")
Back to Journals » Clinical Ophthalmology » Volume 15
A Genotype–Phenotype Analysis of the Bardet–Biedl Syndrome in Puerto Rico
Authors Guardiola GA , Ramos F , Izquierdo NJ, Oliver AL
Received 8 July 2021
Accepted for publication 23 August 2021
Published 7 September 2021 Volume 2021:15 Pages 3757—3764
DOI https://doi.org/10.2147/OPTH.S328493
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Gabriel A Guardiola,1 Fabiola Ramos,2 Natalio J Izquierdo,3 Armando L Oliver2
1Department of Medicine, Universidad Central del Caribe School of Medicine, Bayamon, PR, USA; 2Department of Ophthalmology, University of Puerto Rico School of Medicine, University of Puerto Rico, Medical Sciences Campus, San Juan, PR, USA; 3Department of Surgery, University of Puerto Rico School of Medicine, University of Puerto Rico, Medical Sciences Campus, San Juan, PR, USA
Correspondence: Natalio J Izquierdo 369 De Diego, Torre San Francisco, Suite 310, San Juan, PR, 00923, USA
Tel +1 787 402-0201
Email [email protected]
Background: Bardet–Biedl syndrome is a complex heterogeneous ciliopathy caused by genetic mutations. Although establishing genotype–phenotype correlations has been challenging, some regional variations have been previously reported. Due to its relative geographic isolation, Puerto Rico has a greater prevalence of Bardet–Biedl syndrome than do other regions. We sought to characterize the most frequent genotypic variations in a local cohort of Bardet–Biedl syndrome patients and report any genotypic–phenotypic trends.
Methods: Twenty-seven patients from an ophthalmology clinic in Puerto Rico with genetically confirmed Bardet–Biedl syndrome took a questionnaire inquiring about their most common symptoms. Ophthalmological information was obtained from patient records. The frequencies of the genotypic variations and symptoms were calculated.
Results: In the study population, BBS1 was the most prevalent mutated gene, followed by BBS7. In the BBS1 group, we found homozygotes for c.1169T>G (p.Met390Arg) and c.1645G>T (p.Glu549*), and compound heterozygotes for c.1169T>G (p.Met390Arg) and c.1645G>T (p.Glu549*), with one patient having c.1645G>T (p.Glu549*) and c.432+1G>A (splice donor). All the BBS7 patients were homozygous for c.632C>T (p.Thr211Ile). Compared to BBS7, we found that BBS1 patients generally had a milder ocular and systemic phenotype. However, when analyzing different BBS1 variants, patients with mutations in c.1645G>T (p.Glu549*), both compound heterozygous and homozygous, had more severe systemic phenotypes, overall.
Conclusion: Our study was the first detailed genotype–phenotype analysis of the Bardet–Biedl syndrome in Puerto Rico. Genetic mutations in BBS1 and BBS7 seem to be the most common culprits behind Bardet–Biedl syndrome in this population. Although patients diagnosed with BBS1 are likely to display milder systemic features, this was not the case with our BBS1 patients having the c.1645G>T (p.Glu549*) mutation. Further studies should focus on the c.1645G>T (p.Glu549*) mutation’s impact on the BBS1 gene and protein product.
Keywords: ciliopathy, compound heterozygotes, retinitis pigmentosa, polydactyly
Introduction
Bardet–Biedl syndrome (BBS) is the name given to a group of clinical features and manifestations arising from genetic mutations that induce widespread ciliopathy.1 It is an autosomal recessive condition, and so far, at least 24 different genes associated with it have been described.2 BBS diagnosis is clinical, and patients need to display either four primary features or three primary and two secondary features to be so diagnosed.3 Its primary features include retinal degeneration, polydactyly, truncal obesity, genital anomalies, renal dysfunction, and learning impairment.3 Its secondary features include diabetes mellitus, thyroid disease, hearing loss, heart disease, hypertension, and liver disease, among other diseases, symptoms, and disorders.4,5 However, these criteria overlooks the number of genetically diagnosed BBS patients who present with incomplete phenotypes.6 Genetic testing can confirm the diagnosis in 80% of patients.6,7
The majority of BBS patients have mutations in the genes responsible for the assembly of the BBSome protein aggregate, whose role is crucial for the proper functioning of the primary cilium found in most of the cells of the body.8 While BBS1, 2, 4, 5, 7, 8, 9, and 18 form the BBSome, only BBS2, 7, and 9 are said to compose the BBSome core, and thus, mutations in these genes are believed to have a more significant impact.2,8,9
The estimated BBS prevalence in Puerto Rico is 1:62,000, which places the island among the locations with a relatively high BBS prevalence.10 There is no proper study evaluating the frequency of BBS variants in Puerto Rico. However, Mykytyn et al11 used five Puerto Rican families to identify and locate the BBS1 mutation, concluding that BBS1 is one of the primary genes behind BBS in the North American population. Therefore, BBS1 could be one of the main culprits behind the disease in Puerto Rico.
BBS is a pleiotropic disorder with an ample genetic heterogeneity and complex multigenetic etiologies.2 Regional and ethnic genotypic and phenotypic variations have been previously suggested.12–19 Most of the studies that have explored genotype–phenotype relationships have failed to establish true correlations, yet certain trends have been observed. For instance, Castro-Sanchez et al19 found that, within their Spanish cohort, BBS1 patients seemed to display an overall milder ocular and systemic phenotype than did patients with BBS6, BBS10, or BBS12. In another study, this one by Deveault et al,18 BBS1 and BBS12 patients presented a less severe ocular and systemic phenotype than BBS10 patients did. Although both studies agree on a milder BBS1 phenotype, Deveault characterized BBS12 as having a milder BBS phenotype, as well, while Castro-Sanchez and her team found it to be linked to a more severe phenotype within their cohort.18,19 Regarding visual dysfunction and ocular findings, it has been suggested that the ocular phenotype for BBS1 patients who are homozygous for p.Met390Ar might not be as mild as is commonly reported.19–20
In a meta-analysis of 426 BBS patients, Niederlova et al2 found statistically significant differences between certain genotypes and phenotypes. They reported that their patients with BBS1 had a significantly lower disease burden than did those with BBS2 or BBS7 and proposed that this could have been due to the potentially hypomorphic nature of the type of mutation involved.2 In their study, patients with suspected complete loss-of-function mutations, such as splicing defects or frameshifts, displayed more severe BBS phenotypes than did those with mutations that are generally considered to produce milder alterations in protein function, such as missense mutations. However, after further analysis, the lower frequency of symptoms in BBS1 seems to be ultimately linked to the BBS1 gene and not a specific type of mutation.2
Similarly, in a genotype-phenotype analysis of 25 Italian patients with either BBS1, BBS2, or BBS10, Esposito et al21 reported a milder retinal dystrophy, renal impairment and audiovestibular phenotype in BBS1 patients.
Herein we will present a genotype–phenotype analysis of 27 Puerto Rican BBS patients with the BBS1 and BBS7 mutations. We aim to illustrate the trends related to systemic complications in this population, to guide preventive care and proper management.
Methods
Patients
Twenty-seven patients with a confirmed genetic BBS diagnosis, selected from a single ophthalmology clinic in Puerto Rico, were included in this cross-sectional study. Our group consisted of nine females and 18 males with an average age of 33 (± 13.5) years (range 13–65). The majority of the subjects met the clinical criteria required to confirm a BBS diagnosis.
Individuals had been previously screened for BBS mutations after confirmation of a clinical diagnosis or high clinical suspicion from the ophthalmologist. A folder storing the genetic results from each subject was accessed to create a list with all BBS cases. Patients were contacted over the phone and informed consent was gathered from each one or, when applicable, from their guardian(s), after explaining the purpose of our study and fully disclosing our process for data collection and management. Clinical data was extracted from the electronic medical records and questionnaires administered via phone calls. This investigation was approved by the Universidad Central del Caribe Medical School institutional review board; our work is compliant with the Health Insurance Privacy and Accountability Act. The study adhered to the principles of the Declaration of Helsinki.
Genetic Testing
Results of genetic testing were available prior to study onset. Salivary samples had been collected and sent for analysis using a 248 gene inherited retinal disorders panel through full-gene sequencing and deletion/duplication analysis, including select non-coding variants, coding exons and 10–20 base pairs of adjacent intronic sequence (Invitae Corporation, San Francisco, California, USA).
Clinical Evaluation
Eligible patients were contacted by telephone about participating in this study; those who expressed their willingness to do so were scheduled for an interview, at which they would answer a questionnaire. The questionnaire inquired about pertinent BBS systemic and ocular characteristics. Genital anomaly was defined as micropenis or hypogonadism, truncal obesity as a waist-to-hip ratio greater than 0.9 for men and greater than 0.85 for women, renal anomalies as any structural aberrations or functional insufficiencies, and learning impairment as the need for special education during primary or secondary level education. Clinical and ophthalmologic data, including presenting signs and symptoms and diagnostic images and studies, were collected from the participating patient’s medical records, when such records were available.
Results
Mutational Analysis
All the subjects carried two pathogenic mutations in one of the previously identified BBS causative genes. Specifically, 23 patients had BBS1 (85%), while four patients had BBS7 (15%) (Table 1). Of the 27 patients included, three had an additional heterozygous mutational variant of unclear significance. VUS c.396G>C (p.Gln123His) (BBS9) was found in patient three; VUS c.1463A>G (p.lys488Arg) (BBS10), in patient 24; and Gain (exons 1–2) (BBS9), in patient 26. Within the BBS1 group, 16 patients (70%) were homozygous for either the c.1169T>G (p.Met390Arg) (48%) or the c.1645G>T (p.Glu549*) (22%) mutation. The remaining BBS1 patients were compound heterozygotes, with six patients harboring mutations c.1169T>G (p.Met390Arg) and c.1645G>T (p.Glu549*) and one patient with c.1645G>T (p.Glu549*) and c.432+1G>A (splice donor). All the BBS7 patients were homozygous for the c.632C>T (p.Thr211Ile) mutation.
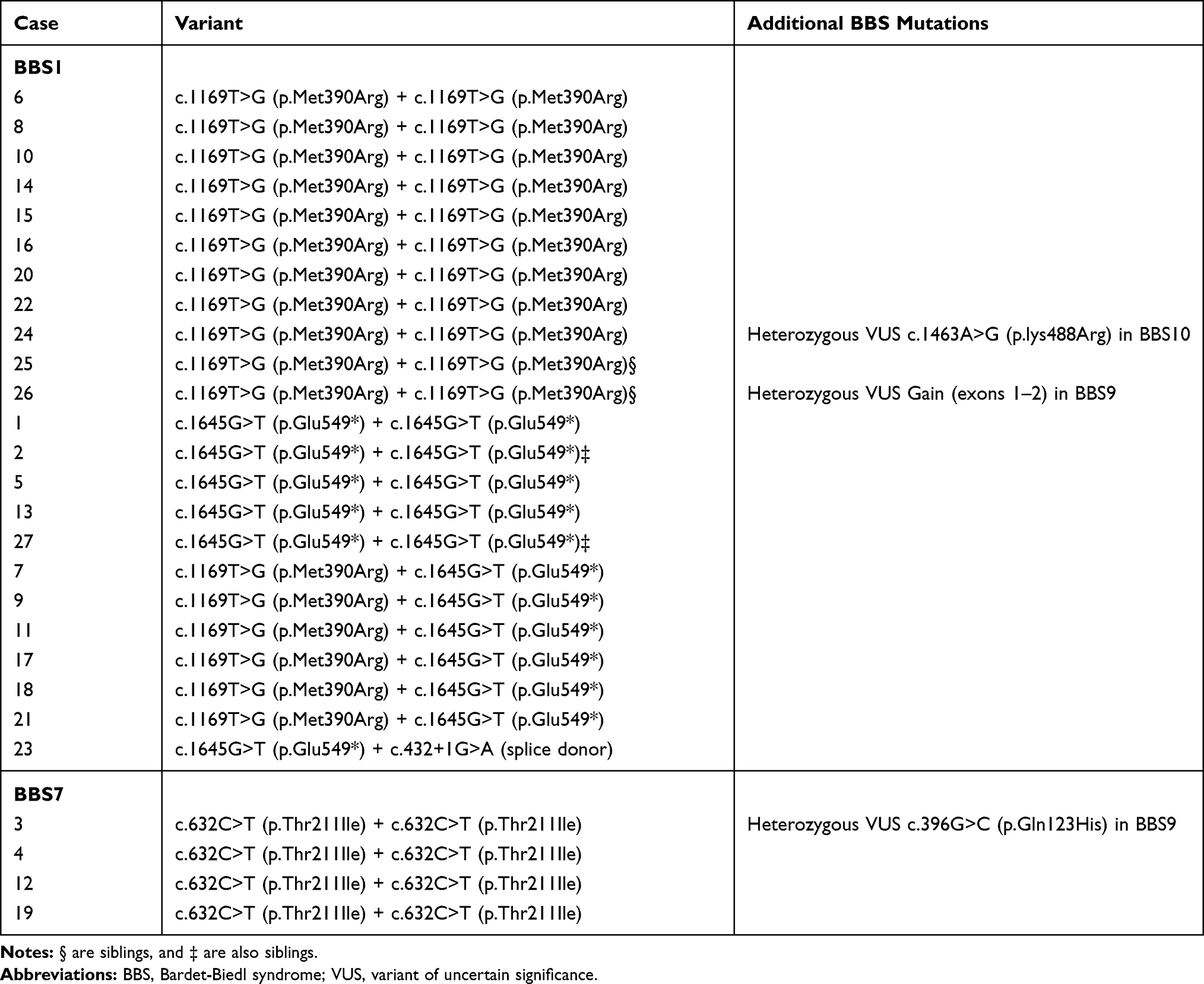 |
Table 1 List of Identified BBS Mutations |
Phenotypes
We could not establish a clear genotype–phenotype correlation in either the BBS1 or BBS7 groups due to the limited sample size. A list of the observed clinical features is provided in Table 2.
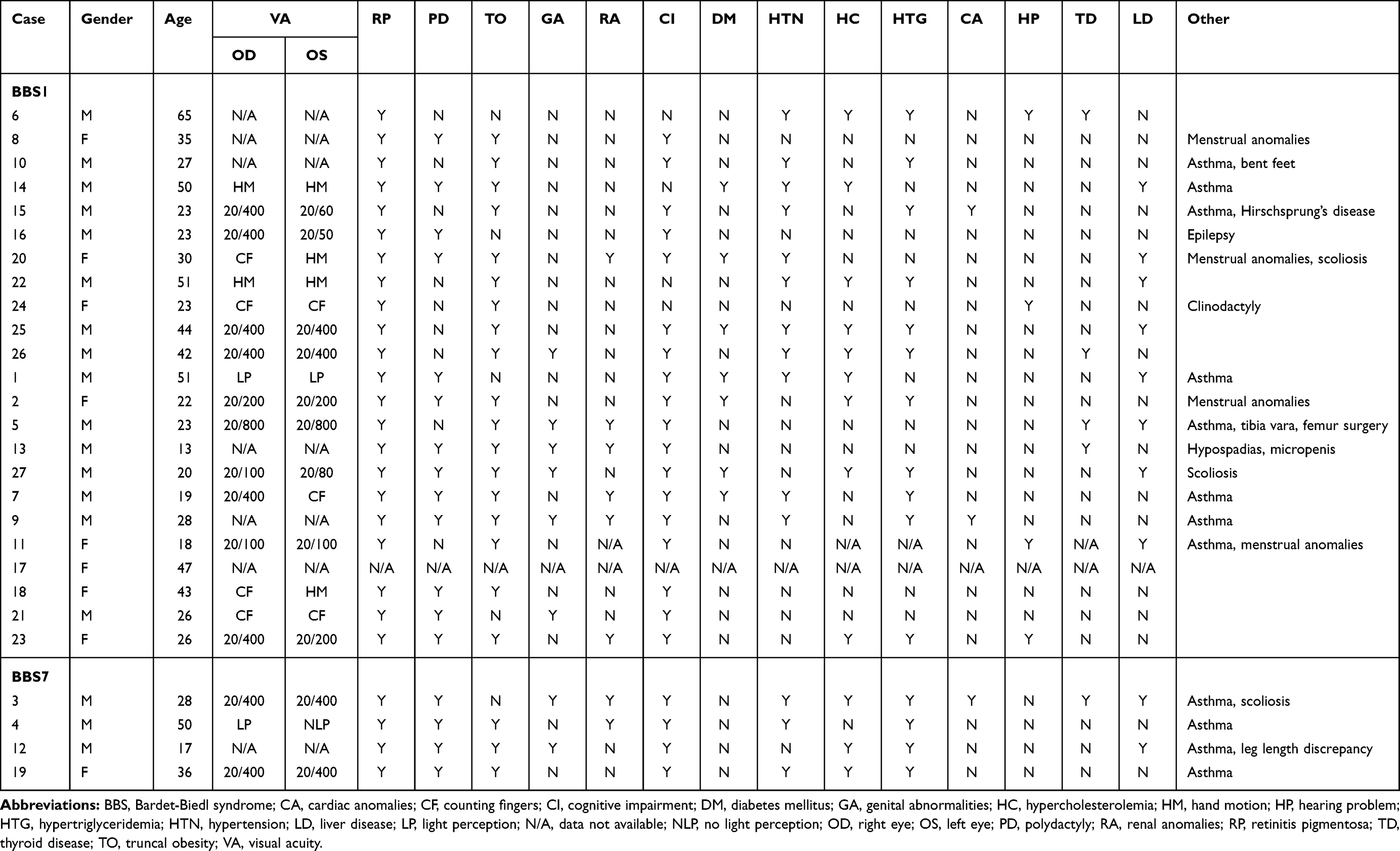 |
Table 2 List of BBS-Associated Characteristics Found in Our Patients |
Most of our patients reported that visual deterioration (ie, night blindness and peripheral vision loss) was the first manifestation of BBS that they detected. As has been seen in previous studies,19 the patients in our study who were homozygous for p.Met390Arg had a relatively later symptom onset, with a mean age of 21 years. Interestingly, all the patients with at least one p.Glu549* allele experienced an earlier onset of visual symptoms (mean age of 8 years) than did any of the patients who were homozygous for p.Met390Arg; however, one should refrain from drawing any conclusions from this fact, considering the small sample size.
Collectively, retinitis pigmentosa, polydactyly, learning impairment, and truncal obesity were the four most common features (Table 3). Retinitis pigmentosa was present in every single subject. Learning impairment, the second most frequent primary feature, was reported in all the BBS7 patients and 82% of the BBS1 patients. It is worth noting that of the BBS1 patients, 64% of those who were homozygous for p.Met390Arg had a learning impairment, while such impairments were reported in 100% of the p.Glu549* homozygotes and the compound heterozygotes with p.Met390Arg and p.Glu549*. Truncal obesity was present in 77% of all the patients. It had a similar frequency across BBS1 subtypes and was observed in three BBS7 patients. Polydactyly was discovered in 62% of all the BBS patients and was identified in all the BBS7 patients and in 59% of the BBS1 patients. Within the BBS1 group, polydactyly was less frequently found in those who were homozygous for p.Met390Arg (36%), while it was seen in 80% of the p.Glu549* homozygotes and compound heterozygotes.
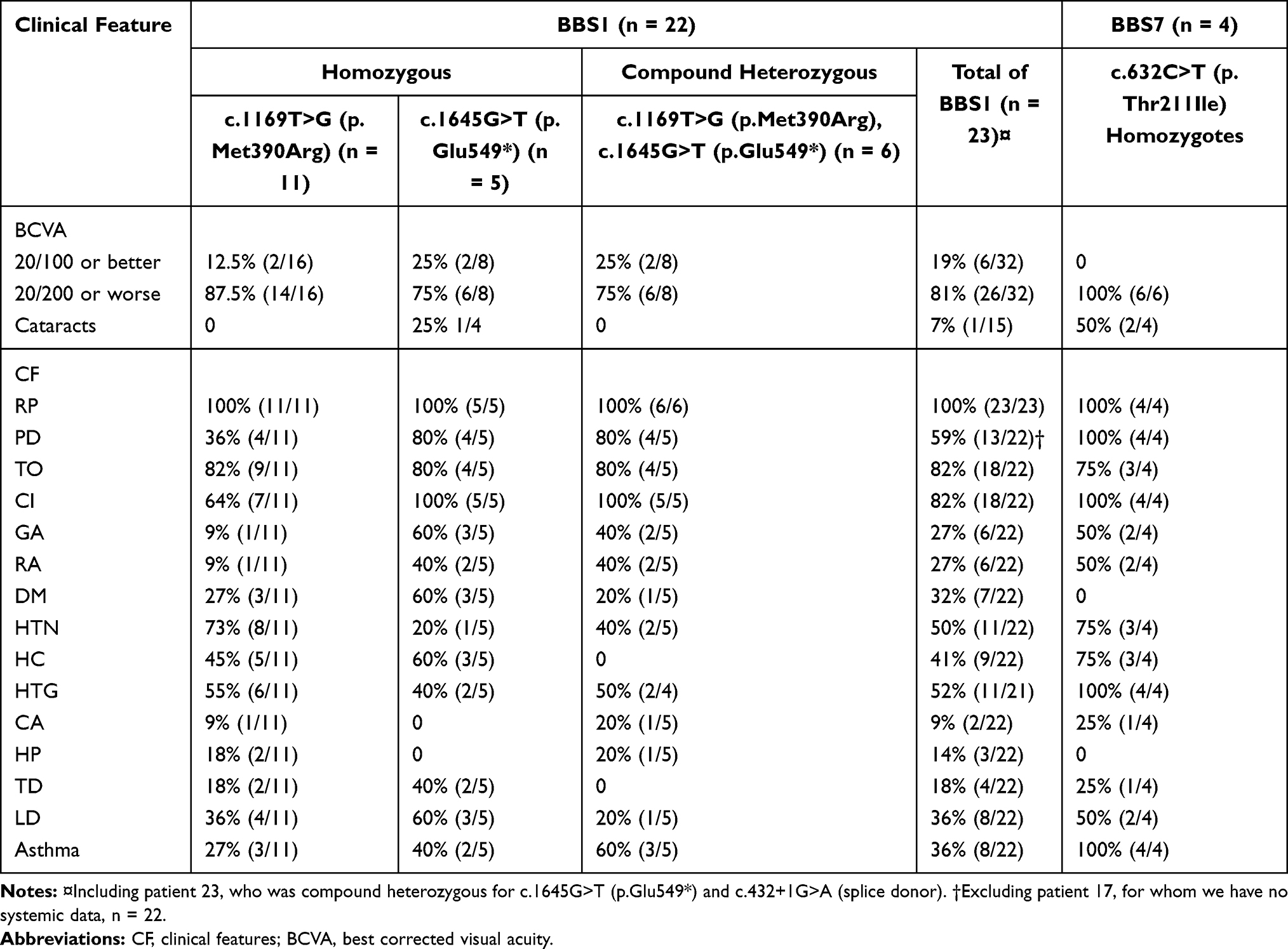 |
Table 3 Frequencies of BBS-Associated Characteristics in Our Patient Sample |
Genital and renal anomalies were more frequently seen in patients with BBS7 (50%) than in those with BBS1 (27%). As with learning impairment, these features were less frequently observed in BBS1 patients who were homozygous for p.Met390Ar. The genital anomalies consisted mainly of hypogonadism and micropenis. The renal anomalies included congenital polycystic kidney disease, chronic kidney disease, and renal insufficiency.
Of the secondary characteristics, hypertension was the most common, with a frequency of 54% in the entire BBS group. Diabetes mellitus (DM) was observed in 32% of the BBS1 patients. In comparison, none of the BBS7 patients presented with DM. Cardiac problems were present in two patients with BBS1 (9%) and in one patient with BBS7 (25%) and included a small myocardial infarction, severe mitral valve defect that needed surgery and a pulmonary vein malformation.
Several systemic complications were also assessed. Hypertriglyceridemia was one of the most frequent complications (54%). It was present in all the BBS7 patients, while it had a frequency of around 50% in the BBS1 patients. Asthma, specifically of childhood-onset, was the second most frequent systemic complication (46%), being present in all the BBS7 patients and being more frequently seen in BBS1 compound heterozygotes (60%) than in BBS1 patients who were homozygous for the p.Met390Arg mutation (27%). Liver disease was also present, with a total frequency of 38%; it was present in half of the patients with BBS7, 60% of those who were homozygous for the p.Glu549* mutation, and 36% of those who were homozygous for the p.Met390Arg mutation. Liver conditions consisted primarily of early-onset fatty liver disease, while one patient had liver cysts.
Interestingly, six of our patients presented with skeletal anomalies (scoliosis, tibia vara, and bent feet). In addition, auditory problems were observed in five patients (all five had BBS1), while three patients experienced menstrual anomalies, two patients had had childhood epilepsy, and one patient had Hirschsprung’s disease.
All the patients presented with retinal changes consistent with retinitis pigmentosa. In terms of visual acuity, 85% of the examined eyes (40) had a best-corrected visual acuity of 20/200 or worse. Cataracts were found in two BBS7 patients (50%), and one BBS1 patient (7%).
Discussion
BBS prevalence in Puerto Rico is estimated to be around 1 in 62,000 people, which more closely resembles the prevalences of places with limited population mobility and high consanguinity.10,22 BBS1 was the most commonly mutated gene in our cohort, which was expected, given previous findings in genetic studies utilizing the Puerto Rican population.11 As has been previously observed,11 the majority of our BBS1 cases were homozygous for a mutation in c.1169T>G (p.Met390Arg), yet we also had homozygous patients with the c.1645G>T (p.Glu549*) mutation, which results in a nonsense mutation. BBS1 compound heterozygotes were also observed with mutations c.1169T>G (p.Met390Arg) and c.1645G>T (p.Glu549*), and c.1645G>T (p.Glu549*) with c.432+1G>A (splice donor).
Attempts to establish a genotype–phenotype correlation of the various BBS subtypes have been challenging due to the high heterogeneity displayed by the disease and the relatively small cohorts from which phenotypical data has been gathered.12–19 Nevertheless, we sought to collect detailed clinical information from our Puerto Rican BBS cohort, since it has been suggested that the island might have one of the highest BBS prevalences, globally.
Although our sample size did not allow us to confirm a clear genotype–phenotype relationship, we did observe some tendencies worthy of mention. Within the group of BBS1 patients, we documented an overall less severe systemic phenotype in the p.(Met390Arg) homozygotes than the phenotypes that were seen in the p.(Glu549*) homozygotes and the compound heterozygotes. Visual symptoms had an earlier onset for homozygotes and compound heterozygotes of the p.(Glu549*) mutation than for p.(Met390Arg) homozygotes. However, the BBS1 p.(Met390Arg) homozygotes had worse visual acuity than did the patients with the other BBS1 subtypes.
Regarding BBS7, all of our patients carried homozygous mutations in p.(Thr211Ile) and had more severe ocular and systemic phenotypes than did the patients with BBS1, as a whole. This finding is consistent with previous research,2 in which it has been suggested that the BBS7 phenotype severity results from the intrinsic role that the BBS7 protein plays as part of the BBSome core. Hence, the phenotypical difference might be accounted for by the particular functions of each protein group.2,23 Yet, there are studies were BBS7 patients present mild phenotypes.24
Compared to BBS2, BBS7, and BBS9, BBS1 has been associated with a low occurrence of renal anomalies.2 Conforming to these findings, we observed renal anomalies in 50% of our patients with BBS7, while it was seen in only 27% of the patients with BBS1. It is worth noting that when analyzing specific BBS1 mutations, renal anomalies were much more frequent in individuals who were homozygous for the p.(Glu549*) mutation and in compound heterozygotes with the p.(Glu549*) and p.(Met390Arg) mutations. This tendency leads us to believe that the type of mutation within a specific BBS variant might have an impact on phenotype severity. However, further analyses are needed if we want to fully explore this observation. Interestingly, although we cannot draw formal conclusions, because of the small sample size, the frequencies of all the primary characteristics of BBS were similar in BBS7 and BBS1 patients, with both possessing at least one p.(Glu549*) mutation.
Previous studies23 suggest that BBS might have a triallelic mode of inheritance, which might explain why some patients carrying an additional heterozygous BBS mutation end up with a more severe phenotype. Of note, the subject displaying the largest number of BBS characteristics in our study was harboring three BBS mutations. Nonetheless, one of the patients who also had an additional mutation did not meet the criteria for a clinical diagnosis of BBS.
Additionally, a significant number reported having had childhood asthma, skeletal issues such as scoliosis, and hearing problems. Childhood epilepsy and Hirschsprung’s disease were also reported.
Some major limitations in our study are the low number of patients and the use of a phone questionnaire for data collection.
Conclusion
In conclusion, our study provides the first detailed genotype–phenotype analysis of the Bardet–Biedl syndrome in Puerto Rico. The tendencies observed should encourage the prompt medical management and careful health monitoring of patients with the p.(Glu549*) BBS1 mutation, since they tend to develop a worse systemic phenotype—similar to what is seen with BBS7—and usually have an earlier onset of ocular symptoms. Furthermore, by comparing the clinical characteristics of p.(Glu549*) homozygotes, (Met390Arg) homozygotes, and compound heterozygotes, our investigation brings attention to the possible phenotypical differences between BBS1 variants.
Data Sharing Statement
In order to protect subject privacy, the databank that resulted from this investigation is not available to the public. If necessary, data requests can be made to the corresponding author.
Ethics Approval
This study was approved by the Institutional Review Board at the Universidad Central del Caribe in Bayamón, Puerto Rico (Protocol number: 2021-05). Informed consent was obtained and all patient safety and confidentiality protocols were strictly followed.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Tsang SH, Aycinena ARP, Sharma T. Ciliopathy: bardet-Biedl syndrome. In: Tsang S, Sharma T editors, Advances in Experimental Medicine and Biology. Vol. 1085. Springer New York LLC; 2018:171–174. doi:10.1007/978-3-319-95046-4_33
2. Niederlova V, Modrak M, Tsyklauri O, Huranova M, Stepanek O. Meta-analysis of genotype-phenotype associations in Bardet-Biedl syndrome uncovers differences among causative genes. Hum Mut. 2019;40(11):2068–2087. doi:10.1002/humu.23862
3. Forsythe E, Kenny J, Bacchelli C, Beales PL. Managing Bardet-Biedl syndrome-now and in the future. Front Pediatr. 2018;6:23. doi:10.3389/fped.2018.00023
4. Torrefranca AB, Santiago APD, Lingao MD, Racoma MJC. Novel compound heterozygous pathogenic BBS5 variants in Filipino siblings with Bardet-Biedl syndrome (BBS). Ophthalmic Genet. 2020;41(6):621–624. doi:10.1080/13816810.2020.1810282
5. Forsyth R, Gunay-Aygun M. Bardet-Biedl syndrome overview. In: Adam MP, Ardinger HH, Pagon RA, editors. GeneReviews®. Seattle: University of Washington, Seattle; 2003.
6. Suspitsin EN, Imyanitov EN. Bardet-Biedl syndrome. Mol Syndromol. 2016;7(2):62–71. doi:10.1159/000445491
7. Forsythe E, Beales PL. Bardet-Biedl syndrome. Eur J Hum Genet. 2013;21(1):8–13. doi:10.1038/ejhg.2012.115
8. Klink BU, Zent E, Juneja P, Kuhlee A, Raunser S, Wittinghofer A. A recombinant BBSome core complex and how it interacts with ciliary cargo. Elife. 2017;6:27434. doi:10.7554/eLife.27434
9. Zhang Q, Seo S, Bugge K, Stone EM, Sheffield VC. BBS proteins interact genetically with the IFT pathway to influence SHH-related phenotypes. Hum Mol Genet. 2012;21(9):1945–1953. doi:10.1093/hmg/dds004
10. Oliveras-Rentas RE, Rodríguez-Irizarry W, Crespo FP, et al. Perfil Neuropsicológico de Niños y adolescentes con el Síndrome de Bardet Biedl en Puerto Rico. Revista Iberoamericana De Neuropsicología. 2020;3(1):1–14.
11. Mykytyn K, Nishimura DY, Searby CC, et al. Identification of the gene (BBS1) most commonly involved in Bardet-Biedl syndrome, a complex human obesity syndrome. Nat Genet. 2002;31(4):435–438. doi:10.1038/ng935
12. Billingsley G, Deveault C, Héon E. BBS mutational analysis: a strategic approach. Ophthalmic Genet. 2011;32(3):181–187. doi:10.3109/13816810.2011.567319
13. Hirano M, Satake W, Ihara K, et al. The first nationwide survey and genetic analyses of Bardet-Biedl syndrome in Japan. PLoS One. 2015;10(9):e0136317. doi:10.1371/journal.pone.0136317
14. Álvarez-satta M, Castro-Sánchez S, Pereiro I, et al. Overview of Bardet-Biedl syndrome in Spain: identification of novel mutations in BBS1, BBS10 and BBS12 genes. Clin Genet. 2014;86(6):601–602. doi:10.1111/cge.12334
15. Hjortshøj TD, Grønskov K, Philp AR, et al. Bardet-Biedl syndrome in Denmark: report of 13 novel sequence variations in six genes. Hum Mutat. 2010;31(4):429–436. doi:10.1002/humu.21204
16. Tao T, Wang L, Chong W, Yang L, Li G. Characteristics of genotype and phenotype in Chinese patients with Bardet–Biedl syndrome. Int Ophthalmol. 2020;40(9):2325–2343. doi:10.1007/s10792-020-01415-3
17. Forsythe E, Sparks K, Best S, et al. Risk factors for severe renal disease in Bardet-Biedl syndrome. J Am Soc Nephrol. 2017;28(3):963–970. doi:10.1681/ASN.2015091029
18. Deveault C, Billingsley G, Duncan JL, et al. BBS genotype-phenotype assessment of a multiethnic patient cohort calls for a revision of the disease definition. Hum Mutat. 2011;32(6):610–619. doi:10.1002/humu.21480
19. Castro-Sánchez S, Álvarez-satta M, Cortón M, Guillén E, Ayuso C, Valverde D. Exploring genotype-phenotype relationships in Bardet-Biedl syndrome families. J Med Genet. 2015;52(8):503–513. doi:10.1136/jmedgenet-2015-103099
20. Cox KF, Kerr NC, Kedrov M, et al. Phenotypic expression of Bardet-Biedl syndrome in patients homozygous for the common M390R mutation in the BBS1 gene. Vision Res. 2012;75:77–87. doi:10.1016/j.visres.2012.08.005
21. Esposito G, Testa F, Zacchia M, et al. Genetic characterization of Italian patients with Bardet-Biedl syndrome and correlation to ocular, renal and audio-vestibular phenotype: identification of eleven novel pathogenic sequence variants. BMC Med Genet. 2017;18(1):10. doi:10.1186/s12881-017-0372-0
22. Moore SJ, Green JS, Fan Y, et al. Clinical and genetic epidemiology of Bardet-Biedl syndrome in Newfoundland: a 22-year prospective, population-based, cohort study. Am J Med Genet A. 2005;132A(4):352–360. doi:10.1002/ajmg.a.30406
23. Daniels AB, Sandberg MA, Chen J, Weigel-DiFranco C, Hejtmancik JF, Berson EL. Genotype-phenotype correlations in Bardet-Biedl syndrome. Arch Ophthalmol. 2012;130(7):901–907. doi:10.1001/archophthalmol.2012.89
24. Aleman TS, O’Neil EC, O’Connor K, et al. Bardet-Biedl syndrome-7 (BBS7) shows treatment potential and a cone-rod dystrophy phenotype that recapitulates the non-human primate model. Ophthalmic Genet. 2021;42(3):252–265. doi:10.1080/1381681020211888132
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.