")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 15
A Genome-Wide Association Study in Early COPD: Identification of One Major Susceptibility Loci
Authors Lee YJ, Choi SH, Kwon SY , Lee Y, Lee JK , Heo EY, Chung HS, Kim DK
Received 24 June 2020
Accepted for publication 23 September 2020
Published 17 November 2020 Volume 2020:15 Pages 2967—2975
DOI https://doi.org/10.2147/COPD.S269263
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Richard Russell
Ye-Jin Lee,1 SeungHo Choi,2 Sung-Youn Kwon,2 Yunhwan Lee,2 Jung Kyu Lee,3 Eun Young Heo,3 Hee Soon Chung,3,4 Deog Kyeom Kim3,4
1Division of Pulmonary, Allergy and Critical Care Medicine, Department of Internal Medicine, Hallym University Kangdong Sacred Heart Hospital, Seoul, Korea; 2Department of Internal Medicine, Healthcare Research Institute, Healthcare System Gangnam Center, Seoul National University Hospital, Seoul 135-984 Korea; 3Division of Pulmonary and Critical Care Medicine, Seoul Metropolitan Government-Seoul National University Boramae Medical Center, Seoul, Korea; 4Department of Internal Medicine, Seoul National University College of Medicine, Seoul, Korea
Correspondence: Deog Kyeom Kim
Division of Pulmonary and Critical Care Medicine, Seoul Metropolitan Government-Seoul National University Boramae Medical Center, Boramae-Gil 41, Dongjak-gu, Seoul 156-707, Republic of Korea
Tel +82-2-870-3207
Fax +82-2-831-0714
Email [email protected]
Background: Identifying the genetic basis of airflow limitation is one of the most interesting issues for understanding chronic obstructive pulmonary disease (COPD) pathophysiology. Several studies have shown that some genetic variants associated with COPD have been identified in genome-wide association study (GWAS), especially in patients with moderate to severe COPD; genetic susceptibility for airflow limitation in the early COPD phase has not been widely studied.
Objective: We investigated the genetic variants in early COPD.
Methods: The present study analyzed Gene-environment interaction and phenotype (GENIE) cohort that included participants who received health screening examination. The association between single nucleotide polymorphism (SNP) and susceptibility to early COPD (FEV1 predicted ≥ 50% and FEV1/FVC < 0.7) was tested.
Results: A total of 130 patients with early COPD and 3478 controls (1700 ever smokers and 1778 never smokers) were recruited. When compared with the total controls, certain SNPs (rs2818103, rs875033, rs9354627, rs34552148) on chromosome 6 were included at the top of our list (p= 5.6 × 10– 7 ∼ 9.6 × 10– 6) although they did not reach genome-wide significance. When compared with the never smoker controls, two SNPs (rs2857210, rs2621419) of the HLA-DQB2 gene class were persistently associated with susceptibility to early COPD.
Conclusion: Certain SNPs located on chromosome 6 or the HLA-DQB2 gene were the top-scoring SNPs for the association with susceptibility to early COPD in the Korean GENIE cohort.
Keywords: early chronic obstructive pulmonary disease, genome-wide association study, single nucleotide polymorphism, SNP, HLA-DQ gene
Introduction
According to the Global Burden of Disease (GBD), COPD patients were estimated at 251 million in 2015, an increase of 44.7% compared with 1990.1 Furthermore, since the number of elderly patients is increasing worldwide, the global prevalence and the socio-economic losses of COPD are expected to increase over the next 30 years.2
Although cigarette smoking is the most important risk factor for developing COPD, several recent studies have implicated a genetic component associated with COPD susceptibility because up to one-third of all COPD cases are made up of never smokers while only 10–20% of heavy smokers have been shown to develop symptomatic COPD.3–5
Similar to A1 antitrypsin deficiency, which is a single gene abnormality that is related to lung function decline and is a well-known genetic factor that predisposes to COPD, several other genetic variants associated with susceptibility to COPD have been discovered in genome-wide association (GWAS) studies.6 Some of these well-established COPD risk genes include HHIP, SERPINA, FAM13A, AGER, CHRNA5, and IL27.6–10 However, these studies included a large number of patients with moderate to severe COPD, with a mean forced expiratory volume in one second (FEV1) of between 25% and 50%.9 There is no study to identify the susceptible genetic risk factors for early COPD. Current studies suggest that early COPD is associated with poor outcomes and early detection of COPD may help to decelerate progression of COPD.11,12 And previous researches have included advanced COPD cases, it is likely to be influenced by the environmental factors as well as the genetic factors. Therefore, the aim of this study is to investigate genetic variants in early COPD. And through this study, we would be able to identify loci that contribute to COPD pathogenesis.
Methods
We analyzed samples from the Gene-environment interaction and phenotype (GENIE) cohort including patients who visited the Seoul National University Hospital Healthcare System Gangnam Center (SNUH-GC) for health check-up between February 2014 and October 2016. Most patients voluntarily came to the hospital for a personal health check-up or came for a health check-up following receipt of financial support from the company. The present study was approved by the Institutional review board (IRB no: H-1804-028-934) of Seoul National University of Hospital and was in accordance with the 1964 and later modifications of the Helsinki declaration on the use of human subjects for research. Informed consents were obtained from all patients in the cohort.
Among the 4524 subjects of the GENIE cohort, a total of 912 subjects who met any one of the following criteria were excluded: (1) under the age of 40 (2) has lung cancer (3) has an unknown pulmonary function test (PFT) or (4) has an unknown smoking status. One hundred and thirty-four patients were classified as COPD and 4 patients were excluded because those had predicted FEV1 of less than 50%. Finally, a total of 130 patients with early COPD and 3478 controls were recruited (Figure 1). Demographics including age, sex, body mass index (BMI), smoking status, and PFT were collected through electronic medical record review at the time of health screening.
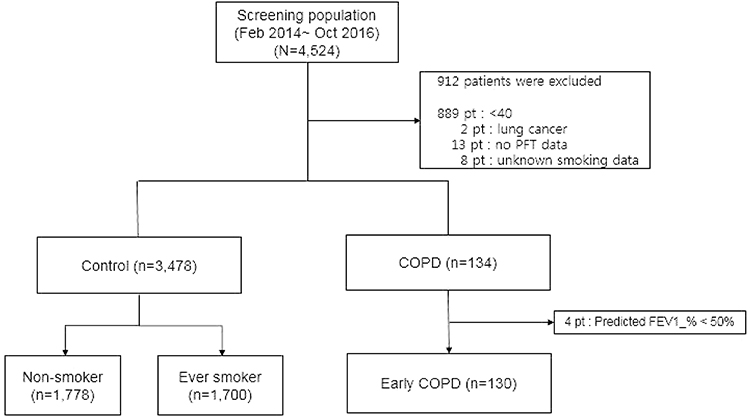 |
Figure 1 Selection criteria used to enroll subjects. |
We obtained PFTs performed by trained professionals in accordance with the standardized ATS guidelines.13 Subjects were labeled control if they had an FEV1/FVC ratio ≥0.7 and an FEV1 ≥80% of the predicted value. Early COPD was defined by an FEV1/FVC ratio <0.7 and FEV1 ≥50% of the predicted value. The primary outcome of our study was to identify functional SNPs associated with early COPD vs the control group.
Genotyping
We collected donated blood samples from the remaining 3608 subjects and analyzed them using the Affymetrix Axiom® Customized Biobank Genotyping Arrays (Affymetrix, Santa Clara, CA, USA), and the PLINK program (ver. 1.07) was used for quality control. Specimens with the following characteristics were excluded from the analyses: low genotype call rate (<97%), sex inconsistency, and related and cryptically related individuals (identical by descent >0.9). SNPs with low call rates (<97%), low minor allele frequency (MAF ≤ 0.05%), or significant deviation from the Hardy-Weinberg equilibrium permutation test (HWE P < 1.0 × 10−5) were excluded. After performing the quality control evaluations for this study, 345,072 autosomal SNPs were retained for the association analyses.
Statistical Analyses
The SNPs associated with early COPD were evaluated with multiple linear or logistical regression methods with adjustments for age, sex, BMI, and smoking status (ever or never smokers), which had the possibility to alter outcomes. A total of 345,072 SNPs that passed the quality control assessment were used for the GWAS. The R software package (version 3.1.1., R Development Core Team; R Foundation for Statistical Computing, Vienna, Austria) was used for statistical analyses and to draw the Manhattan–log10 plots. The significant SNPs (P < 1.45 × 10−7, value derived from the 345,072 QC-qualified SNPs and Bonferroni correction). We performed 1:10 match using propensity score matching (PSM) using Stata 12 software (StataCorp LP, College Station, TX) with the age, sex, and BMI between early COPD and control, never smoker among early COPD and never smoker among the control group, respectively. With this PS matching, we can show what SNPs are associated with early COPD susceptibility regardless of differences (age, sex, and BMI) between early COPD and normal group.
Results
Participant Details
The baseline characteristics of the study population are shown in Table 1. Subjects in the early COPD group were older, had a higher proportion of ever smokers when compared with the control group. When compared with the never smoker control group, the early COPD group had a significantly higher proportion of males and were likely to have poorer pulmonary functions including FEV1, FVC, and FEV1/FVC ratio.
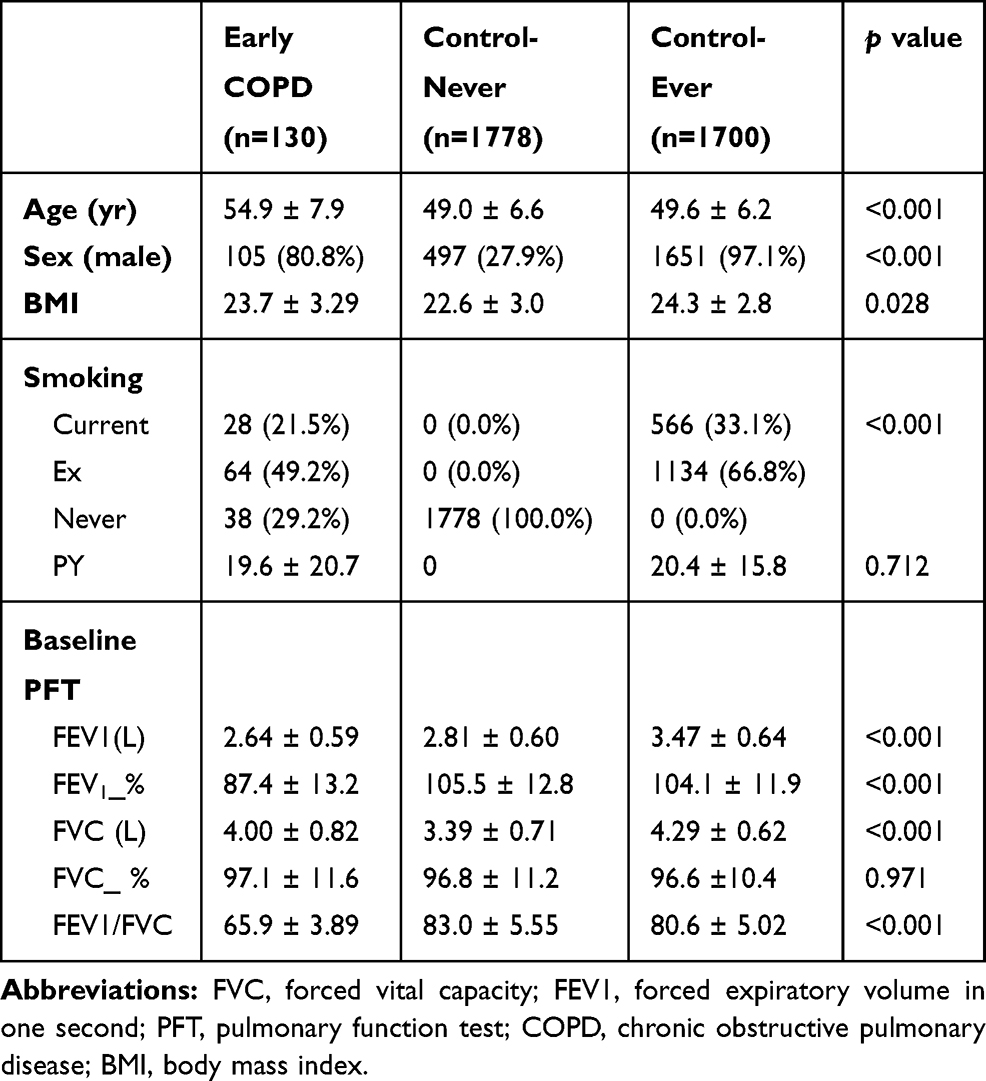 |
Table 1 Baseline Characteristics of Subjects |
Top SNPs
In our study, we found no significant SNP association with early COPD in the GENIE cohort. The SNP most associated with early COPD was the rs2818103 SNP and the second most associated SNP was the rs875033 SNP, yielding p =5.6 x 10−7 and p = 2.7 x 10−6, respectively. Both SNPs were located on chromosome 6. The top 10 most significant p-values obtained after analysis are presented in Table 2. The Manhattan and the Q-Q plots showing the distribution of the observed p-values from the GENIE cohort is shown in Figure 2. These results were obtained after adjusting for age, sex, BMI, and smoking status (ever vs never smoker).
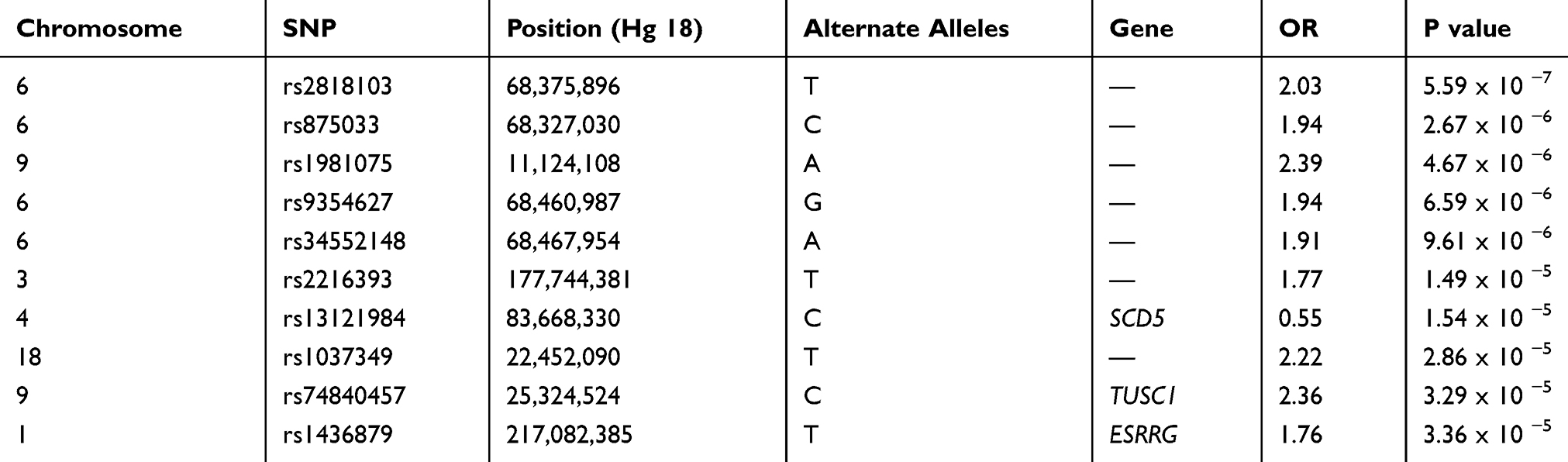 |
Table 2 Association Results for the Top 10 Selected SNPs with Early COPD Compared with Control |
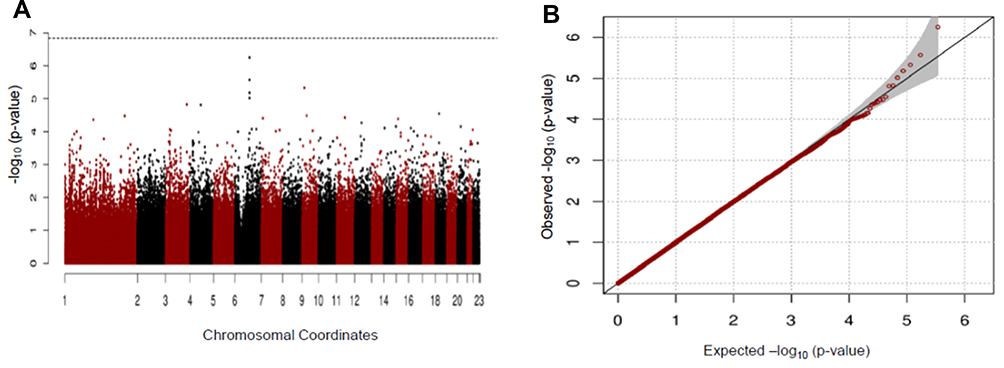 |
Figure 2 Manhattan (A) and Q-Q (B) plots for GWAS analysis of early COPD based on the GENIE cohort. |
In PS matching (age, sex and BMI) analysis, the results were similar in that the rs2818103 SNP on chromosome 6 was just below the genome-wide significance threshold (p = 1.9 x 10−7) and several other SNPs located on chromosome 6 had trends for association with early COPD (Table 3 and Figure 3). This chromosome 6 was detected five times among the top 10 most significant SNPs found in this study.
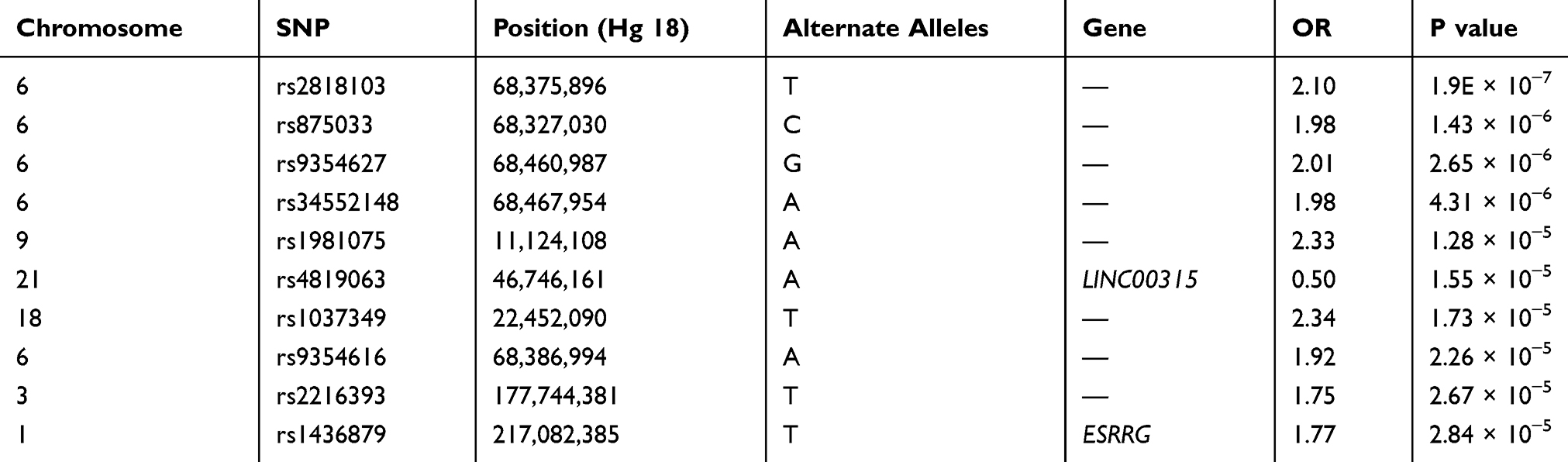 |
Table 3 Association Results for the Top 10 Selected SNPs with Early COPD Compared with Control After Propensity Score Matching Using Age, Sex, and BMI |
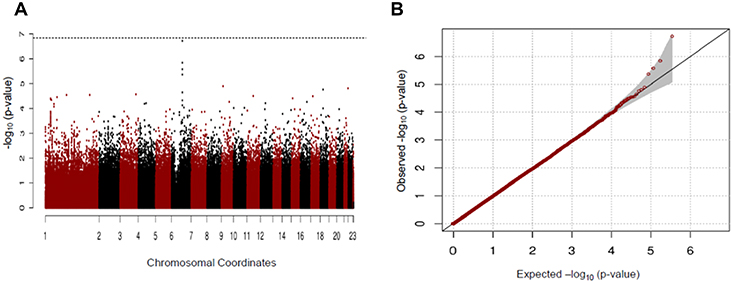 |
Figure 3 (A) Manhattan and (B) Q-Q plots for GWAS analysis of early COPD based on the GENIE cohort after propensity score matching using age, sex, and BMI. |
There was a consistent association of chromosome 6 with never smoker among early COPD, when compared with never smoker control. Although no novel variants reached genome-wide significance, but several SNPs (rs2857210 and rs2621419) on this chromosome 6 were just below the genome-wide significance threshold (P = 7.90 × 10−6, 8.56 × 10−6, respectively) (Table 4). These two SNPs and other SNPs on chromosome 6 were located within HLA (Human Leukocyte Antigen)-DQB2 (Figure 4). After SNPs in chromosome 6, the next most significant SNP in never smoker among early COPD patients, when compared with never smokers, was the rs74013641 SNP which lies the mitogen-activated protein kinase 6 (MAPK6) (P = 1.52 × 10−5) gene. The top 10 SNPs (with the most significant P values) associated with never smoker among early COPD when compared with the never smoker control is presented in Table 4 and Figure 4.
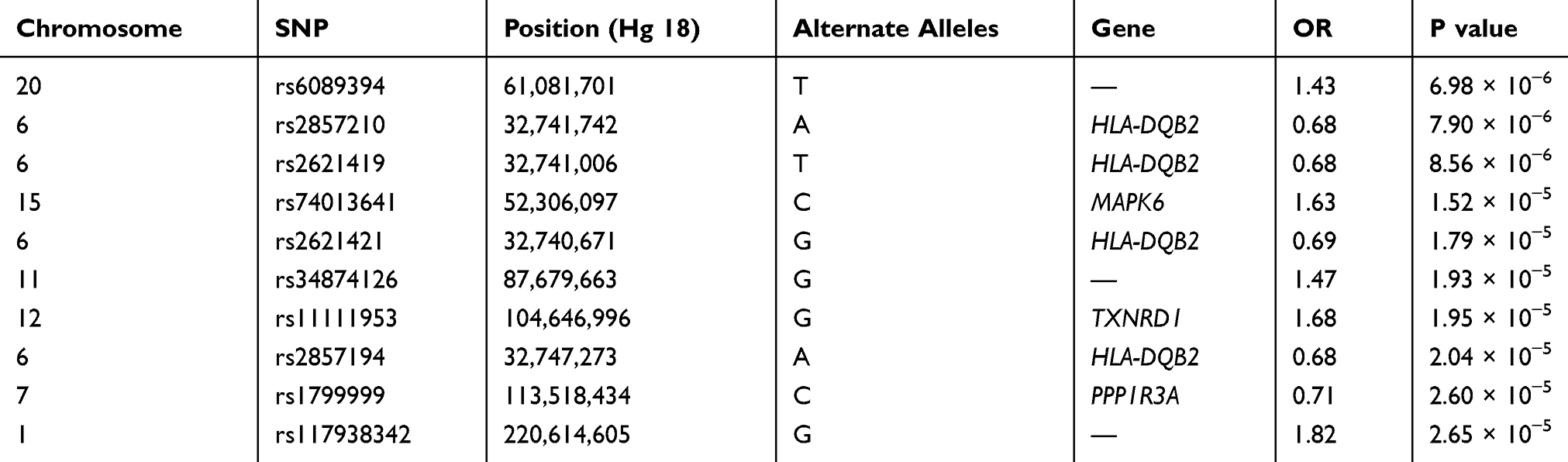 |
Table 4 Association Results for the Top 10 Selected SNPs with Never Smoker Among Early COPD Compared with Those in Control |
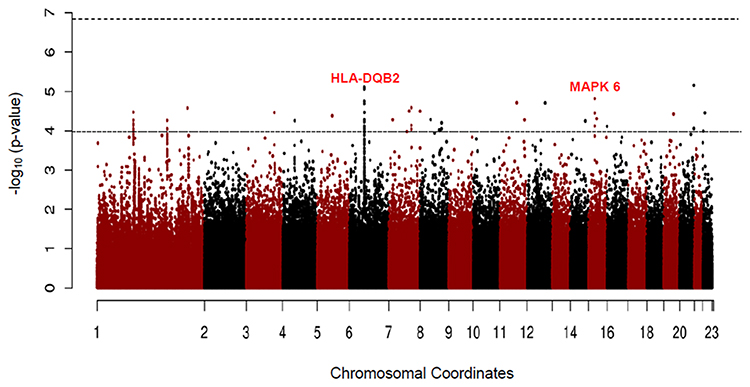 |
Figure 4 Manhattan plot for GWAS analysis of never smoker among early COPD based on 38 cases and 1778 never smoker controls. |
In addition, to exclude confounding effect of smoking, which is by far the most important cause of COPD, we compared the never smokers in the early COPD group with the never smokers in the control group. After PSM, 38 never smoker among early COPD patients and 1294 never smoker controls were included in each group, respectively. In this analysis, the rs2777774 SNP located in the transducin-like enhancer of split 1 (TLE1) gene on chromosome 9 was associated with patients in the early COPD group (despite having no history of smoking) but no association was observed with the never smoker controls. The second most associated SNP associated with the never smokers in the early COPD group was the rs117744226 SNP located in the pellino E3 ubiquitin protein ligase family number 2 (PELI2, on chromosome 14). Unfortunately, these SNPs did not reach statistical genome-wide significance (Table 5 and Figure 5).
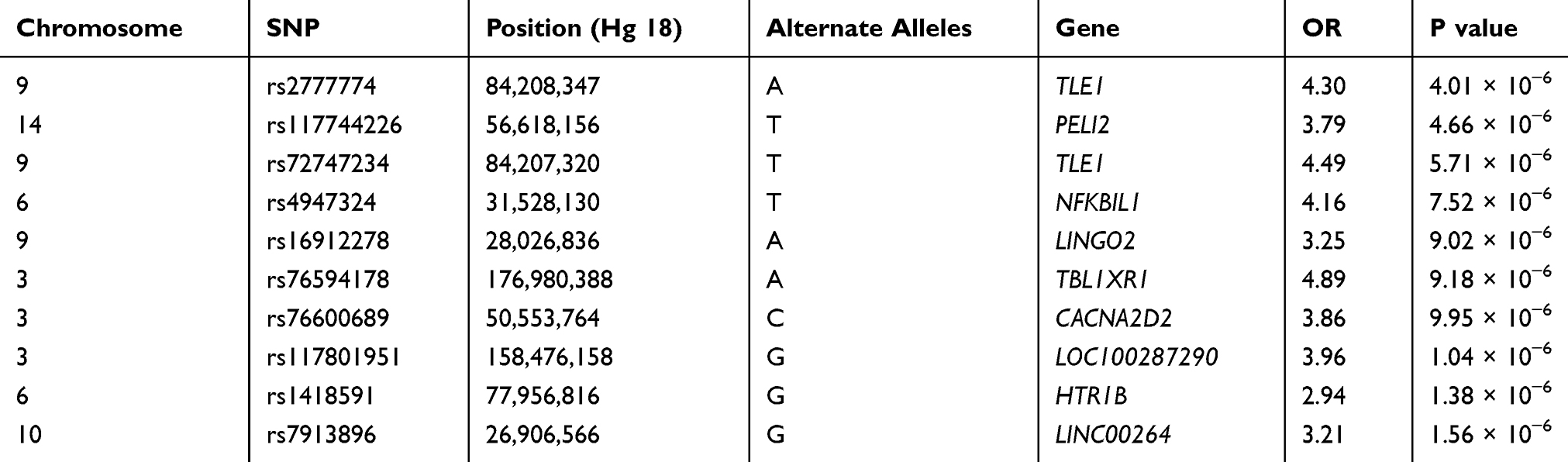 |
Table 5 Association Results for the Top 10 Selected SNPs with Never Smoker Among Early COPD Compared with Those in Control After Propensity Score Matching Using Age, Sex, and BMI |
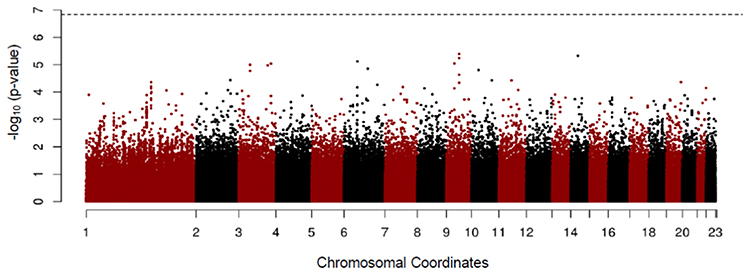 |
Figure 5 Manhattan plot for GWAS analysis of never smoker among early COPD based on 38 cases and 1294 those among control after propensity score matching using age, sex, and BMI. |
Discussion
In this Korean GENIE cohort study, we have shown that the rs2818103 and rs875033 SNPs on chromosome 6 are associated, after adjusting for smoking, age, sex, and BMI, with genetic susceptibility to early COPD although genome-wide significance was not met. After PSM for age, sex, and BMI, these SNPs were also observed to be associated with susceptibility to early COPD. Furthermore, we identified that the rs2857210 and rs2621419 SNPs in the HLA-DQB2 loci on chromosome 6 are associated with susceptibility to early COPD but not with the never smoker controls.
As we mentioned above in the introduction briefly, the previous researches have been focused on the major susceptibility loci of patients with moderate to severe COPD and they adjusted for smoking behavior and pack-years of smoking but did not show significant susceptible SNPs among never smoker separately. Our study showed not only the susceptible SNPs in early COPD after adjustment smoking behavior, but also demonstrated the GWAS result of never smoker early COPD patients.
To date, only a few studies have looked at the relationship between COPD and HLA. Two previous studies reported that HLA-Bw6014 and HLA-DRB1*1415 were associated with COPD. A small number of GWAS have demonstrated that there is a COPD susceptibility locus in the HLA region on chromosome.7,16,17 In addition, one other study, using microRNAs, identified HLA-DQB1-AS1 as a key player in smoking-related COPD.17 Although the association described in these studies is with advanced COPD, emphysema,17 and smoking-related COPD,18 these findings support previously described associations of COPD with HLA-DQ, nevertheless. Autoimmunity may contribute to COPD pathogenesis since COPD occurs among never smokers and, more frequently, in patients with rheumatoid arthritis.19 The HLA molecules, particularly HLA class II, are associated with autoimmune diseases, for example, type 1 diabetes and systemic lupus erythematous.20 This study, therefore, raises the possibility of an association between COPD and HLA class II, mainly HLA-DQ.
The third most significant SNP in our cohort is the rs74013641 SNP found in the intron of MAPK6 which is one of the MAP kinase. The MAPK6 is one of the MAP kinases which is known to be activated in fibroblasts upon treatment with serum or phorbol esters21 and this gene encodes the extracellular signal-regulated kinase 3 (Erk3). This gene is known to be activated in fibroblasts upon treatment a gene associated with marked pulmonary hypoplasia in mice.22 Other studies have shown that MAPK6, as a mediator of antenatal steroid action, plays a key role in producing surfactant protein.23,24 The role of MAPK6 in the pathogenesis of COPD or in lung development in human remains unknown. Hence, our finding is the first to demonstrate a potential novel COPD susceptibility locus in the MAPK6 gene on chromosome 15.
In the analysis excluding smoking (a well-known, most powerful risk factor for the development of COPD) the TLE1 gene was found to be associated with the never smokers in the early COPD group. TLE1 is a member of the Groucho/TLE gene family encoding transcriptional corepressors and it interacts with DNA-binding transcriptional factors.25,26 This gene has been described as an important player in synovial sarcoma,27,28 lung cancer29 and a major negative regulator of inflammation involving macrophages by regulating the NF-κB pathway leading to lung hypoplasia.30 However, no literature reported the association of TLE1 with COPD. Large sample studies are needed to validate our findings and to evaluate for a possible association of TLE1 with COPD patients with no previous smoking history.
PELI2 gene, which was associated with early COPD next to the TLE genes in our result, involved in the toll-like receptor and interleukin-1 signaling pathways and is an activator of the MAP kinase. However, like TLE1, there is no study to evaluate the association between PELI2 and COPD.
More than 70% of patients with COPD have GOLD stage 1 (mild) or 2 (moderate) with no apparent respiratory symptoms such as dyspnea on exertion.31 Previous studies showed that Tiotropium could ameliorate the annual decline in the FEV1 and lower the frequency of acute exacerbation when compared with placebo-treated patients with GOLD stage 1 or 2 COPD.32,33 Therefore, earlier detection of COPD could lead to early effective intervention, reducing disease progression and socioeconomic burden. Such early detection has been the focus in areas like in the United States America (USA) where COPD monitoring is performed through the National Lung Health Education Program.
It is cost effective to perform selective PFT with bronchodilator response only to patients with genetic susceptibility to COPD, rather than to all patients. Also, considering that the development of COPD is not only linked to genetic susceptibility but also to exposure to various environmental factors, a GWAS of early COPD is needed to determine the genetic factors affecting COPD. Despite substantial progress in evaluating the genetic susceptibility to COPD using GWAS, there has been no study that investigated the genetic risk factors for early COPD. Thus, our study has provided meaningful insights into this genetic risk association.
Our study has several limitations. First, our top SNPs did not approach genome-wide significance probably due to the small patient sample size when compared with the control sample size. Therefore, a replication cohort study with a large sample size to support our study is needed. Second, the definition of COPD is based on pre-bronchodilator data. Indeed, it is possible that asthma patients might have been included among those defined as having COPD. Third, we did not perform functional investigation of whether these single genetic variants affect early COPD in lung tissue samples. Finally, the prevalence of COPD in this study (4.1%) is lower than expected among Korean adults over 40 years old (13.4%).34 A possible explanation for this discrepancy is that the current cohort consists of participants who are healthy, have a high socioeconomic status than the general population and have regular check-ups.
In conclusion, our study, using the GENIE Korean cohort, suggests that several SNPs including the rs2818103 and rs875033 SNPs on chromosome 6 and the rs2857210 and rs2621419 SNPs in loci HLA-DQB2 are possibly associated with early COPD. We also identified that the MAPK6 and TLE1 genes might have a role in susceptibility to early COPD development, particularly in lung hypoplasia. Further studies are needed to validate our findings.
Statement of Ethics
All study participants provided informed consents. The present study was approved by the Institutional review board (IRB no: H-1804-028-934)
Author Contributions
All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Funding
This study received funding from the Seoul National University Hospital Research Fund, grant number 2720170100 (2017-3376).
Disclosure
The authors have no conflicts of interest to declare.
References
1. Adeloye D, Chua S, Lee C, et al. Global and regional estimates of COPD prevalence: systematic review and meta-analysis. J Glob Health. 2015;5(2). doi:10.7189/jogh.05.020415.
2. Lopez A, Shibuya K, Rao C, et al. Chronic obstructive pulmonary disease: current burden and future projections. Eur Respir J. 2006;27(2):397–412. doi:10.1183/09031936.06.00025805
3. Bascom R. Differential susceptibility to tobacco smoke: possible mechanisms. Pharmacogenet Genomics. 1991;1(2):102–106. doi:10.1097/00008571-199111000-00008
4. Mannino DM. COPD: epidemiology, prevalence, morbidity and mortality, and disease heterogeneity. Chest. 2002;121(5):121S–126S. doi:10.1378/chest.121.5_suppl.121S
5. Mannino DM. Chronic obstructive pulmonary disease: definition and epidemiology. Respir Care. 2003;48(12):1185–1193.
6. Silverman EK, Sandhaus RA. Alpha1-antitrypsin deficiency. N Engl J Med. 2009;360(26):2749–2757. doi:10.1056/NEJMcp0900449
7. Pillai SG, Ge D, Zhu G, et al. A genome-wide association study in chronic obstructive pulmonary disease (COPD): identification of two major susceptibility loci. PLoS Genet. 2009;5(3):e1000421. doi:10.1371/journal.pgen.1000421
8. Cho MH, Boutaoui N, Klanderman BJ, et al. Variants in FAM13A are associated with chronic obstructive pulmonary disease. Nat Genet. 2010;42(3):200. doi:10.1038/ng.535
9. Hobbs BD, Parker MM, Chen H, et al. Exome array analysis identifies a common variant in IL27 associated with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2016;194(1):48–57. doi:10.1164/rccm.201510-2053OC
10. Ferhani N, Letuve S, Kozhich A, et al. Expression of high-mobility group box 1 and of receptor for advanced glycation end products in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;181(9):917–927. doi:10.1164/rccm.200903-0340OC
11. General USJWDoH, Services H. The Health Benefits of Smoking Cessation. Washington: Department of Health and Human Services; 1990.
12. Soriano JB, Polverino F, Cosio B. What is early COPD and why is it important? Eur Respir J. 2018;52(6):1801448. doi:10.1183/13993003.01448-2018
13. American Thoracic Society. Standardization of spirometry, 1994 update. Am J Respir Crit Care Med. 1995;152(3):1107–1136. doi:10.1164/ajrccm.152.3.7663792
14. Maranetra N, Chandanayingyong D, BoYornklttl S. HLA antigen and ventilatory drive in Thais with chronic obstructive pulmonary disease. Asian Paci J Allerg Immunol. 1990;8(2):137.
15. Faner R, Cruz T, Agusti A. Immune response in chronic obstructive pulmonary disease. Expert Rev Clin Immunol. 2013;9(9):821–833. doi:10.1586/1744666X.2013.828875
16. Qiu W, Cho MH, Riley JH, et al. Genetics of sputum gene expression in chronic obstructive pulmonary disease. PLoS One. 2011;6(9):e24395. doi:10.1371/journal.pone.0024395
17. Hardin M, Cho M, McDonald M-L, et al. The clinical and genetic features of COPD-asthma overlap syndrome. Eur Respir J. 2014;erj02160–02013.
18. Qian Y, Mao Z-D, Shi Y-J, Liu Z-G, Cao Q, Zhang Q. Comprehensive analysis of miRNA-mRNA-lncRNA networks in non-smoking and smoking patients with chronic obstructive pulmonary disease. Cell Physiol Biochem. 2018;50(3):1140–1153. doi:10.1159/000494541
19. Dhital R, Basnet S, Paudel P, Acharya YP, Poudel DR. Prevalence of chronic obstructive pulmonary disease (COPD) among rheumatoid arthritis: results from national inpatient database. J Commun Hosp Internal Med Perspect. 2018;8(4):211–214. doi:10.1080/20009666.2018.1485460
20. Klein J, Sato A, Mackay IR, Rosen FS. The HLA system. N Engl J Med. 2000;343(10):702–709. doi:10.1056/NEJM200009073431006
21. Pearson G, Robinson F, Beers Gibson T, et al. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev. 2001;22(2):153–183.
22. Klinger S, Turgeon B, Lévesque K, Wood GA, Aagaard-Tillery KM, Meloche S. Loss of Erk3 function in mice leads to intrauterine growth restriction, pulmonary immaturity, and neonatal lethality. Proc Natl Acad Sci. 2009;106(39):16710–16715. doi:10.1073/pnas.0900919106
23. Pew BK, Harris RA, Sbrana E, et al. Structural and transcriptomic response to antenatal corticosteroids in an Erk3-null mouse model of respiratory distress. Am J Obstet Gynecol. 2016;215(3):
24. Guaman MC, Sbrana E, Shope C, et al. Administration of antenatal glucocorticoids and postnatal surfactant ameliorates respiratory distress syndrome–associated neonatal lethality in Erk3−/− mouse pups. Pediatr Res. 2014;76(1):24. doi:10.1038/pr.2014.54
25. Chen G, Courey AJ. Groucho/TLE family proteins and transcriptional repression. Gene. 2000;249(1–2):1–16. doi:10.1016/S0378-1119(00)00161-X
26. Jennings BH, Ish-Horowicz D. The Groucho/TLE/Grg family of transcriptional co-repressors. Genome Biol. 2008;9(1):205. doi:10.1186/gb-2008-9-1-205
27. Terry J, Saito T, Subramanian S, et al. TLE1 as a diagnostic immunohistochemical marker for synovial sarcoma emerging from gene expression profiling studies. Am J Surg Pathol. 2007;31(2):240–246. doi:10.1097/01.pas.0000213330.71745.39
28. Knösel T, Heretsch S, Altendorf-Hofmann A, et al. TLE1 is a robust diagnostic biomarker for synovial sarcomas and correlates with t (X; 18): analysis of 319 cases. Eur J Cancer. 2010;46(6):1170–1176. doi:10.1016/j.ejca.2010.01.032
29. Yao X, Ireland SK, Pham T, et al. TLE1 promotes EMT in A549 lung cancer cells through suppression of E-cadherin. Biochem Biophys Res Commun. 2014;455(3):277–284. doi:10.1016/j.bbrc.2014.11.007
30. Ramasamy S, Saez B, Mukhopadhyay S, et al. Tle1 tumor suppressor negatively regulates inflammation in vivo and modulates NF-κB inflammatory pathway. Proc Natl Acad Sci. 2016;113(7):1871–1876. doi:10.1073/pnas.1511380113
31. Zhong N, Wang C, Yao W, et al. Prevalence of chronic obstructive pulmonary disease in China: a large, population-based survey. Am J Respir Crit Care Med. 2007;176(8):753–760.
32. Zhou Y, Zhong N-S, Li X, et al. Tiotropium in early-stage chronic obstructive pulmonary disease. N Engl J Med. 2017;377(10):923–935. doi:10.1056/NEJMoa1700228
33. Welte T, Vogelmeier C, Papi A. COPD: early diagnosis and treatment to slow disease progression. Int J Clin Pract. 2015;69(3):336–349. doi:10.1111/ijcp.12522
34. Hwang YI, Park YB, Yoo KH. Recent trends in the prevalence of chronic obstructive pulmonary disease in Korea. Tuberc Respir Dis (Seoul). 2017;80(3):226–229. doi:10.4046/trd.2017.80.3.226
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.