")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 13
A functional SNP upstream of the ADRB2 gene is associated with COPD
Authors Li JX , Fu WP, Zhang J, Zhang XH, Sun C, Dai LM, Zhong L, Yu L, Zhang YP
Received 8 September 2017
Accepted for publication 3 November 2017
Published 16 March 2018 Volume 2018:13 Pages 917—925
DOI https://doi.org/10.2147/COPD.S151153
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Chunxue Bai
Jin-Xiu Li,1,2,* Wei-Ping Fu,3,* Jing Zhang,4 Xiao-Hua Zhang,1,2 Chang Sun,1,5 Lu-Ming Dai,3 Li Zhong,1,5,6 Li Yu,1,2 Ya-Ping Zhang1,7
1State Key Laboratory for Conservation and Utilization of Bio-Resource in Yunnan, 2Key Laboratory for Animal Genetic Diversity and Evolution of High Education in Yunnan Province, School of Life Sciences, Yunnan University, 3Department of Respiratory Critical Care Medicine, 4Department of Thoracic Surgery, The First Affiliated Hospital of Kunming Medical University, Kunming, 5College of Life Sciences, 6Provincial Demonstration Center for Experimental Biology Education, Shaanxi Normal University, Xi’an, 7State Key Laboratory of Genetic Resources and Evolution, and Yunnan Laboratory of Molecular Biology of Domestic Animals, Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming, China
*These authors contributed equally to this work
Background: Previous studies have suggested that β2-adrenergic receptor (ADRB2) is associated with COPD. However, the role of genetic polymorphisms in ADRB2 on COPD has not been evaluated yet.
Methods: In this study, SNaPshot genotyping, luciferase assay, chromatin immunoprecipitation and real-time polymerase chain reaction were adopted to investigate the association between ADRB2 genetic polymorphisms and COPD, comprehensively.
Results: One single nucleotide polymorphism (rs12654778), located upstream of ADRB2, showed a significant association with COPD by the logistic regression analysis after adjusting for age, sex and smoking history (p=0.04) in 200 COPD patients and 222 controls from southwest Chinese population. Furthermore, the luciferase assay indicated that rs12654778-A allele reduced the relative promoter activity by ~26% compared with rs12654778-G allele (p=0.0034). The chromatin immunoprecipitation analysis demonstrated that rs12654778 modulated the binding affinity of transcription factor neurofibromin 1. In addition, a significantly reduced expression of ADRB2 in COPD patients was observed, compared with normal controls (p=0.017).
Conclusion: Our findings suggest a previously unknown mechanism linking allele-specific effects of rs12654778 on ADRB2 expression to COPD onset, for the first time.
Keywords: β2-adrenergic receptor, ADRB2, FEV1, lung, polymorphism
Introduction
COPD, one of the most common respiratory diseases in old people, is characterized by airflow limitation, that is, a chronic persistent inflammatory process that is not fully reversible. Nowadays, COPD has become the third source of morbidity in the world.1 Although tobacco smoking has been suggested to be the predominant environmental factor for COPD, only ~10%–20% smokers develop airway obstruction.2 This phenomenon, together with the familial clustering in COPD patients,3,4 indicates that genetic factors might play an important role in the development of COPD. Recent genome-wide association studies have identified multiple COPD susceptibility genes,8 for example, Hedgehog interacting protein (HHIP). So far, only α1-antitrypsin (SERPINA1) has been confirmed to be a genetic risk factor for COPD. However, the mutant protease inhibitor Z homozygote of the gene, which could increase individual susceptibility to COPD, is extremely rare across worldwide populations (0.001%–4.5%), especially in Asians (<0.004%), and accounts for only 2% of COPD patients.5,6 Thus, additional genes were assumed to also play a crucial role in the predisposition to COPD and remained to be identified.7
A major cause for COPD is airflow obstruction in the lung and respiratory parenchyma maintained by airway smooth muscle cells. Chronic obstructive abnormality followed by airway remodeling increases the thickness of the airway and causes airflow obstruction.9 Therefore, genes involved in the regulation of airway smooth muscle tone are good candidates for the genetic predisposition to COPD.
β2-adrenergic receptor (ADRB2) is a G protein-coupled transmembrane receptor located on airway smooth muscle cells,10 and increase in ADRB2 gene expression was observed in COPD patients compared to patients with mild/moderate asthma,11 leading to the speculation that it is a candidate gene for COPD.12 Thus, many previous studies have focused on the relationship between single nucleotide polymorphisms (SNPs) in the coding region of ADRB2 and COPD in the Asian population, for example, rs1042713 (Arg16Gly)13–20 and rs1042714 (Gln27Glu),13,15,17,18,21 since these SNPs are thought to be affecting the construction of ADRB2 or altering the function of the translation product. However, there is still controversy in this issue,13–15 and more genotyping data and a meta-analysis are indispensable to resolve this conflict.
In addition, growing knowledge has shown that noncoding SNPs, especially the ones in the promoter region, are more important and might be functional, for example, affecting transcription factors’ (TFs) binding affinity and further influencing gene expression.22 Thus, investigation of the variants in the promoter of ADRB2 is valuable not only to focus on how the gene expression of ADRB2 is regulated, but also to discover the causal molecular mechanism of COPD. There are several SNPs, rs1801704 (−20T/C), rs1042711 (−47T/C), rs11959427 (−367T/C), rs11168070 (−468C/G), rs12654778 (−654G/A), rs2053044 (−1023G/A), rs2400707 (−1343A/G) and rs2895795 (−1429T/A), identified in the promoter region of ADRB2, which contain a number of putative regulatory elements.23,24 Previous studies focused on rs1042711 and found that this SNP could introduce a non-conservative amino acid change (Arg→Cys) at the 19th amino acid, further influencing the gene expression of ADRB2,24,25 since it lies within a 19 amino acid peptide (referred to as β upstream peptide) in the 5′ leader region.25 However, the crucial issue of whether other SNPs affect the expression of ADRB2 or the onset of COPD has never been scrutinized.
Due to the pivotal role of ADRB2 in lung function, we hypothesized that noncoding SNPs of ADRB2 could be important for its expression. To address this hypothesis, we validated whether its expression could be associated with COPD. Subsequently, a comprehensive evaluation to identify the causal variants and investigate the functional impact of this SNP on COPD pathogenesis was undertaken. This study would provide new insight into the potential molecular basis for COPD.
Materials and methods
Study population and lung tissues
A total of 422 adult subjects (200 unrelated patients with COPD and 222 healthy smokers) were recruited from the First Affiliated Hospital of Kunming Medical University (Kunming, China) for genotyping. All smokers belonged to Han nationality to minimize the potential sampling bias due to population stratification. COPD patients were diagnosed based on the results from multiple examinations, including the ratio of forced expiratory volume in 1 second/forced vital capacity (FEV1/FVC ratio <70% and FEV1 <80% predicted), according to the Global Initiative for Chronic Obstructive Lung Disease criteria.26 The healthy smokers exhibited normal pulmonary function (FEV1/FVC ratio >70% and FEV1 >80% predicted) and a smoking history of ≥10 pack-years. In addition, they were excluded from the possibility of COPD by chest computed tomography (CT).
To investigate whether the expression of ADRB2 could be related with COPD, human lung tissue samples from 18 COPD patients as case subjects (FEV1 <80%) and 24 control subjects with normal lung function were also collected from the same hospital.
This study was approved by the institutional ethics committee of the First Affiliated Hospital of Kunming Medical University, and all participants were contacted by telephone to obtain verbal informed consent. Detailed information of patients and healthy smokers is presented in Table 1.
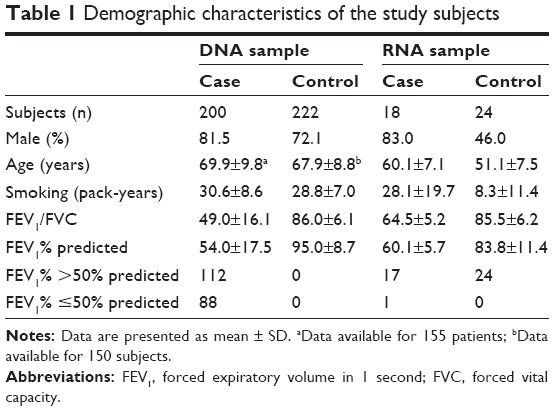 | Table 1 Demographic characteristics of the study subjects |
Transcription analysis
Total RNAs were isolated by Trizol (Thermo Fisher Scientific, Waltham, MA, USA) from human lung tissues stored in RNAlater (Thermo Fisher Scientific) solution. cDNA was synthesized by SuperScript® III First-Strand Synthesis System (Thermo Fisher Scientific). Transcript levels for ADRB2 gene in the lung were measured by real-time polymerase chain reaction (PCR) with SYBR green (Kapa Biosystems, Wilmington, MA, USA) and primers (forward primer: 5′-AGGCAGCTCCAGAAGATTG-3′ and reverse primer: 5′-CCAGCAGAGGGTGAAAGTG-3′). All samples were tested on ABI PRISM® 7000 Sequence Detection System (Thermo Fisher Scientific) with three replications under the following cycling conditions: 2 min at 50°C, 10 min at 95°C, 40 cycles of 15 s at 95°C and 1 min at 60°C. The average cycle number at threshold (Ct) was normalized by glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The expression level of ADRB2 was calculated based on the 2−ΔΔCt method.
Tag SNPs selection
SNPs within the ~8 kb region (chr5: 148203156–148211197, relative to build 37) containing the entire ADRB2 gene in East Asian population (Han Chinese in Bejing, China [CHB] and Japanese in Tokyo, Japan [JPT], n=89) were downloaded from 1,000 genomes project (www.internationalgenome.org/).27 Tag SNPs were chosen by ldSelect software with r2>0.8.28 Among the 32 SNPs identified in the East Asian population, eight tag SNPs were chosen for genotyping (Supplementary Figure 1).
Sample size power calculation
To assess whether our sample size for the association study was enough, genetic power calculation was used in this study,29 with 7.3% disease prevalence in the Chinese population30 and α=0.05, for a variant with 0.05 minor allele frequency in a dominant model. In the calculation of genetic power, ≥80% is a general threshold for acceptable power.
Genotyping of tag SNPs in ADRB2
Genomic DNA was extracted from peripheral blood by phenol–chloroform method. The genotype of each tag SNP for COPD patients and healthy smokers was screened by SNaPshot according to the manufacturer’s protocol (Thermo Fisher Scientific). In brief, multiplex PCR was performed by primers given in Supplementary Table 1 with FastStart Taq DNA polymerase (Roche, Basel, Switzerland). After alkaline phosphatase (Shrimp, Takara-Bio Inc., Kusatsu, Japan) and exonuclease I (Takara Bio Inc.) clean-up, single base extension was performed by SNaPshot Multiplex Ready Reaction Mix (Thermo Fisher Scientific) and the products were analyzed on ABI PRISM 3730 sequencer (Thermo Fisher Scientific). The genotypes of some random samples were confirmed by resequencing in 3730 sequencer.
Cell culture
Human bronchus normal epithelial cells Beas-2B (#CRL-9609; American Type Culture Collection, Manassas, VA, USA) were cultured in 1640 medium (Thermo Fisher Scientific) with 10% fetal bovine serum (Thermo Fisher Scientific) in 5% CO2 at 37°C.
Luciferase reporter assay
ADRB2 promoter ~1.38 kb region (chr5: 148205009–148206387, relative to build 37) was amplified using primers 5′-CAGTCGCTAGCTTTGGTAAGTCACAGACGCCAG-3′ and 5′-CAGTCAAGCTTAGTCTGGCAGGTGAGCGCAC-3′, which introduced restriction sites for NheI and HindIII (New England Biolabs, Ipswich, MA, USA), respectively. PCR was performed by Pfu DNA polymerase (recombinant) enzyme (Thermo Fisher Scientific) to avoid artificial mutation. After digestion, the segment was cloned into the compatible sites of the pGL3-basic vector (Promega, Madison, WI, USA). A plasmid with another allele (G) for rs12654778 was generated through mutagenesis with Phusion Site-Directed Mutagenesis Kit (Thermo Fisher Scientific) and primer pair 5′-TCGGTATAAGTCTAAGCATGTCTGCC-3′ and 5′-ACCACAGCCATAGACACTGAGACAC-3′, according to the manufacturer’s protocol. Plasmid DNA was sequenced to exclude any PCR errors and check the orientation of the haplotypes prior to transfection.
Plasmid constructs with rs12654778-A and G (475 ng) were transfected into Beas-2B cells by Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer’s recommendations. After 24 hours transfection, cells were harvested and luciferase activity was measured by Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer’s protocol. Plasmid pRL-TK (25 ng; Promega) was co-transfected as an internal control and the promoter activity was expressed as the ratio between firefly and Renilla luciferase. Independent transfection and reporter assays were performed six times.
Chromatin immunoprecipitation (ChIP)-PCR assay
ChIP was carried out with EZ-ChIP Assay Kit (EMD Millipore, Billerica, MA, USA) according to the manufacturer’s protocol. Briefly, Beas-2B cells were grown to reach sub-confluency. Approximately 1×107 cells were cross-linked for 10 min with formaldehyde (1% final concentration) at room temperature, which was followed by addition of glycine for 5 min to end the cross-linking. After washing twice with ice-cold phosphate-buffered saline (Thermo Fisher Scientific) containing protease inhibitor cocktail, cells were scraped, lysed and sonicated to obtain 200–800 bp fragments in the Sonicator (Branson, Medford, NY, USA). The chromatin solution was diluted 10-fold with dilution buffer and precleared with protein G beads. After centrifuging and transferring the supernatant, 1% sample was stored as input and the remaining protein/chromatin complex was subjected to immunoprecipitation with mouse monoclonal NF-1 antibody (Santa Cruz Biotechnology, Dallas, TX, USA) or normal mouse IgG as a negative control, and precipitated by protein G beads. After washing with low salt, high salt, LiCl and Tris-EDTA buffer (twice), the immunoprecipitated protein/chromatin complex was resuspended in elution buffer and the cross-links were reversed. Protein was digested by proteinase K and DNA was recovered. The obtained DNA from ChIP preparation was quantified by real-time PCR to evaluate the enrichment using SYBR green with primers 5′-TGTGTTGGACAGGGGTGACTT-3′ and 5′-ACATTCGGAAGGAAACGAGAGT-3′. In the ChIP assay, relative enrichment was normalized by input DNA. Data are presented as the mean ± SD of triplicate experiments.
Statistical analysis
Age, smoking history and pulmonary function data are displayed as mean ± SD. Hardy–Weinberg equilibrium was evaluated by a goodness-of-fit chi-square test with one degree of freedom. The frequencies of each SNP between patients and controls were compared by two-tailed chi-square tests. To assess the independent effect of each SNP on COPD, a logistic regression analysis with tag SNPs (rs17108803, rs1432623, rs12654778, rs1042713, rs1042714, rs1042717, rs1042719 and rs1042720) as independent variables adjusted for age, sex and smoking history was also performed. For comparing the ADRB2 expression between cases and controls in the lung tissues and the luciferase activity, independent t-test was performed. All statistical tests were performed in SPSS 13.0 (SPSS Inc., Chicago, IL, USA). Odds ratios and 95% CIs were also calculated to assess the relative disease risk. In this study, the significance level was accepted when p (probability) value was <0.05.
Transcription factor-binding site prediction
For evaluating whether rs12654778 would alter the binding affinity of the TF, a putative TF-binding site was analyzed by using the web-based TRANSFAC database (http://www.gene-regulation.com/cgi-bin/pub/programs/match/bin/match.cgi).
Results
ADRB2 expression in lung tissues from cases and controls
We firstly assessed whether ADRB2 gene expression was altered in the lung tissues from COPD subjects. We measured ADRB2 expression in the lung tissues of COPD subjects and smokers who had normal lung function. By real-time PCR, we found that mRNA levels of ADRB2 were significantly reduced ~28% in COPD subjects compared with control subjects (p=0.017; Figure 1), confirming that ADRB2 is differentially expressed between pathologic and normal tissues and the decreased ADRB2 expression is associated with COPD development or onset.
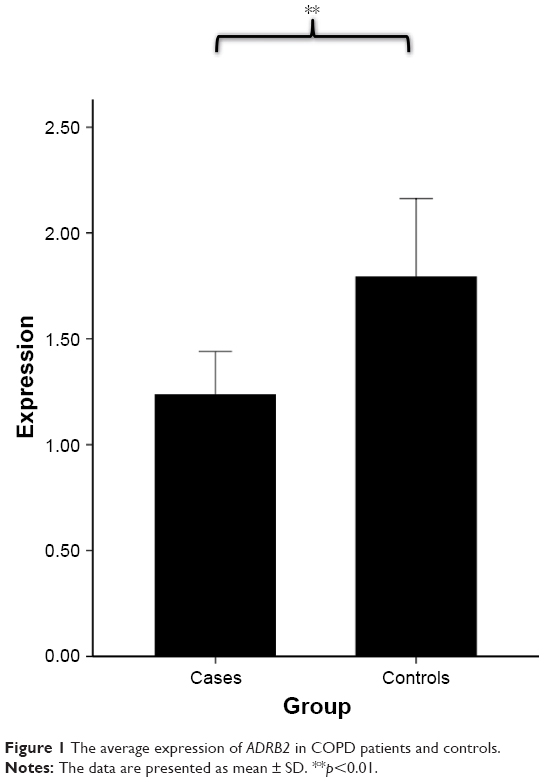 | Figure 1 The average expression of ADRB2 in COPD patients and controls. |
Tag SNPs selection
To develop a comprehensive list of common genetic variants for ADRB2, the genotype data for East Asian population were obtained from 1,000 genomes project. In this ~8 kb region, 32 SNPs were identified, among which 12 were in the upstream region of ADRB2, six were in the 5′ untranslated region, six were in the 3′ untranslated region, five were synonymous and three were missense mutations in the coding region. Subsequently, 14 blocks were observed (Supplementary Figure 1; Supplementary Table 2). Eight blocks showed minor allele frequency >5%, and thus, eight tag SNPs (rs17108803, rs1432623, rs12654778, rs1042713, rs1042714, rs1042717, rs1042719 and rs1042720) were selected from each block for further association study.
Association study between SNPs in ADRB2 and COPD
To assess the relationship between these tag SNPs of ADRB2 and COPD, 200 COPD subjects and 222 normal controls were recruited in this study (Table 2). There was no significant difference in sex, age or smoking history between cases and controls (p>0.05). In contrast, a significant difference was found in the baseline FEV1 percentage predicted and FEV1/FVC between cases and controls.
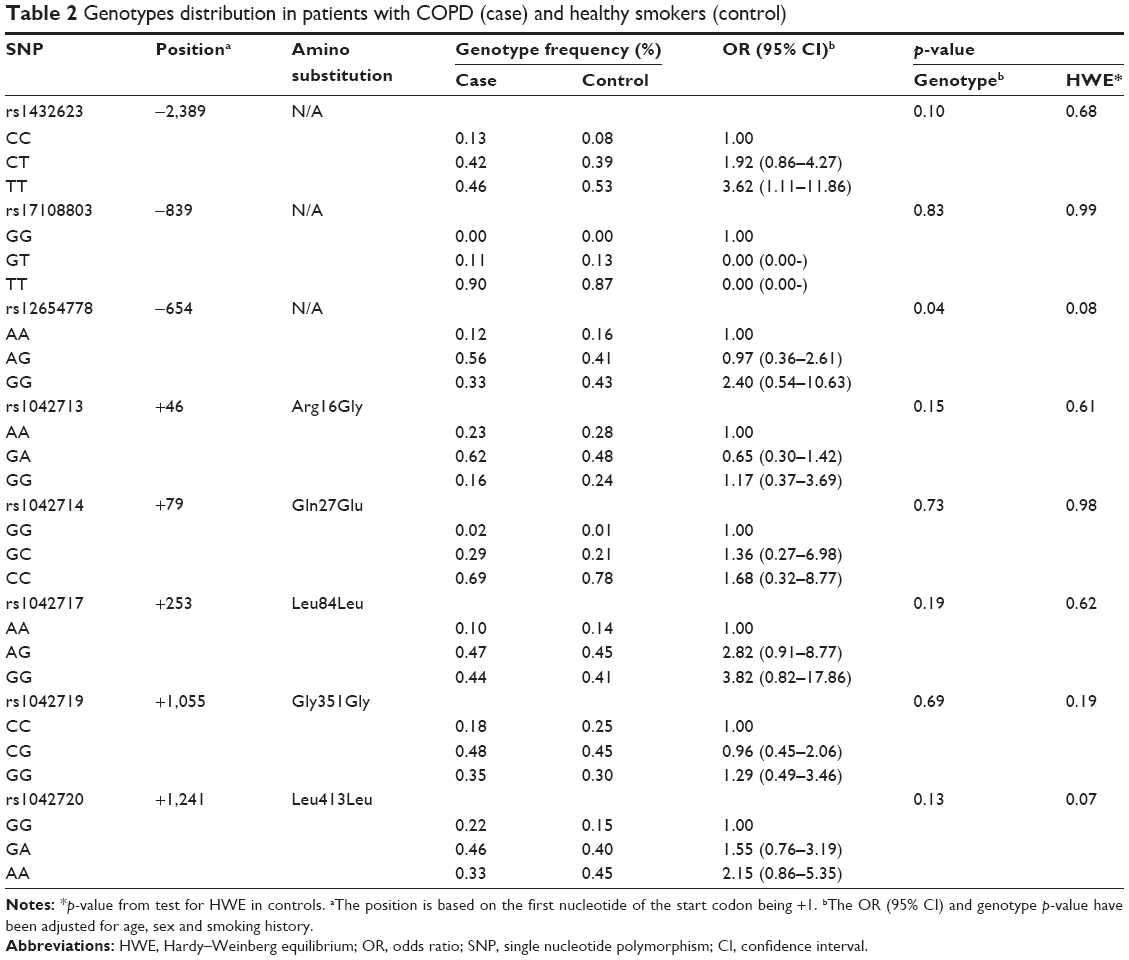 | Table 2 Genotypes distribution in patients with COPD (case) and healthy smokers (control) |
Although our study sample size was moderate, our sample size provided >80% power to detect a genetic relative risk (or odds ratio) of 2.35, by using genetic power calculator,29 with 7.3% disease prevalence in the Chinese population30 and α=0.05, for a variant with 0.05 minor allele frequency in a dominant model.
The tag SNPs from these eight blocks were genotyped in 422 subjects and the result is presented in Table 2. All eight SNPs were under Hardy–Weinberg equilibrium in controls (p>0.05). As shown in Table 2, rs1042713 (Arg16Gly), which was detected in most previous studies,13–19,31–34 was also investigated in our study, and the genotype (AA, AG, GG) frequency was 23%, 62% and 16% in COPD and 28%, 48% and 24% in controls, respectively, indicating that there was no statistically significant difference (p=0.15) after adjusting for age, sex and smoking history. The same analysis was performed on rs1042714 (Gln27Glu) and similar results were obtained (p=0.73; Table 2). These results were consistent with those of Brogger et al,31 while they disagree with Ho et al’s results.13 To resolve this conflict, a total of 13 previous studies on rs1042713 and rs104271413–21,31–34 were collected and reanalyzed by meta-analysis (Supplementary Table 3). Besides the publication of Niu et al,35 four more studies (Wang et al,19,21 Vacca et al33 and our genotyping data) were involved. In total, 2,908 cases and 2,946 controls were included in this meta-analysis and no significant difference was observed in rs1042713 or rs1042714 under allele model (p>0.05, Supplementary Figure 2), consistent with a previous meta-analysis in 2012.35
However, one SNP (rs12654778), located in the promoter region of ADRB2 (−654), presented a significant difference between cases (AA 12%, AG 56%, GG 33%) and controls (AA 16%, AG 41%, GG, 43%; p=0.04) adjusted by age, sex and smoking history. Lung function (FEV1 value) of COPD patients with the different genotype of rs12654778 was also investigated. Although the result was not significant, FEV1 value in individuals with AA genotype was higher than that of other genotypes (GA and GG; Supplementary Figure 3), suggesting that this SNP should be a candidate site for regulating ADRB2 gene expression and should be further associated with COPD or FEV1.
Promoter activity of different alleles for rs12654778
There are two SNPs (rs12654778 and rs17778257) in this block, and rs12654778 is closer to ADRB2 translation start codon than rs17778257 (~2 kb upstream). Considering that rs17778257 is far away from the regulatory region of ADRB2 and as no TF binding near this site (Supplementary Figure 4) was found by searching the ENCODE database, rs12654778 was chosen for further functional analysis. We proposed that rs12654778 may influence the transcriptional activity of ADRB2. To verify this hypothesis, we generated the plasmids containing different alleles of rs12654778 and transiently transfected them into Beas-2B cell lines. As shown in Figure 2, the cloned region showed ~100-fold higher luciferase expression compared with pGL3-basic plasmid, demonstrating the strong promoter activity of this region in the lung tissue. Moreover, rs12654778-A allele showed a ~21.8% reduction in promoter activity compared to the rs12654778-G allele (p=0.0034, Figure 2), suggesting that this SNP could regulate ADRB2 gene expression in the lung tissue.
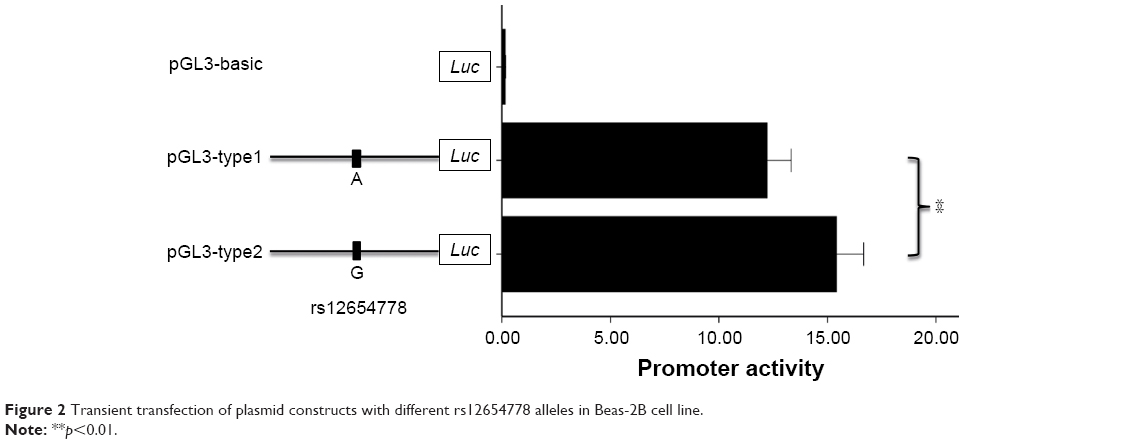 | Figure 2 Transient transfection of plasmid constructs with different rs12654778 alleles in Beas-2B cell line. |
Transcription factor neurofibromin 1 (NF1) binding rs12654778 surrounding region
Considering that rs12654778 is located within a conserved CCAAT box-like motif,36 which may function as a canonical binding site for NF1 based on TRANSFAC prediction, we hypothesized that NF1 is involved in the transcription of ADRB2 and this SNP might affect the binding affinity of NF1 to this region. To investigate whether NF1 binds the upstream region of ADRB2, we performed ChIP assays in Beas-2B cells using an anti-NF1 antibody and quantified the enrichment in the predicted NF1 binding site by real-time PCR. As shown in Figure 3, the chromatin immunoprecipitated by NF1 antibody was significantly enriched in the region surrounding rs12654778 compared with IgG (p=0.0021), suggesting that NF1 binds this region in the lung tissue.
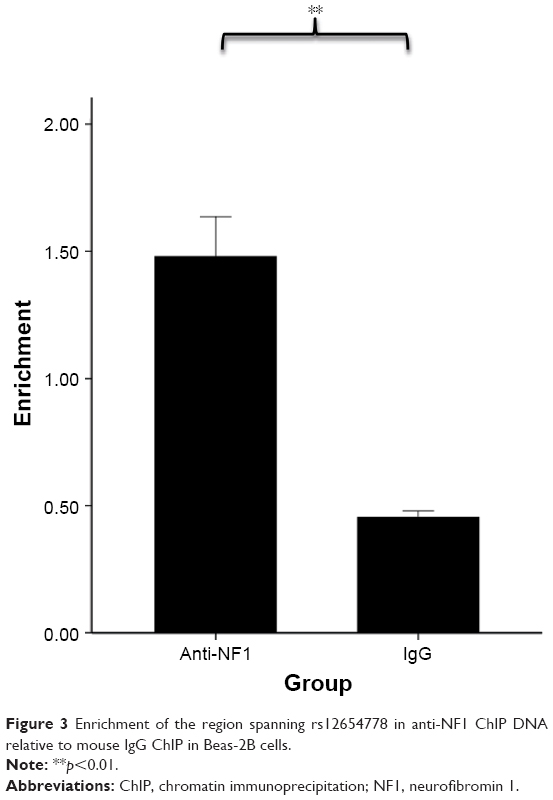 | Figure 3 Enrichment of the region spanning rs12654778 in anti-NF1 ChIP DNA relative to mouse IgG ChIP in Beas-2B cells. |
Discussion
COPD is a common respiratory disease caused by interaction of environmental risk factors with genetic background.37 While several relevant environmental risk factors of COPD have been identified, the genetic risk factors are less well understood. ADRB2, which encodes the β2-adrenergic receptor, is expressed in airway smooth muscle cells and is considered as an important pharmacologic target in the management of COPD, thus leading to speculation of its contributions to the onset of COPD. Previous studies have found that several genetic variants of ADRB2 are associated with COPD,13 but the real function of these variants is not elaborated, especially the noncoding variants, which might play a more important role in altering ADRB2 expression and further resulting in COPD. So, it is essential and necessary to use new data and multilevel surveys, including genetic association study and functional analysis, to further elucidate the role of ADRB2 noncoding variant in COPD, comprehensively. Several lines of evidence in this study implicated a noncoding SNP of ADRB2 as a COPD-susceptibility variant. Firstly, we confirmed that the expression level of ADRB2 was significantly reduced in COPD lung tissues. Secondly, a noncoding SNP (rs12654778), located upstream of ADRB2, was associated with COPD. Thirdly, functional analysis indicated that rs12654778 could modulate the binding affinity of TF (NF1) and its risk allele could reduce the transcriptional activity of ADRB2 gene expression. Taking all evidence together, our results are the first to reveal that the differential NF1 binding at rs12654778 could lead to reduced ADRB2 expression level in the lung tissue and increased susceptibility to COPD.
There are several studies showing that mutations in the coding region could affect the disease, for example, SERPINA1, PiMZ heterozygote produced by α1-antitrypsin defciency.38 However, most common variants identified by the association panel are located in noncoding regions, especially by the genome-wide association studies, and might be cis-regulatory elements for the nearby gene.39 Indeed, successful identification of functional variants in these promoter regulatory elements has been reported for β-thalassemia40 and pyruvate kinase deficiency.41 Considering that the study of regulatory elements is very important and it is difficult to identify functional genetic variants in the regulatory regions, it is worth performing intensive investigation. We contend that the identification of functional variants in such regions is an extremely important requisite for at least two reasons. On one hand, the identification of functional variants can conclusively prove which gene is actually involved in disease susceptibility. On the other hand, study of functional variants can lead to new insights into the pathophysiological mechanisms of diseases.
Since rs12654778 is located in the ADRB2 gene regulatory region, another important concern is about the potential mechanism by which this SNP in ADRB2 is associated with COPD susceptibility. The rs12654778 is located ~654 bp upstream of ADRB2, which is with the histone modification H3K27Ac, H3K4me1 and H3K4me342 (Supplementary Figure 4). Since H3K27Ac and H3K4me3 are usually correlated with activation of chromatin, it was reasonable to hypothesize that this region is pivotal for ADRB2 gene expression. Here, we firstly showed that rs12654778 could reduce ADRB2 expression level in the lung tissue by altering TF NF1 binding with ADRB2 promoter region, and contributed to the COPD onset, and this SNP has been identified with other diseases in several studies.43,44 Meanwhile, ADRB2 is also expressed in lymphoblastoid cells,10 and it is interesting to know whether this variant is correlated with the expression of ADRB2 in lymphoblastoid cells. To address this issue, we utilized eQTL browser (http://eqtl.uchicago.edu/cgi-bin/gbrowse/eqtl/), which collected published eQTL data of lymphoblastoid cell lines (LCL) from four HapMap populations to search for potential association. Interestingly, a significant association between rs12654778 and expression of ADRB2 in LCL was observed in Yoruban in Ibadan, Nigeria (YRI), Utah residents with Northern and Western European ancestry from the CEPH collection and two samples which were treated as a single analysis panel of 90 Asians populations45 (data not shown). We further evaluated the effect of this SNP on ADRB2 expression in LCL from 726 HapMap3 individuals in GENe Expression VARiation (http://www.sanger.ac.uk/resources/software/genevar/).46 The AA genotype of rs12654778 presented a significantly reduced expression of ADRB2, compared with the other genotypes AG or GG in most populations (CHB, Gujarati Indians in Houston, TX, USA, JPT, Mexican ancestry in Los Angeles, CA, USA, Maasai in Kinyawa, Kenya and YRI, shown in Supplementary Figure 5), which was consistent with our result.
In addition, β2-adrenoceptors, through their extracellular domain, can bind to Gs and prevent adenylyl cyclase from activating the cAMP signaling pathway,47 a critical pathway for embryonic lung development. Decreased ADRB2 expression leads to overactivation of the cAMP pathway in multiple types of breast tumor, which in turn contributes to uncontrolled cellular proliferation.48 In our study, we found that ADRB2 expression was decreased in the lung tissue from COPD cases compared with control subjects with normal lung function, indicating that lower ADRB2 expression may exacerbate COPD pathogenesis. This was inconsistent with Selivanova et al’s study,11 which might be due to the difference in the control group. In our study, the control group consisted of people with normal lung function, while in Selivanova et al’s study, the control group consisted of mild/middle asthma patients.11 Further mechanistic studies on the cAMP pathway in the context of smoking may provide novel insights into the pathogenesis of COPD.
Conclusion
Our study is the first to demonstrate that a functional SNP (rs12654778), upstream of ADRB2, was significantly associated with increased risk for COPD. These results offer valuable insights into the signaling, maintenance and regulatory mechanisms of ADRB2 in lung and its further correlation with COPD.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (grant no 31200940) and Yunnan University (grant no 2011ZD01). The authors wish to thank all the patients for allowing them to collect blood samples and tissues samples for genetic association studies over the years.
Disclosure
The authors report no conflicts of interest in this work.
References
Jiang Z, Knudsen NH, Wang G, et al. Genetic control of fatty acid beta-oxidation in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2017;56(6):738–748. | ||
Weiss ST. Lung function and airway diseases. Nat Genet. 2010;42(1):14–16. | ||
Patel BD, Coxson HO, Pillai SG, et al. Airway wall thickening and emphysema show independent familial aggregation in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2008;178(5):500–505. | ||
Zhou Y, Wang C, Yao W, et al. [A cross-sectional survey of familial aggregation of chronic obstructive pulmonary disease: in seven provinces/cities in China.] Chin J Intern Med. 2014;53(5):354–358. Chinese. | ||
de Serres FJ, Blanco I, Fernandez-Bustillo E. PI S and PI Z alpha-1 antitrypsin deficiency worldwide. A review of existing genetic epidemiological data. Monaldi Arch Chest Dis. 2007;67(4):184–208. | ||
Silverman EK, Sandhaus RA. Clinical practice. Alpha1-antitrypsin deficiency. N Engl J Med. 2009;360(26):2749–2757. | ||
Sampsonas F, Karkoulias K, Kaparianos A, Spiropoulos K. Genetics of chronic obstructive pulmonary disease, beyond a1-antitrypsin deficiency. Curr Med Chem. 2006;13(24):2857–2873. | ||
Cho MH, McDonald ML, Zhou X, et al; NETT Genetics, ICGN, ECLIPSE and COPDGene Investigators. Risk loci for chronic obstructive pulmonary disease: a genome-wide association study and meta-analysis. Lancet Respir Med. 2014;2(3):214–225. | ||
Nardini S, Camiciottoli G, Locicero S, et al. COPD: maximization of bronchodilation. Multidiscip Respir Med. 2014;9(1):50. | ||
Barnes PJ. Beta-adrenergic receptors and their regulation. Am J Respir Crit Care Med. 1995;152(3):838–860. | ||
Selivanova PA, Kulikov ES, Kozina OV, et al. Differential expression of the beta2-adrenoreceptor and M3-cholinoreceptor genes in bronchial mucosa of patients with asthma and chronic obstructive pulmonary disease. Ann Allergy Asthma Immunol. 2012;108(1):39–43. | ||
Liggett SB. Polymorphisms of the beta2-adrenergic receptor and asthma. Am J Respir Crit Care Med. 1997;156(4 Pt 2):S156–S162. | ||
Ho LI, Harn HJ, Chen CJ, Tsai NM. Polymorphism of the beta (2)-adrenoceptor in COPD in Chinese subjects. Chest. 2001;120(5):1493–1499. | ||
Hegab AE, Sakamoto T, Saitoh W, et al. Polymorphisms of IL4, IL13, and ADRB2 genes in COPD. Chest. 2004;126(6):1832–1839. | ||
Ma L, Feng DX, Zhang XY, et al. Association between the genetic polymorphisms of β2-adrenergic receptor and the chronic obstructive pulmonary disease. Chin J Pract Intern Med. 2006;26(4):267–269. | ||
Chen JX, Shi YK, Li SL, et al. The Study of β2-AR polymorphisms and chronic obstructive pulmonary disease on relations. Acta Acad Med Weifang. 2008;30(1):63–65. | ||
Shi YK, Ma J, Yuan ZJ, et al. Investigation on the relation between polymorphisms of β2 adrenergic receptor and the chronic obstructive pulmonary disease. Shandong Med J. 2008;48(13):9–11. | ||
Cao X, Li QQ, Chen GZ, et al. Polymorphism of IL-4, IL-13, and ADR Beta 2 genes in patients with chronic obstructive pulmonary disease. Med J Wuhan Univ. 2009;30(2):219–223. | ||
Wang C, Tan F, Yang AL, et al. Association between the Arg16Gly β2-adrenoceptor polymorphisms and the older-aged chronic obstructive pulmonary disease with hypertension (in Chinese). J Pract Med. 2011;28(7):4442–4444. | ||
Wang W, Yu YJ, Qian R, et al. Association between the susceptibility of COPD and IL-13, IL-4, polymorphisms of beta2-adrengic receptor. J Clin Intern Med. 2011;28(5):332–334. | ||
Wang C, Yang AL, Li H, et al. Association between the Gln27Glu β2-adrenoceptor polymorphisms and the older-aged chronic obstructive pulmonary disease with hypertension (in Chinese). J Pract Med. 2012;28(7):1100–1103. | ||
Edwards SL, Beesley J, French JD, Dunning AM. Beyond GWASs: illuminating the dark road from association to function. Am J Hum Genet. 2013;93(5):779–797. | ||
Heinemeyer T, Wingender E, Reuter I, et al. Databases on transcriptional regulation: TRANSFAC, TRRD and COMPEL. Nucleic Acids Res. 1998;26(1):362–367. | ||
Scott MG, Swan C, Wheatley AP, Hall IP. Identification of novel polymorphisms within the promoter region of the human beta2 adrenergic receptor gene. Br J Pharmacol. 1999;126(4):841–844. | ||
McGraw DW, Forbes SL, Kramer LA, Liggett SB. Polymorphisms of the 5′ leader cistron of the human beta2-adrenergic receptor regulate receptor expression. J Clin Invest. 1998;102(11):1927–1932. | ||
Vogelmeier CF, Criner GJ, Martinez FJ, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive lung disease 2017 report: GOLD executive summary. Eur Respir J. 2017;49(3). pii: 1700214. | ||
1000 Genomes Project Consortium, Abecasis GR, Altshuler D, Auton A, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467(7319):1061–1073. | ||
Carlson CS, Eberle MA, Rieder MJ, Yi Q, Kruglyak L, Nickerson DA. Selecting a maximally informative set of single-nucleotide polymorphisms for association analyses using linkage disequilibrium. Am J Hum Genet. 2004;74(1):106–120. | ||
Purcell S, Cherny SS, Sham PC. Genetic power calculator: design of linkage and association genetic mapping studies of complex traits. Bioinformatics. 2003;19(1):149–150. | ||
Yin P, Wang H, Vos T, et al. A subnational analysis for mortality and prevalence of chronic obstructive pulmonary disease in China 1990 to 2013: findings from Global Burden of Disease Study (GBD) 2013. Chest. 2016;150(6):1269–1280. | ||
Brogger J, Steen VM, Eiken HG, Gulsvik A, Bakke P. Genetic association between COPD and polymorphisms in TNF, ADRB2 and EPHX1. Eur Respir J. 2006;27(4):682–688. | ||
Matheson MC, Ellis JA, Raven J, Johns DP, Walters EH, Abramson MJ. Beta2-adrenergic receptor polymorphisms are associated with asthma and COPD in adults. J Hum Genet. 2006;51(11):943–951. | ||
Vacca G, Schwabe K, Duck R, et al. Polymorphisms of the beta2 adrenoreceptor gene in chronic obstructive pulmonary disease. Ther Adv Respir Dis. 2009;3(1):3–10. | ||
Papatheodorou A, Makrythanasis P, Kaliakatsos M, et al. Development of novel microarray methodology for the study of mutations in the SERPINA1 and ADRB2 genes their association with obstructive pulmonary disease and Disseminated Bronchiectasis in Greek patients. Clin Biochem. 2010;43(1–2):43–50. | ||
Niu LM, Liang Y, Xu M, Zhang YY, Zhang Y, He B. Effect of polymorphisms in the beta2-adrenergic receptor on the susceptibility and pulmonary function of patients with chronic obstructive pulmonary disease: a meta-analysis. Chin Med J (Engl). 2012;125(12):2213–2218. | ||
Heinemeyer T, Chen X, Karas H, et al. Expanding the TRANSFAC database towards an expert system of regulatory molecular mechanisms. Nucleic Acids Res. 1999;27(1):318–322. | ||
Corhay JL, Frusch N, Louis R. [COPD: genetics and environmental interactions]. Rev Med Liege. 2012;67(5–6):292–297. French. | ||
Molloy K, Hersh CP, Morris VB, et al. Clarification of the risk of chronic obstructive pulmonary disease in alpha1-antitrypsin deficiency PiMZ heterozygotes. Am J Respir Crit Care Med. 2014;189(4):419–427. | ||
Pazin MJ. Using the ENCODE Resource for Functional Annotation of Genetic Variants. Cold Spring Harb Protoc. 2015;2015(6):522–536. | ||
Hardison RC, Chui DH, Giardine B, et al. HbVar: a relational database of human hemoglobin variants and thalassemia mutations at the globin gene server. Hum Mutat. 2002;19(3):225–233. | ||
van Wijk R, van Solinge WW, Nerlov C, et al. Disruption of a novel regulatory element in the erythroid-specific promoter of the human PKLR gene causes severe pyruvate kinase deficiency. Blood. 2003;101(4):1596–1602. | ||
Rosenbloom KR, Dreszer TR, Long JC, et al. ENCODE whole-genome data in the UCSC Genome Browser: update 2012. Nucleic Acids Res. 2012;40(Database issue):D912–D917. | ||
Hocking LJ, Smith BH, Jones GT, Reid DM, Strachan DP, Macfarlane GJ. Genetic variation in the beta2-adrenergic receptor but not catecholamine-O-methyltransferase predisposes to chronic pain: results from the 1958 British Birth Cohort Study. Pain. 2010;149(1):143–151. | ||
Cai W, Yin L, Cheng J, et al. Relationship between the single nucleotide polymorphisms of beta(2)-adrenergic receptor 5′-regulatory region and essential hypertension in Chinese Kazakh ethnic minority group. Int J Clin Exp Pathol. 2015;8(7):8358–8366. | ||
Veyrieras JB, Kudaravalli S, Kim SY, et al. High-resolution mapping of expression-QTLs yields insight into human gene regulation. PLoS Genet. 2008;4(10):e1000214. | ||
Stranger BE, Montgomery SB, Dimas AS, et al. Patterns of cis regulatory variation in diverse human populations. PLoS Genet. 2012;8(4):e1002639. | ||
Wang WC, Pauer SH, Smith DC, et al. Targeted transgenesis identifies Galphas as the bottleneck in beta2-adrenergic receptor cell signaling and physiological function in airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2014;307(10):L775–L780. | ||
Bruzzone A, Sauliere A, Finana F, Sénard JM, Lüthy I, Galés C. Dosage-dependent regulation of cell proliferation and adhesion through dual beta2-adrenergic receptor/cAMP signals. FASEB J. 2014;28(3):1342–1354. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.