese PC families are presented here. In Family A, the proband was a 13-year-old male presenting with severely thickened, dystrophic nails with dark coloration on all nails since birth (Figure 1A). Oral leukokeratosis was mainly observed on the buccal mucosa (Figure 1B). In Family B, a 26-year-old male presented with varying degrees of thickening and discoloration of all the nails that had been present from two years of age (Figure 1C). This was followed by palmar hyperhidrosis (Figure 1D) and focal plantar keratoderma. The latter sometimes manifested as plantar pain when walking (Figure 1E). Both probands had been diagnosed with PC-1. Consistent with an autosomal dominant inheritance pattern, some family members of each PC pedigree presented with similar symptoms.
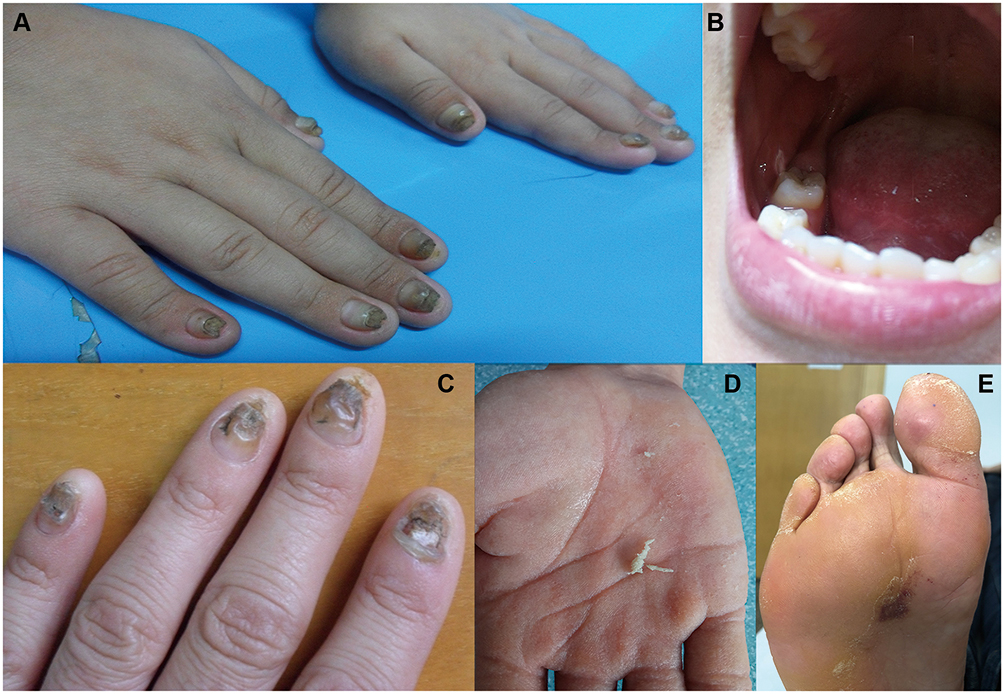 |
Figure 1 Clinical manifestations of patients. (A) Thickened and dark fingernails. (B) Oral leukokeratosis. (C) Thickened and discolored fingernails. (D, E) Palmar hyperhidrosis and palmoplantar keratoderma. |
All of participants signed the informed consent. This study was approved by the Ethics Committee at the Shanxi Medical University. The principles of the 1964 Helsinki declaration and its later amendments were followed. Genomic DNA (QIAGEN, Germany) was extracted from peripheral blood from all available family members and 100 unrelated healthy controls. The exonic and flanking region sequences of the KRT6A, KRT16, KRT17, KRT6B, and KRT6C genes were amplified through PCR and Sanger sequencing. The primer sequences were synthesized by Shanghai Sangon Biotech Co., Ltd., Shanghai, China and are summarily included in Supplementary Table 1.
In Family A, a previously reported c.1393T>C (p.Tyr465His) mutation in exon 7 of KRT6A10, (Supplementary Figure 1A, was identified. In Family B, a novel c.1237G>C heterozygous missense mutation, which produces a transition from Glu at position 413 to Gln in the expressed protein, was identified in exon 6 of the KRT16 gene (Supplementary Figure 1B). No mutations were detected when DNA sequencing was extended to the parental and control samples.
Discussion
PC encompasses a group of rare inherited ectodermal dysplasia disorders. Three of the most common clinical features associated with PC cases are thickened toenails, plantar keratoderma (mostly focal), and plantar pain. There is considerable overlap between PC1 and PC211–13 In 2010, a new variant classification was proposed at the International Pachyonychia Congenita Consortium (IPCC) Symposium. This included PC-6a, PC-6b, PC-6c, PC-16, and PC-17 for a patient with mutations in the KRT6A, KRT6B, KRT6C, KRT16, and KRT17 keratin genes.14 As of October 2020, 383 and 319 patients registered in the IPCRR had confirmed KRT6A and KRT16 mutations, respectively. While plantar keratoderma was common between PC-K6a and PC-K16 cases, a high number of fingernails/toenails and oral leukokeratosis were significantly associated with PC-K6a. Comparatively, palmar keratoderma was common with PC-K16. The demographics and distinguishing clinical symptoms among PC-6a and PC-16 cases are described in Supplementary Table 2. The conclusion from our statistical analysis was consistent with a recently published large cohort PC study.15
The c.1393T>C (p.Tyr465His) KRT6A mutation found in family A was first reported in a Chinese pedigree in 2008.16 According to the American College of Medical Genetics and Genomics (ACMG; http://acmg.cbgc.org), the p.Tyr465His substitution was predicted as being probably damaging. Similarly, the variant was described as being probably damaging with a polyphen2 score of 1.000 (sensitivity 0.00; specificity 1.00), and as deleterious with a SIFT score of −4.651. The novel heterozygous c.1237G>C (p.Glu413Gln) missense mutation in exon 6 of the KRT16 gene was reported in family B. The p.Glu413Gln substitution was rated according to ACMG standards as being probably damaging, probably damaging by polyphen2 with a score of 0.999, and deleterious by SIFT with a score of −2.766.
Keratin is the major structural protein of all epithelia as well as a diverse group of cytoskeletal scaffolding proteins that form intermediate filament (IF) networks. This provides structural support for keratinocytes to maintain the integrity of the skin.17 The c.1393T>C (Y465H) mutation is located in the highly conserved TYRKLLEGE protein motif hotspot region (residues 464–472). Residues exposed on the surface are an important biochemical anchor for coiled-coil dimers during the first stage in filament assembly.18 We hypothesize that the p.Tyr465His mutation disrupts with coiled-coil dimers during filament assembly (Figure 2A), resulting in cell fragility and cytoskeleton destruction. The c.1237C>C (E413Q) mutation was located in the highly conserved 2B domain in keratin 16 (K16; Figure 2B); a c.1237G>A (Glu413Lys) mutation at the same location was registered in the IPCRR. The 2B helical region is critically important for end-to-end association of proteins in the elongation phase of filament assembly.19 Amino acid substitutions are reported to affect the 2B helical structures, which are essential to form IF.20 A different mutation—p.E478Q, in the 2B domain of keratin 1 (K1)—leads to cytoskeleton destruction through the loss of hydrogen bonds between p.478E of K1 and p.450R of keratin 10 (K10).20 We hypothesize that the p.Glu413Gln mutation causes destruction of the hydrogen bond and electrostatic interaction by changing the acidic amino acid residue to a neutral form. This results in the presence of an incorrect keratin heterodimer, causing cell fragility and cytoskeleton destruction.21
 |
Figure 2 The KRT6A and KRT16 gene domains. (A) The mutation in the hotspot TYRKLLEGE protein motif region (residues 464–472 of KRT6A). Since the helical region serves a mechanical role in forming stiff bundles of fibers, mutations located in this region lead to the lateral IF association. (B) The position of the mutation in the end of the highly conserved 2B helical domain of KRT16. The PC-K16 phenotype is due to substitution of the acidic glutamic acid residue to a neutral glutamine residue. This affects the IF and destroys the integrity of the keratin. |
Mutations in KRT16 can also cause focal non-epidermolysis plantar keratoderma (FNEPPK), which manifests as mild to severe focal plantar keratoderma with absent or subtle nail changes (such as bending nails, hyperkeratosis).22 While a combination of genotypic and phenotypic data confirmed the diagnosis, this clinical description overlaps with the phenotype of patients in this study. Previous studies have shown that a case with the c.374A>G (p.N125S) mutation in KRT16 caused a FNEPPK phenotype with subtle change in the nails, while a different case with a c.373A>G (p.N125D) mutation in KRT16 was diagnosed with PC-1 due to the severe hypertrophic nail dystrophy.23 Different amino acid substitutions within the same locus lead to different clinical symptoms. This further highlights the correlation between the type of amino-acid substitution and clinical phenotype.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethics Committee at the Shanxi Medical University. The patients/participants provided their written informed consent to participate in this study and consented the publishment of case details and accompanying images.
Acknowledgments
We are most grateful to the PC patients and their family members for participating in this study.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Chiriac A, Murgu A, Rusu C, et al. First report of pachyonychia congenita type PC‐K6a in the Romanian population. Maedica. 2017;12(2):
2. Leachman SA, Kaspar RL, Fleckman P, et al. Clinical and pathological features of pachyonychia congenita. J Investig Dermatol Symp Proc. 2005;10(1):3–17. doi:10.1111/j.1087-0024.2005.10202.x
3. Shah S, Boen M, Kenner-Bell B, Schwartz M, Rademaker A, Paller AS. Pachyonychia congenita in pediatric patients: natural history, features, and impact. JAMA Dermatol. 2014;150:146–153. doi:10.1001/jamadermatol.2013.6448
4. Bowden PE, Haley JL, Kansky A, et al. Mutation of a type II keratin gene (K6a) in pachyonychia congenita. Nat Genet. 1995;10(3):363–365. doi:10.1038/ng0795-363
5. Jackson AD, Lawler SD. Pachyonychia congenita; a report of six cases in one family, with a note on linkage data. Ann Eugen. 1951;16:142–146. doi:10.1111/j.1469-1809.1951.tb02468.x
6. Irwin MWH, David HC, Eliason Mark J, et al. The phenotypic and molecular genetic features of pachyonychia congenita. J Invest Dermatology. 2011;131(5):1015–1017. doi:10.1038/jid.2011.59
7. Leachman Sancy A, Kaspar Roger L, Philip F, et al. Clinical and pathological features of pachyonychia congenita. J Invest Dermatology Symp Proce. 2005;10(1):3–17.
8. Herrmann H, Strelkov SV, Burkhard P, Aebi U. Intermediate filaments: primary determinants of cell architecture and plasticity. J Clin Invest. 2009;119(7):1772–1783. doi:10.1172/JCI38214
9. Eliason M, Feng BJ, Leachman SA, Feng BJ, Schwartz ME, Hansen CD. A review of the clinical phenotype of 254 patients with genetically confirmed pachyonychia congenita. J Am Acad Dermatol. 2012;67(4):
10. Katkar DP, Kadam SB, Mote BN, Adhav AS. Subblock level monitoring in leprosy programme: need of the hour. Indian J Dermatol Venereol Leprol. 2017;83(1):
11. Bai Z-L, Feng Y-G, Tan -S-S, et al. Mutations of KRT6A are more frequent than those of KRT16 in pachyonychia congenita type 1: report of a novel and a recently reported mutation in two unrelated Chinese families. Br J Dermatology. 2008;159(1):238–240. doi:10.1111/j.1365-2133.2008.08603.x
12. Forrest CE, Casey G, Mordaunt DA, Thompson EM, Gordon L. Pachyonychia congenita: a spectrum of KRT6a mutations in australian patients. Pediatr Dermatol. 2016;33(3):337–342. doi:10.1111/pde.12841
13. Fu T, Leachman SA, Wilson NJ, et al. Genotype-phenotype correlations among pachy -onychia congenita patients with K16 mutations. J Invest Dermatol. 2011;131(5):1025–1028. doi:10.1038/jid.2010.373
14. Samuelov L, Smith FJ, Hansen D, et al. Pachyonychia congenita: a case-cohort study of 815 patients. Br J Dermatol. 2019;182(3):738–746. doi:10.1111/bjd.18794
15. Smith FJ, Liao H, Cassidy AJ, et al. The genetic basis of pachyonychia congenita. J Investig Dermatol Symp Proc. 2005;10:21–30.
16. Bai ZL, Feng YG, Tan SS, et al. Mutations of KRT6A are more frequent than those of KRT16 in pachyonychia congenita type 1: report of a novel and a recently reported mutation in two unrelated Chinese families. Br J Dermatol. 2008;159(1):238–240.
17. Chamcheu JC, Siddiqui IA, Syed DN, Adhami VM, Liovic M, Mukhtar H. Keratin gene mutations in disorders of human skin and its appendages. Arch Biochem Biophys. 2011;508(2):123–137. doi:10.1016/j.abb.2010.12.019
18. Wilson AK, Coulombe PA, Fuchs E. The roles of K5 and K14 head, tail, and R/KLLEGE domains in keratin filament assembly in vitro. J Cell Biol. 1992;119(2):401–414. doi:10.1083/jcb.119.2.401
19. Ghazawi FM, Hassani-Ardakani K, Henriques L, et al. Identification of a novel substitution mutation (R103C) in the rod domain of the keratin 17 gene associated with pachyonychia congenita type 2. Int J Dermatol. 2019;58(2):233–236. doi:10.1111/ijd.14082
20. Xu Q, Zhang Q, Tang L, et al. A KRT16 mutation in the first Chinese pedigree with Pachyonychia congenita and review of the literatures. J Cosmet Dermatol. 2019;18(6):1930–1934. doi:10.1111/jocd.12905
21. Banerjee S, Wu Q, Ying Y, et al. In silico predicted structural and functional insights of all missense mutations on 2B domain of K1/K10 causing genodermatoses. Oncotarget. 2016;7(33):52766–52780. doi:10.18632/oncotarget.10599
22. Perez‐Perez L, Allegue F, Alfonsin N, Caeiro JL, Fabeiro JM, Zulaica A. Identification of two recurrent mutations in keratin genes in three cases with pachyonychia congenita. J Eur Acad Dermatol Venereol. 2009;23(2):
23. Liao H, Sayers JM, Wilson NJ, et al. A spectrum of mutations in keratins K6a, K16 and K17 causing pachyonychia congenita. J Dermatol Sci. 2007;48(3):
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.