")
Back to Journals » Cancer Management and Research » Volume 15
A Focused Clinical Review of Lynch Syndrome
Authors Georgiou D, Monje-Garcia L, Miles T, Monahan K , Ryan NAJ
Received 5 October 2022
Accepted for publication 23 December 2022
Published 18 January 2023 Volume 2023:15 Pages 67—85
DOI https://doi.org/10.2147/CMAR.S283668
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Kattesh Katti
Demetra Georgiou,1 Laura Monje-Garcia,2,3 Tracie Miles,4 Kevin Monahan,2,5 Neil AJ Ryan6,7
1Genomics and Personalised Medicine Service, Charing Cross Hospital, London, UK; 2The St Mark’s Centre for Familial Intestinal Cancer Polyposis, St Mark’s Hospital, London, UK; 3School of Public Health, Imperial College, London, UK; 4South West Genomics Medicine Service Alliance, Bristol, UK; 5Department of Gastroenterology, Imperial College, London, UK; 6Department of Gynaecological Oncology, Royal Infirmary of Edinburgh, Edinburgh, UK; 7The College of Medicine and Veterinary Medicine, The University of Edinburgh, Edinburgh, UK
Correspondence: Neil AJ Ryan, Department of Gynaecology Oncology, Royal Infirmary of Edinburgh, Edinburgh, UK, Email [email protected]
Abstract: Lynch syndrome (LS) is an autosomal dominant condition that increases an individual’s risk of a constellation of cancers. LS is defined when an individual has inherited pathogenic variants in the mismatch repair genes. Currently, most people with LS are undiagnosed. Early detection of LS is vital as those with LS can be enrolled in cancer reduction strategies through chemoprophylaxis, risk reducing surgery and cancer surveillance. However, these interventions are often invasive and require refinement. Furthermore, not all LS associated cancers are currently amenable to surveillance. Historically only those with a strong family history suggestive of LS were offered testing; this has proved far too restrictive. New criteria for testing have recently been introduced including the universal screening for LS in associated cancers. This has increased the number of people being diagnosed with LS but has also brought about unique challenges such as when to consent for germline testing and questions over how and who should carry out the consent. The results of germline testing for LS can be complicated and the diagnostic pathway is not always clear. Furthermore, by testing only those with cancer for LS we fail to identify these individuals before they develop potentially fatal pathology. This review will outline these challenges and explore solutions. Furthermore, we consider the potential future of LS care and the related treatments and interventions which are the current focus of research.
Keywords: genetic counselling, Lynch syndrome, mainstreaming
Introduction
Lynch syndrome (LS) is the most common inherited cancer predisposition syndrome; it is thought to affect over 1:280 people.1 The condition arises due to germline pathogenic variants within the mismatch repair genes, namely MLH1, MSH2 (EPCAM), MSH6 and PMS2 leading to a lifelong haploinsufficiency. After a somatic second hit, individuals with LS develop a defective DNA repair machinery which decreases DNA fidelity during cellular replication. This machinery, known as the mismatch repair system, is shown as a schema in Figure 1. Typically, in LS numerous insertion, deletion, and mis-incorporation mutations occur with a propensity for such errors in the microsatellites of the DNA.2 Microsatellites are tandem repeats of DNA in which sequences of bases reoccur such as AAAA or TCTCTC. Without a functional mismatch repair these microsatellites corrupt which in turn has a deleterious effect on protein function and cellular metabolism. As these errors accumulate over time, somatic mutations arise that can lead to carcinogenesis.3
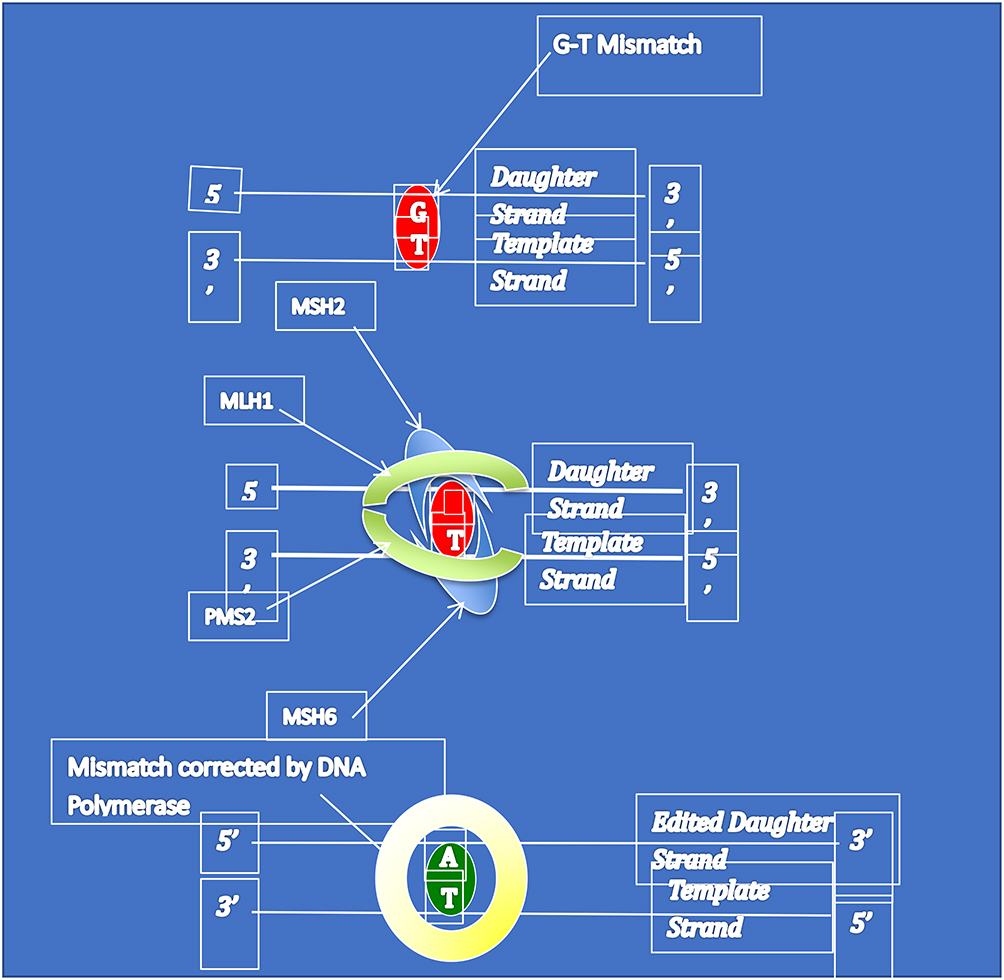 |
Figure 1 The MMR system. This graphic, in a simplified manor, explains the mechanism of the mismatch repair system. A mismatch repair error occurs, in this case a G-T base pairing. This is identified by the dimer of proteins MSH2/MSH6 identifies the error and recruits MLH1/PMS2 to excise the error. This allows DNA polymerase to insert the correct base leading to a A-T pairing. |
These molecular changes enable clinicians to offer accurate tumour-based screening for LS and definitive germline testing to confirm LS. Immunohistochemistry is a technology that uses antibodies to bind and stain specific proteins. However, if a protein is misshapen or absent the antibody will fail to bind and the stain is lost.4 This enables pathologists to quickly screen cancers for mismatch repair deficiency (MMRd) as they will fail to stain. As a specific protein will be absent, immunohistochemistry can also help clinicians identify the gene that is likely to contain the pathogenic variant. In addition, microsatellite instability is detectable by polymerase-chain-reaction based analysis. This therefore can be used to screen cancers for LS. Unlike immunohistochemistry, microsatellite instability does not indicate which gene could be affected. It has also been shown that the accuracy of microsatellite instability is reliable in colorectal cancer; it may be less accurate in endometrial cancer.5 Therefore, in endometrial cancer immunohistochemistry is preferentially used. Both methods do not diagnose LS as they are performed on the tumour and so can only speak to the tumours’ DNA. Therefore, germline testing is needed to confirm LS. The number of people sent for germline testing can be reduced by reflex testing for MLH1 hypermethylation or in colorectal cancer BRAF V600E testing.6 If either MLH1 hypermethylation of a BRAF V600E mutation is found, the individual is very unlikely to have LS.
Diagnosing LS is of clinical importance. Those affected by LS are at an increased lifetime risk of a constellation of cancers. The most closely associated are cancers of the colorectum, endometrium, ovaries, and urinary tract.7 However, these cancers are in part avoidable with the application of colonoscopic surveillance, aspirin chemoprophylaxis, risk reducing surgery and lifestyle modifications.8,9 Once correctly diagnosed, those with LS can be enrolled in the risk reducing strategies and cascade testing of their relatives can begin. Cascade testing enables clinicians to find more LS carriers within a family before they develop cancer.10 Those found to have LS can also be offered risk reducing interventions with the aim of preventing them from ever developing a potentially fatal cancer.
However, which cancers should be screened for LS, who should be offered LS testing, how patients should be counselled for LS testing, what should and can be done to reduce cancer risk in LS are all still, to some degree, matters for debate. In this article, these questions will be discussed with a summary of the current evidence base.
Diagnostic vs Predictive Testing vs Prenatal Diagnosis
Genetic testing processes vary considerably depending on health history and life stage of the individual. With a wide range of risk management and screening options available, identification of LS carriers has been on the forefront of several health systems through universal screening.11 LS genetic testing can be available in diagnostic, predictive and reproductive stage. In individuals with prior history of LS-related cancer, a test is diagnostic and is thought to help explain the cause of their disease. In the United Kingdom, such tests are often reflex and offered to all patients who have colorectal and endometrial carcinoma. These are often available through secondary care where results are used for ongoing patient management as well as for future health planning.
Cascade Screening
Economically, viability of universal screening programmes for LS relies on cascade screening, which is the onward testing of 1st degree relatives of newly diagnosed individuals.12–15 It is expected that unaffected family members who are informed of their positive carrier status through cascade genetic testing (also referred to as pre-symptomatic or predictive testing), will engage in risk reducing activities, uptake screening and risk management options and will have a reduced risk of cancer overall or an earlier stage at diagnosis. Menko et al and Griffin et al found that uptake of cascade testing was not very high in LS families; partly due to gender barriers and inadequate patient education.16,17 As such, one needs to place a lot of attention on factors that facilitate cascade screening; patient awareness, family communication, health system accessibility and national standardisation.13 A more novel suggestion is the health system-led contact of relatives (with patient consent) that would complement the current traditional method of patient-led dissemination.17,18
Predictive Testing
In healthy, unaffected adults, a genetic test is predictive as it is performed to predict whether an individual has an increased risk of LS-related cancers. A predictive test for late onset cancer susceptibilities such as LS has more psychosocial and practical implications (such as life insurance) hence the counselling approach needs more consideration in this context.19,20 Counselees need to be informed, supported, and offered an assessment by the genetics clinician to ensure they would be well equipped to manage the psychosocial impact and distress linked with an unfavourable result before they consent to a genetic test. Godino et al suggest a multistep approach enabling communication, decision making and action at appropriate timepoints.21 Given LS is a late onset disease predictive testing in childhood is not available, although families are encouraged to discuss the familial LS status with their children throughout their lives.11
Reproductive Options
Prenatal diagnosis (PND) and Preimplantation Genetic Diagnosis (PGD) have broadly been available for LS patients. Chorionic Villus Sampling and Amniocentesis are well established techniques that assist the molecular diagnosis of a foetus, where pregnant individuals are informed of the pregnancy status to make an informed decision on whether to continue with a pregnancy. These have risks to the pregnancy and are known to have limitations in practice (difficult to detect/establish level of mosaicism/sample quality dependent). A further challenge with PND comes with the outcomes of pregnancies identified as positive carriers of genetic disease when parents decide to continue with pregnancy. This is ethically challenging as no individual should have a late onset pre-symptomatic test without their involvement and consent (https://www.rcplondon.ac.uk/file/13314/download). Women and couples undergoing invasive prenatal testing should be offered genetic counselling to help understand the risks, weigh their options, and plan their decisions.
PGD has been available for the last few decades and has supported many individuals with hereditary conditions to conceive an unaffected embryo. This service is offered to couples with a serious hereditary condition; in the United Kingdom LS is licenced for PGD. Although these methods are widely available and offered by healthcare professionals in the context of childhood disease or early onset cancer susceptibility; it is clear that healthcare professionals feel it is less acceptable to offer these options to families with late onset conditions where some form of prevention/management is available.22 The increasing availability of risk management options for LS as well as lower penetrance (especially when associated with variants in PMS2), have led to ongoing consultations in United Kingdom as to if PND and PGD should be offered. These definitions are summarised in Table 1.
 |
Table 1 Definitions of Different Forms of Genetic Testing |
When to Test for Lynch Syndrome
Population Level Testing
LS is thought to affect up to 1:280 people, although large biobank studies put this figure closer to 1:400.23 Around 95% of those with LS are unaware of their diagnosis.24 Indeed, with a prevalence of 1:400 we would expect around 200,000 people in the United Kingdom to be affected by LS, however only 10,000 tests for LS have been recorded by NHS digital. With so many undiagnosed and at risk of developing cancer, an argument has been made to introduce population level testing in which there are no restrictions are applied on LS testing.25 In this scenario, everyone would be eligible to have LS testing; this would either be on a volunteered basis or by a governmental screening program such as the blood spot test to screen for metabolic diseases of the new born.26 Population level testing has been shown to be cost-effective for high risk ovarian cancer genes.27 In addition, testing 30yr olds for LS could also be cost-effective.28
To date, no country offers state funded population level LS testing. Population-based germline testing has already been carried out in the confines of research programmes.29 Furthermore, private companies offer LS testing to those who can pay for it without any pre-selection. Therefore, there exists a degree of inequality to access LS testing; those who can pay or those who are enrolled into research can be tested, whereas those who are not can only access LS testing if they meet prespecified criteria.
The cited barriers to population-level LS testing are around informed consent and resource allocation.30,31 The current model of consent for germline testing involves a lengthy discussion with genetics professional before the test is undertaken. If this was to be rolled out across a population, genetics services would be overwhelmed. In addition, laboratory capacity is limited. With population level testing, demand would increase beyond current capacity leading to systemic issues.32 Therefore, for the time being, population-level testing for LS remains limited to research projects or to those who pay.
Cancer Testing
LS is closely associated with an increased risk of certain cancers, namely colorectal and endometrial cancer.33 Therefore, colorectal and endometrial cancer populations are enriched, when compared to the general population, for LS. As such, there is a clear argument to screen those with endometrial and colorectal cancer for LS as has become common practice in North America and Europe.1 Around 3% of colorectal34 and endometrial5 cancers are caused by LS. It has been demonstrated that the universal screening in these groups is cost-effective.35–37 Therefore, there has been a move away for selected screening, such as based on age or family history, in these cancers. Indeed, universal screening is recommended by international guidelines.8,9
LS is also associated with ovarian cancers, however only around 1% of ovarian cancers are associated with LS.38 Therefore, currently it is not clear if the universal screening of ovarian cancer is cost-effective. Selective screening of ovarian cancer, namely those of an endometrioid histotype or in women less than 50 years old, has been recommended by international guidelines.8 There is no clear consensus as to if other cancers should also be screened for LS, as again the prevalence of LS is thought to be low outside of colorectal and endometrial cancers.
Screening cancer populations for LS has an innate disadvantage: the individual must have developed a cancer before they are tested. This is counterintuitive as the reason to diagnose LS is to try and prevent cancers. For endometrial cancers an argument can be made. Women often survive endometrial cancer.39 For women with LS, endometrial cancers are often the sentinel cancer that they develop.40 Therefore, there is a diagnostic opportunity as they can be diagnosed with LS during their endometrial cancer treatment, go on to survive, and be enrolled in preventative measures that would decrease their risk of a potentially fatal colorectal cancer. Sadly, the universal screening of colorectal cancer is unlikely to have a significant impact on the index case. Yet it does provide the opportunity to enable cascade testing which can in turn find those relatives who are LS carriers before they develop a cancer. On average around 3 LS carriers are found for each index case.10 Therefore, cancer screening can still identify LS carriers who can benefit from intervention.
Criterion Based Testing
Several different score systems exist that can be used to identify individuals at high risk of being carriers for LS. These can be used in either the general population or to pre-select those with cancers associated with LS. Indeed, it was on the basis of family linage that LS was first described by Aldred Warthin and later Henry Lynch.41 The Bethesda42 and Amsterdam43 criteria were devised in the 1990s as a means to identify those thought to have LS, and as the technology developed, identify those for germline testing. However, these scores suffer from a low sensitivity especially for pathogenic variant carriers of MSH6 and PMS2.44 More modern criteria have tried to address this such as PREMM5,45 MMRpredict46 and MMMRpro.47 These scores report reasonable sensitives but still fail to reliably identify MSH6 and PMS2 pathogenic variant carriers.1 With these caveats, they can be deployed as to help identify those in the general population who should go on to have germline testing.
Mainstreaming v Traditional Model
As molecular biomarkers become a routine part of the diagnostic work up for the colorectal and endometrial cancers the clinical community is looking at the feasibility and utility of mainstreaming genetic testing in these tumour sites.
The term mainstreaming in the genetic testing describes pre-test counselling and consent process being undertaken by a member of the clinical cancer team caring for the patient instead of referring to a clinical genetics professional. This mainstream approach saves time, is fiscally advantageous and importantly provides continuity of knowledge for both clinician and patient.
The concept of mainstreaming is not new, with the advent of targeted therapies, for example poly(ADP-ribose) polymerase (PARP) inhibitors being used in ovarian cancers, the genetic testing for BRCA1 & BRCA2 being a necessary companion test to inform prescribing. A study looking at implementing rapid, robust, cost-effective, patient centred and routine genetic testing in ovarian cancer patients was undertaken by George et al with excellent outcomes demonstrating transferability and scalability to other tumour sites and cancer services.48
Since this seminal study in ovarian cancer, the principles have been reproduced with similar positive outcomes,49 with transferability and scalability in breast and BRCA testing50 to the endometrial pathway10 looking at Lynch testing, with a systematic review addressing the feasibility of implementing mainstreaming germline genetic testing in cancer care51 supporting these outcomes.
There are several important elements to achieving a robust mainstreaming service:
- genomic literacy of the clinical team enabled by bespoke education/training programmes.
- job planning to accommodate the mainstreaming consulting time.
- pathway mapping of mainstream service, to include timing, return of results, collaboration with clinical genetics.
In the United Kingdom, these issues are being addressed with the advent of the Genomic Medicine Service Alliances. This organisation is charged, amongst other things, to bring mainstreaming across the National Health Service (NHS) in England.
As genomics now moves from niche to necessity with the nursing and midwifery colleges recognising the importance of genomic literacy in the workforce, cancer nurse specialists are now well placed to take up mainstreaming as part of their role. This will need workforce task analysis and role description to include genomic literacy in the skill set and is being looked at by MacMillan and the leadership of the National Health Service to support this role evolution. Third sector colleagues Macmillan have already undertaken a survey review of cancer nurse specialists attitudes and needs (to include capacity and knowledge current limitations) in order to safely practice and offer genomic support to their patients, we anticipate publication in 2023. The education piece is superbly supported by the Lynch Transformation Programme, Health Education England and The Cancer Alliances have been afforded funding to put Lynch Nurse Specialists in place to across the health service to ensure equity of rollout of mainstreaming across the country, working closely with the national Lynch transformation team.
In summary, the concept of mainstreaming “right test, right place, right time” is proven. Now is the right time for the clinical community to move at a faster pace to implement as the tools in the form of education, training and workforce planning have arrived, with importantly funding support.
Diagnostic Genetic Testing
Diagnostic testing in LS is the genetic testing performed on an individual with history of cancer to confirm or exclude LS. Diagnostic genetic testing is now mainstreamed in routine oncology clinics. The Health Education England’s Genomic Education Program (https://www.genomicseducation.hee.nhs.uk) developed competency frameworks to facilitate genomic testing and communicating germline genomic results that can support health care professionals to gain the skills necessary to provide genomic testing. It’s important to take into consideration that the frameworks should be interpret in the context in which is going to be used, and not all competencies will be applicable to all situations. In addition, it acknowledges that healthcare professionals might already be competent in some of the themes. These frameworks are not intended to be used as an assessment tool, but rather as a resource to support learning and practice. Figure 2 illustrates some of the specific aspects of diagnostic genetic testing for LS not covered by these frameworks.
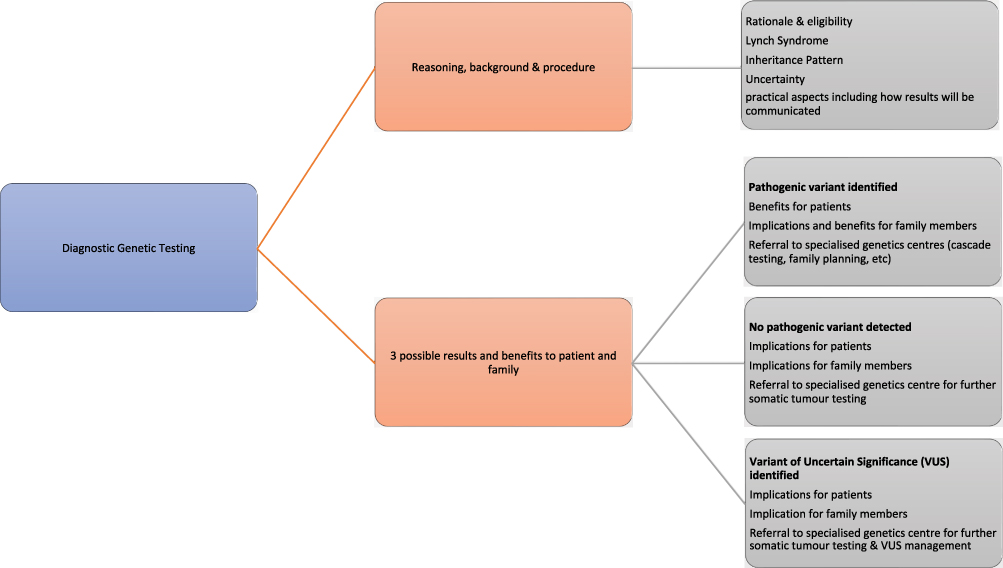 |
Figure 2 A flow diagram illustrating specific aspects of diagnostic genetic testing for LS not covered by current Health Education England’s Genomic Education Program (GEP, 2022) competency frameworks. |
In combination with the above-mentioned frameworks, there are a wide variety of resources produced as part of the National Lynch Syndrome Project that describes the genomic test consultation, benefits to patients and their family members and the rationale for referring all patients, regardless of their genetic result, to specialised genetic centres. It also explains the responsibilities of specialised genetic centres following results, such as cascade testing should a pathogenic variant be identified, or further somatic tumour testing when a pathogenic variant has not been detected to clarify if the patient has LS.52 Patients in which LS cannot be ruled out are classified as “Lynch-like syndrome”. These patients and their first-degree relatives are eligible for 2 yearly colonoscopic surveillance from the age of 25.53
What to Do About the Variant of Uncertain Significance
There are three potential results from diagnostic testing for LS, the identification of a pathogenic variant in an MMR gene (the definition of a diagnosis of LS), no variant detected, or a variant of uncertain significance (VUS). With a VUS the clinical significance of a genetic variant is uncertain, it may represent healthy or disease-associated variation in an MMR gene. With the advent of next generation sequencing platforms high volume genomic analysis has resulted in large scale variant identification which need to be catalogued and categorised systematically. VUS results are dealt with by specialists who will assign the degree of concern for that specific VUS.
Identified pathogenic variants and VUSs in MMR genes should be submitted to the international variant registry supported by InSIGHT (https://www.insight-group.org/variants/databases/), where such variants are curated, may be assigned pathogenicity, and therefore this database may inform interpretation of results.54 In 2015, the American College of Medical Genetics and Genomics (ACMG) has produced a guideline to support DNA sequence variant interpretation which is widely used in clinical practice worldwide - this utilises clinical information including evidence of tumour MMR status, and segregation within families.55 We would also recommend that diagnostic services maintain a registry of variants which they might clinically review at regular intervals by working with variant review boards. Coordinated national approaches such as the Cancer Variant Interpretation Group (https://canvaruk.org) in the United Kingdom may facilitate systematic variant review by linking genomics laboratories to multidisciplinary clinical networks.
Barriers to Lynch Syndrome Testing
Given the benefits to discovering that someone has LS, not just for the patient but for their immediate family members, it is expected that most people would welcome genetic testing for LS. In fact, there is good number of studies that suggest that hypothetically there is a high interest in finding out this information.56,57 By contract, there are good reports that show that there is a high number of people that decline testing.58 Few studies to date have looked at why some people decline genetic testing. Keogh and her team conducted a couple of qualitative studies looking at this phenomenon in Austria. In the final study conducted in 2017 the team divided the people who declined genetic testing into four groups: people who are uniformed, who have a weak intention, who conditionally decline, and unconditionally decline.59
Poor knowledge can be a barrier that can be overcome if identified. The uninformed participants declined due to not being aware that they were eligible for genetic testing or misinterpreting the information that has been given to them. An important factor affecting their risk perception and decision making was the belief their family’s cancers have been caused by lifestyle factors, and the information or the test was inappropriate for them. Following the clarification and education provided as part of the research they changed their mind.
There was a second group that was passive or unsure and delayed their result. Barriers in this group were poor knowledge, and fear of changes in their cancer surveillance or insurance discrimination. This second group was classified as having a “weak intention” and could be divided into the level of knowledge they have. People with partial knowledge were undecided because they misunderstood their risk of cancer, believed that the result was not applicable to them at that time and could be delayed. People with fair knowledge were already under colonoscopic surveillance and were afraid of losing their cancer screening or were concerned about insurance discrimination.
The third group called “Conditional decliners” was firm about declining; however, they kept the option open to find out the result in the future. The main barriers in this group were their inability to see the value of genetic testing as if the result wouldn’t alter anything, and only cause anxiety. In addition to this, fears found in previous group such as fear of losing their cancer surveillance or insurance discrimination was more evident. Their levels of knowledge range from partial to complete and stated that they would change their mind if the results provide clear benefits, for instance, remove concerns about insurance, if there any signs of cancer, or their children wanted the results. This group was older and was already under surveillance.
The fourth and last group called “unconditional decliners” was firm about declining and would be unlikely that they change their mind in the future. Barriers include the same as previous group, but their belief and confidence was stronger. They expressed a strong need to avoid the anxiety related to a genetic diagnosis. Some talked about their experiences of seeing other family members going through cancer and found the thought of going through it unbearable. People in this group was also under colonoscopic surveillance.
We find that the barriers found in these studies are also pertinent in the United Kingdom and provide important information to guide genetic counselling conversations.59 The consultation needs to address misconceptions and concerns so patients can make informed decisions. Benefits of testing need to be made clear to patients. For the people who are already under surveillance, the additional surveillance offered as part of LS diagnosis needs to be highlighted, as well as contraindications for people who no longer need it. Lastly, addressing anxieties and fears or living with LS as well as exploring significant family history of cancer, and providing emotional support will be key. The essential information to share with a patient before offering genetic testing is outlined in Box 1.
 |
Box 1 Essential Information to Share with Patients Before Offering a Diagnostic Test |
Figure 3 illustrates some of the more salient barriers and facilitators for germline genetic testing. Some are unique to predictive genetic testing on asymptomatic patients that are not applicable to mainstreaming. However, they should be taken into consideration when offering germline genetic testing as they affect risk-perception and decision making.59 In addition to these barriers, there are other external barriers that can affect decision making, like for instance family communication and healthcare barriers (eg, lack of specialised services and variability in surveillance packages) that can exacerbate uncertainty and anxiety13,60 and should be explored during the consultation. On the other hand, other external facilitators to testing are the work undertaken by the Genomic Medicine Service Alliances and the National Lynch syndrome project in mainstreaming, education, and reduction in variation of care.
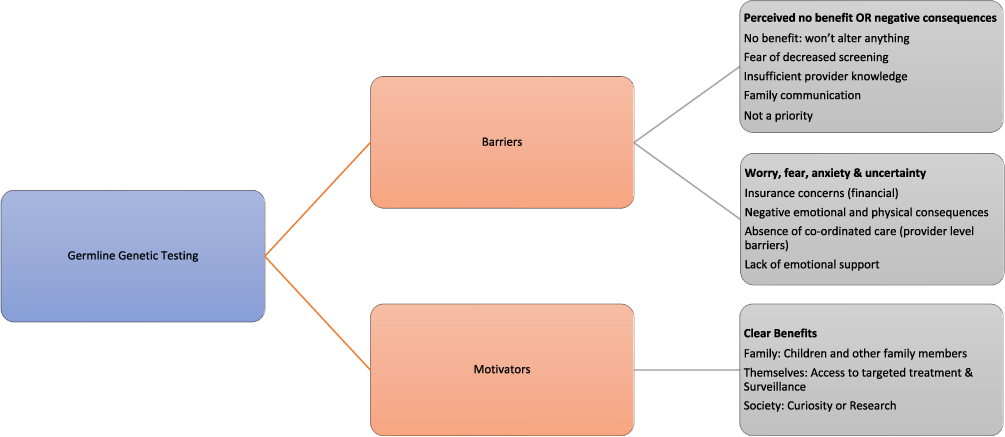 |
Figure 3 An illustration of barriers and facilitators for germline genetic testing. |
Cancer Risk Reduction
Colorectal Cancer
Colonoscopy
The cumulative lifetime incidence of CRC in people with LS is gene-specific ranging between 14% in PMS2 carriers and over 40% in MLH1 or MSH2 carriers, considerably higher than average population risk.5 LS patients have an accelerated pathway to carcinogenesis compared to the general population.14 Colonoscopy reduces the incidence and mortality associated with CRC in LS.15 As CRC risk is gene-specific, with earlier age diagnoses with higher risk genotypes, colorectal surveillance with routine colonoscopy every 2 years should start at 25 years for MLH1 and MSH2 carriers or at 35 years for MSH6 and PMS2 gene carriers.53,61
Despite adherence to appropriate, good quality endoscopic surveillance there are still high rates of interval CRC in LS. Although the prevalence of colonic polyps in patients with LS seems may not be higher than in the general population, the dMMR pathway to CRC seems to be accelerated at 3–3.5 years compared to 10–15 years in sporadic CRCs.62 Ahadova et al recently described a “3 pathways” model in LS CRCl carcinogenesis: dMMR usually represents an early and possibly initiating event, while CTNNB1 and TP53 mutations occurs in tumours lacking evidence of non-flat morphology polypoid growth.62
There is an association with intervals CRCs and quality of colonoscopy. A high proportion of post-colonoscopy cancers in LS are caused by missed lesions as a result of inadequate examination, lack of adherence to surveillance recommendation, and incomplete polyp resection.63 Reassuringly, a large multicentre study recently showed that rates of post colonoscopy CRC in centres complying with current guidance was 1.2%.64 Newton et al demonstrated that different hospital recall systems along with clinician and patient factors resulted in variable compliance with the recommended surveillance intervals for LS with a significant risk to patients not on well managed surveillance.65 In this study, the cumulative incidence of colorectal cancer to the age of 70 was 25% in the surveillance population and 81% in genetically diagnosed LS patients not undergoing colonoscopic surveillance.
In 2016, a United Kingdom multi-society meeting recommended the development of a quality-assured colonoscopic surveillance programme for people with LS,66 and from April 2023 the national screening programme in England will deliver this surveillance. This new programme will deliver registration, episode recall and high-quality colonoscopy for people with LS.
Aspirin
Meta-analyses of observational data amongst populations taking aspirin revealed an absolute risk reduction of 20% in all cancers, within gastrointestinal cancers the benefit was up to 34% risk reduction. A difference in CRC risk manifests in people taking aspirin compared to a population not taking aspirin after a lag-period of approximately 7–8 years after commencing aspirin.67
The CAPP2 trial recruited 861 people with LS. who were randomised to receive either 600mg of aspirin or placebo. Although the primary outcome of a difference in CRC incidence at 5 years was not confirmed, cancer outcomes at a mean of 10 years showed that 9% (40/427) in aspirin group developed CRC and 13% (58/434) in placebo group, in line with the observed “lag period” noted in previous observational meta-analyses.35 Adverse events were similar across the two groups. Data from CAPP2 suggest that aspirin should be taken daily for at least 2 years, and up to 5 years in total, after which it may be discontinued. In recent years United Kingdom guidelines recommended that aspirin be offered to people with LS with a linked decision aid developed by NICE to allow people with LS understand the risks and benefits of prophylactic aspirin.53
Data from the ASPREE trial suggests a more cautious approach in older patients. This study performed in people aged over 70 years starting aspirin demonstrated an increase of cancer diagnoses, and adverse effect on cancer stage at diagnosis. Although it is unclear, this observation may be due to aspirin suppressing the inflammatory response and facilitating metastasis.68
Gynaecological Cancer
Endometrial and ovarian cancers are closely associated with LS. The only proven way of reducing the risk of these cancers in women with LS is prophylactic surgery.69 This is in the form of hysterectomy and bilateral salpingo-oophorectomy. The age of surgery is one that should be decided on in consultation with the patient. Those who carry pathogenic variants in MSH6 or PMS2 can wait until after 45 years of age.70 However, women who carry pathogenic variants in MLH1 or MSH2 should consider risk reducing surgery once their families are complete. If there is a history of early onset gynaecological cancer within the family, this should be taken into account and surgery at an earlier age considered. For most women, risk reducing surgery can be done laparoscopically which allows a short hospital admission.71 After surgery, those premenopausal women found not to have an occult cancer, should be offered hormone replacement therapy.8
The role of gynaecological cancer surveillance in women with LS has been discussed in detail.72 There is no good evidence to support its use. However, such surveillance is offered in the United Kingdom albeit in an ad hoc fashion.73 Women with LS should be seen by a gynaecologist every year to two years as to discuss red flag symptoms of gynaecological cancer, family planning and the time of risk reducing surgery.8 Where there are red flag symptoms, such as irregular or heavy menstrual bleeding, investigations should be considered.74 In addition, those who can, should be advised to take aspirin. The CAPP2 found that those women taking aspirin had a 50% reduction in the incidence of endometrial cancer; however, it should be noted the study was not powered to explore this outcome and so this finding should be interpreted cautiously.75 The use of hormonal therapy to reduce endometrial and ovarian cancer risk in women with LS remains controversial. No meaningful trial data exists to support the hypothesis that hormonal therapy reduces gynaecological cancer risk in women with LS. However, in non-LS populations, there is clear observational evidence that the use of the combined pill reduces the risk of endometrial and ovarian cancer.76,77 In addition, the levonorgestrel intrauterine system has also been shown to greatly reduce endometrial cancer in non-LS populations.39 Therefore, many clinicians do advise women with LS to use hormonal forms of contraception as to reduce their cancer risk on the assumption that the evidence based within the non-LS population is applicable to women with LS.73 There is limited evidence that this maybe so, a small prospective biomarker study did find endometrial cellular proliferation was reduced with the use of progesterone/progestins in women with LS.78
Other Cancers
There is no reliable evidence as to how best to reduce the risk of the other cancers associated with LS. The use of prostate-specific antigen (PSA) as a means of surveillance for prostate cancer in men with LS is currently being investigated. The results of a prospective screening programme round found that male MSH2 and MSH6 pathogenic variant carriers have a higher incidence of prostate cancer.79 The overall positive predictive value of a PSA threshold of 3.0 ng/mL was 32.1% (20.3–46.0). However, these results are only from the first round of screening and further data is needed from more rounds of screening before the utility of PSA-based surveillance can be decided. Therefore, PSA screening for prostate cancer in men with LS should only be used in the context of a research trial. Another potential technology that could enable urinogenital surveillance in LS is urine cytology.80,81 However, there is insufficient evidence currently for this to be recommended.
Lifestyle risk factors for cancers should be addressed in those with LS. Namely, those with LS should be advised to exercise, maintain a healthy weight, and eat a healthy diet.82 In addition, they should not smoke and avoid other environmental carcinogens.83
The cancers associated with LS are summarised in Table 2.
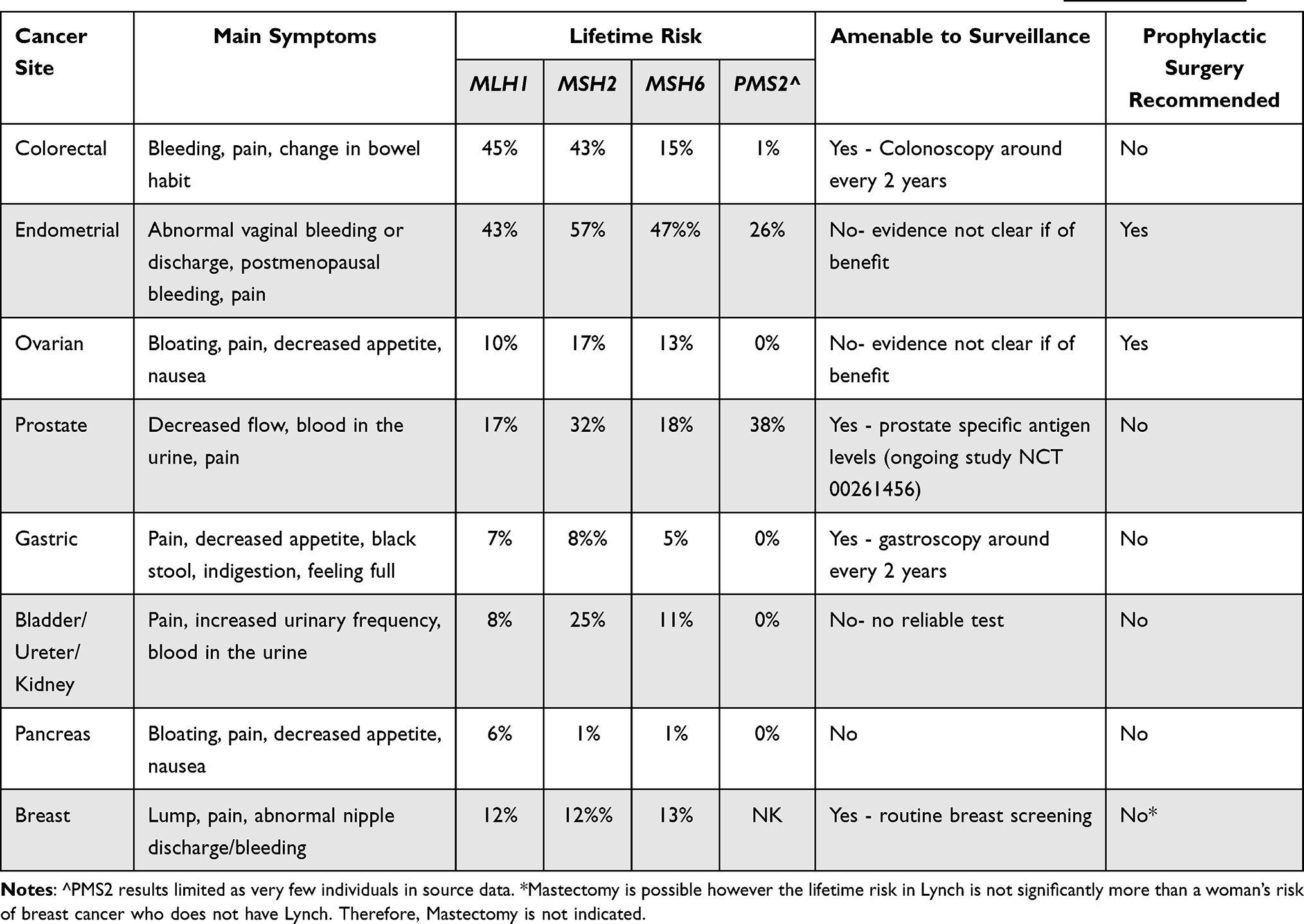 |
Table 2 Salient Clinical Features of Lynch Syndrome Associated Cancers. Sources Data Taken from PLSD (http://www.plsd.eu) |
The Global Perspective
As the authors practice in the United Kingdom, the perceptive of this article has been focused on clinical practice in the United Kingdom. Herein we detail key areas of practice variation seen globally. Furthermore, clinicians must be culturally aware when applying the guidance in this article. Patients must be viewed and treated holistically and in the context of their culture. Clinicians should seek to follow local guidelines where possible. A summary of the guidelines mentioned is provided in Table 3; this is not an exhaustive list of guidelines however a summary guidance from major professional bodies regionally.
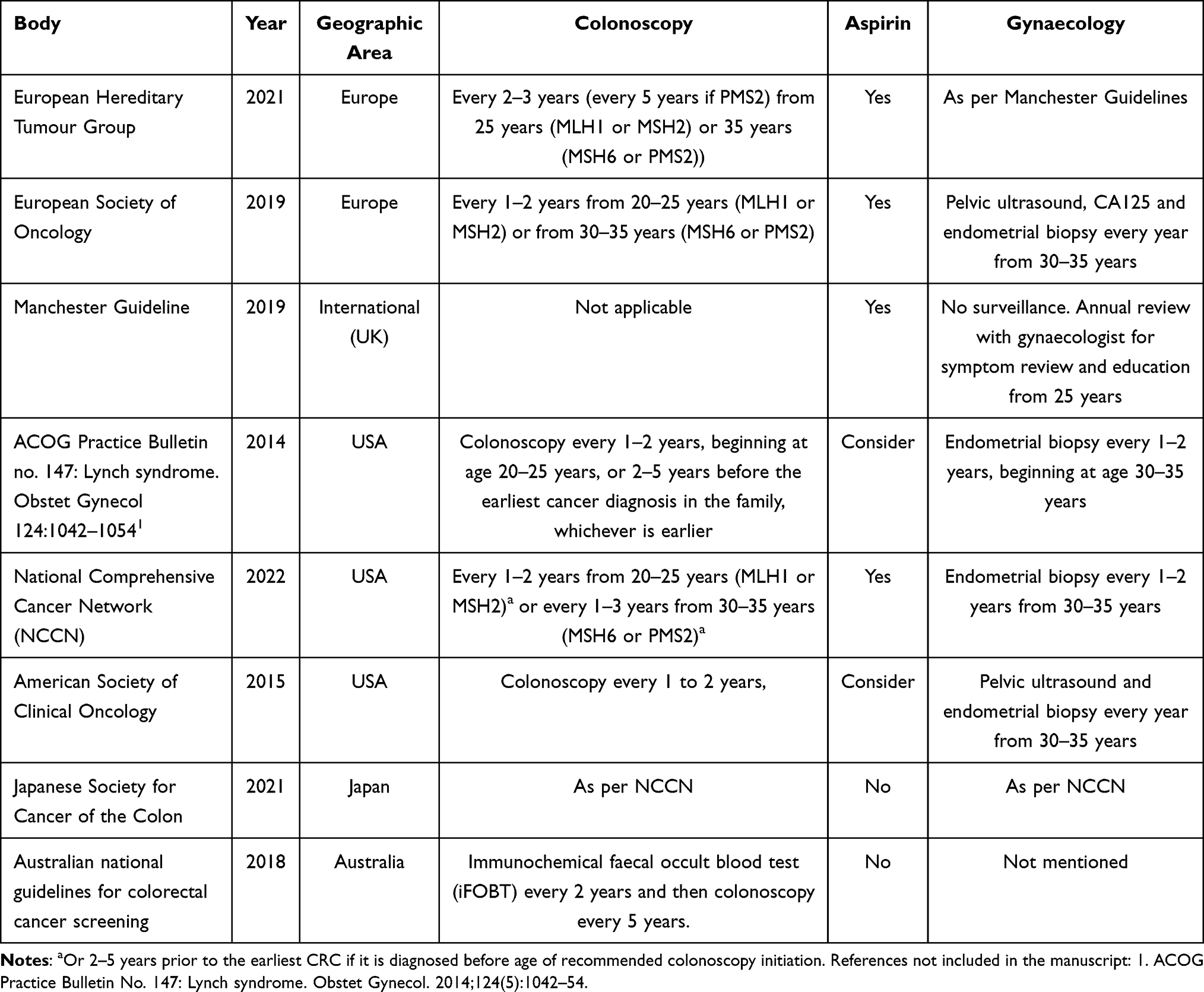 |
Table 3 Summary of Key Clinical Guidelines on Lynch Syndrome from Around the World |
North America
Healthcare in North America is mostly delivered through an insurance-based model. For many, their healthcare will be funded by private providers who charge a premium. One of the barriers not seen in a socialised health care model (such as the National Healthcare Service) is the implications of a positive LS test on insurance premiums. Indeed, the impact of increased premiums secondary to positive LS testing has been cited as a key barrier to testing.84 Furthermore, there are numerous private labs providing germline testing leading to an array of different report formats and terminology along with varying quality assurance. This in turn can make the interpretation of reporting difficult.85 Currently in North America, there is no movement towards a national registry of those with LS or nationally directed care.
The universal testing of endometrial and colorectal cancer is recommended by National Comprehensive Cancer Network (NCCN), however it may not be covered by an individual’s insurance meaning that the actual coverage is not universal.86 Conversely, those with more comprehensive insurance may find the spectrum of cancers that undergo MMR testing is broader than currently seen in the United Kingdom.87 Whether someone can access PGD is also determined by their level of insurance cover.
Regarding cancer surveillance, the recommendations for colonoscopy are equitable to those discussed within this article although once more, access is determined by an individual’s level of insurance. Those women with sufficient provision are often offered gynaecological cancer surveillance with a variety of methods used. This is supported by local guidelines from American Society of Clinical Oncology (ASCO) and NCCN which include gynaecological cancer surveillance for women with LS with either an ultrasound or/and an endometrial biopsy.88,89 Risk reducing surgery for women is offered around the same time as it is in the United Kingdom.
Europe
European healthcare is tapestry of private and state provided. The issues and barriers with private healthcare discussed above apply to those also paying premiums in Europe. Guidelines for the care of those with LS have been published by two continent wide bodies: The European Hereditary Tumour Group (EHTG) and The European Society for Medical Oncology (ESMO).9,90
Regarding gynaecological surveillances, the EHTG guidelines accepted the recommendations made in the United Kingdom Manchester Guidelines, and therefore does not recommend surveillance for gynaecological cancer].8 However, ESMO guidelines do support the use of annual endometrial biopsy, transvaginal ultrasound and CA125 for gynaecological cancer surveillance from 30 years of age.90 It should be noted that the authorship of the ESMO guidelines did not include a gynaecologist. Colonoscopy is recommended every 1–2 years but otherwise is in line with the United Kingdom.
Asia/Australasia
Once more, Asia has a broad range of healthcare systems operating in a diverse spectrum of cultures and economies. The Japanese Society for Cancer of the Colon have published a comprehensive guideline on the clinical management of Lynch syndrome. They accept the recommendations of the NCCN as discussed above and support the universal testing of endometrial and colorectal cancers for MMRd. They do not support the use of Aspirin for chemoprophylaxis in LS. Australian guidelines concentrate on colorectal screening.91 They recommend a novel approach in that 35–44yrs, those with LS are offered immunochemical faecal occult blood test (iFOBT) every 2 years and then colonoscopy every 5 years. No mention is made to extracolonic cancers.
The Rest of the World
No specific guidelines could be found from South America. The Jerusalem workshop was held in 2010 however this was organised and attended by clinicians from the USA and Europe.92 The Middle East Network on Hereditary Colorectal Cancer (HCCN-ME) have not produced their own guidelines but have endorsed those of other professional bodies.93
The Future
LS is now the focus of a gambit of research that looks to prevent, screen, and treat the cancers associated with the condition.
Vaccination
LS-associated cancers arise in the background of numerous insertion, deletion, and mis-incorporation mutations because of a dysfunctional mismatch repair system. This leads to numerous frameshifts within DNA transcription and translation.94 The resulting neo-peptides that are produced are immunogenic acting as antigens. Therefore, many LS associated cancers are associated with a strong immune response which will often lead to the destruction of the cancer.95 The neo-peptides produced by LS-associated cancers are to some degree predictable.96 As such, similar neo-peptides could be used as the basis of vaccine to inoculate LS carriers so that their immune system recognises cancers early and are able to clear them before they become clinically meaningful. Sadly, no trial data currently exists to support vaccination as a preventative measure in LS. The use of such vaccines in a murine model did lead to a significant decrease in tumour size which is encouraging and could also suggest vaccination could form part of the treatment for those with advanced LS associated cancers.97 Therefore, there is real excitement about the development of LS cancer vaccinations soon.98 Phase I and II trials have been undertaken; these have demonstrated LS associated cancers are well tolerated by patients and have an acceptable side effect profile.99 In addition, immunological analysis from these studies suggests a meaningful immune response secondary to vaccination; however, it is not clear if this will translate into meaningful clinical outcomes such as improved overall survival or a lower incidence in cancer.99 Vaccines are also being explored as an adjuvant therapy to complement more conventional cancer treatments. These studies are in a pre-clinical stage but have shown overall promise.97
New Screening Technologies
Currently, those with LS undergo a colonoscopy every two years.9 This procedure is uncomfortable, requires bowel preparation and associated with complications; one in 1000 colonoscopies end in visceral perforation.100 In addition, colonoscopy can miss cancers and provide false reassurance; around 7–10% of people who have undergone a colonoscopy are diagnosed with a colorectal cancer within 3 years.101 For women there is currently no method that can be reliably used to detect either ovarian cancer or endometrial cancer.72
However, new technologies are currently be evaluated to provide non-invasive and reliable methods of cancer surveillance. Faecal immunochemical testing (FIT) is a non-invasive screening test for colorectal cancer which is used in the general population to stratify people for colonoscopy. The test works by detecting occult blood in the stool through an antibody-based assay for human globin.102 FIT is currently being evaluated for use in the LS population which, if successful, could greatly limit the number of colonoscopies those with LS undergo.103 For women, cytology-based methods of endometrial cancer surveillance are currently being evaluated in the general population.104,105 These could be a non-invasive alternative for women with LS, however this is yet to be evaluated. In addition, novel microsatellite analysis of urine has to been used to detect cancers of the urogenital tract and endometrium in those with LS with some success.106 Larger studies are needed however, to confirm if this could be a viable non-invasive means of cancer surveillance. Finally, the development of technologies that utilise cell-free DNA could mean in the near future a blood test could be used to screen for all LS-associated cancers through the detection of molecular markers.107,108 These technologies are still unproven in the general population and further work would be needed before they could be applied to high-risk groups like LS.
Immunotherapy
LS-associated cancers arise within a dysfunctional mismatch repair system and therefore the tumours have a high mutational burden.109 This leads to a high number of immunogenic peptides that stimulate an anti-cancer immune response.110 To overcome this, LS-associated cancer develops immune escape mechanisms namely they utilise the programmed death-ligand 1 (PD-L1)/programmed death-1 (PD-1) axis.111 This is a druggable mechanism; the development of checkpoint immune inhibitors has revolutionised the treatment of cancers with mismatch repair deficiency.112 Seminal work by Le et al demonstrated a significant survival benefit in mismatch repair cancers that are treated with immune checkpoint inhibitors.113 These treatments have now become the mainstay of therapy for advanced mismatch repair tumours.114 As we come to better understand the utility of these treatments, through trial data, it maybe they become maintenance therapies.115 As our understanding of the immune landscape improves, we may be able to find novel targets for cancers in LS that prove resistant to immune checkpoint inhibition.
Conclusion
LS is a complicated clinical entity. The diagnosis requires people to come forward for testing which is difficult as there are no accurate means by which to identify healthy LS carriers. Universal testing is potentially expensive, will put pressure on already under resourced laboratories and it is not clear how best to take informed consent on such a large scale. Therefore, currently we screened those with LS associated cancers for LS by way of testing their tumours for features of mismatch repair deficiency. As we move to mainstreaming, those more likely to have LS will be offered germline testing by their clinical team. Therefore, only those with confirmed LS will need to be seen by clinical genetics who can go on to organise cascade testing and find those relatives who are healthy LS carriers. They can also aid with the interpretation of VUS results. Not all people wish to undergo testing for LS and having an appreciation of how to communicate risk and the barriers to testing is important so that clinicians can support individuals through the LS testing process.
Once diagnosed with LS, people need to be encouraged and supported to take evidenced-based measures that can reduce their risk of cancer. Sadly, many of these are invasive such as colonoscopy or risk reducing surgery and therefore individualised care is vital based on the individual’s risk and preferences. Further research is needed and needs to be funded to improve the care of those with LS. Vaccines hold hope of lifelong cancer prevention. New technologies could mean cancers that do develop are detectable on a blood test and could be treated with immunotherapies before they even became clinically relevant. This future is only realisable if the current focus on LS-related research is maintained, and funding bodies seek to fund it.
In summary, LS care has come a long way over the last twenty years. We know understand the individual cancer risk to inform consent, tests to accurately diagnoses LS and ways by which we can reduce cancer risk. However, more needs to be done to find those who are undiagnosed, develop less invasive cancer surveillance methods and develop new vaccinations and treatments.
Data Sharing Statement
All data contained here within this manuscript is available on request from the corresponding author.
Ethics Statement
All studies were approved by the respective institutional review boards and conducted with appropriate ethical criteria in each country and in accordance with the Declaration of Helsinki.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
No specific funding was used for this study.
Disclosure
Dr Tracie Miles and Dr Neil AJ Ryan report personal fees from Glaxo Smith Kline, during the conduct of the study. The authors declare no other conflicts of interest in this work.
References
1. Ryan NA, McMahon RF, Ramchander NC, Seif MW, Evans DG, Crosbie EJ. Lynch syndrome for the gynaecologist. Obstet Gynaecol. 2021;23(1):9–20. doi:10.1111/tog.12706
2. Martín-López JV, Fishel R. The mechanism of mismatch repair and the functional analysis of mismatch repair defects in Lynch syndrome. Fam Cancer. 2013;12(2):159–168. doi:10.1007/s10689-013-9635-x
3. Imai K, Yamamoto H. Carcinogenesis and microsatellite instability: the interrelationship between genetics and epigenetics. Carcinogenesis. 2007;29(4):673–680. doi:10.1093/carcin/bgm228
4. Wong H-L, Christie M, Gately L, et al. Mismatch repair deficiency assessment by immunohistochemistry: for Lynch syndrome screening and beyond. Future Oncol. 2018;14(26):2725–2739. doi:10.2217/fon-2018-0319
5. Ryan NAJ, Glaire MA, Blake D, Cabrera-Dandy M, Evans DG, Crosbie EJ. The proportion of endometrial cancers associated with Lynch syndrome: a systematic review of the literature and meta-analysis. Genet Med. 2019;21(10):2167–2180. doi:10.1038/s41436-019-0536-8
6. Toon CW, Walsh MJ, Chou A, et al. BRAFV600E immunohistochemistry facilitates universal screening of colorectal cancers for Lynch syndrome. Am J Surg Pathol. 2013;37(10):1592. doi:10.1097/PAS.0b013e31828f233d
7. Møller P, Seppälä T, Bernstein I, et al. Cancer incidence and survival in Lynch syndrome patients receiving colonoscopic and gynaecological surveillance: first report from the prospective Lynch syndrome database. Gut. 2017;66(3):464–472. doi:10.1136/gutjnl-2015-309675
8. Crosbie EJ, Ryan NAJ, Arends MJ, et al. The Manchester International Consensus Group recommendations for the management of gynecological cancers in Lynch syndrome. Genet Med. 2019;21(10):2390–2400. doi:10.1038/s41436-019-0489-y
9. Seppälä TT, Latchford A, Negoi I, et al. European guidelines from the EHTG and ESCP for Lynch syndrome: an updated third edition of the Mallorca guidelines based on gene and gender. Br J Surg. 2021;108(5):484–498. doi:10.1002/bjs.11902
10. Ryan NA, Donnelly L, Stocking K, Evans DG, Crosbie EJ. Feasibility of gynaecologist led Lynch syndrome testing in women with endometrial cancer. J Clin Med. 2020;9(6):1842. doi:10.3390/jcm9061842
11. Metcalfe A, Coad J, Plumridge GM, Gill P, Farndon P. Family communication between children and their parents about inherited genetic conditions: a meta-synthesis of the research. Eur J Hum Genet. 2008;16(10):1193–1200. doi:10.1038/ejhg.2008.84
12. Snowsill T, Coelho H, Huxley N, et al. Molecular testing for Lynch syndrome in people with colorectal cancer: systematic reviews and economic evaluation. Health Technol Assess. 2017;21(51):1–238. doi:10.3310/hta21510
13. Srinivasan S, Hampel H, Leeman J, et al. Stakeholder perspectives on overcoming barriers to cascade testing in Lynch syndrome: a qualitative study. Cancer Prev Res. 2020;13(12):1037–1046. doi:10.1158/1940-6207.CAPR-20-0141
14. Salikhanov I, Heinimann K, Chappuis P, et al. Swiss cost-effectiveness analysis of universal screening for Lynch syndrome of patients with colorectal cancer followed by cascade genetic testing of relatives. J Med Genet. 2022;59:jmedgenet-2021–108062.
15. Jasperson K. Colorectal cancer: cascade genetic testing in Lynch syndrome: room for improvement. Nat Rev Gastroenterol Hepatol. 2013;10(9):506–508. doi:10.1038/nrgastro.2013.122
16. Griffin NE, Buchanan TR, Smith SH, et al. Low rates of cascade genetic testing among families with hereditary gynecologic cancer: an opportunity to improve cancer prevention. Gynecol Oncol. 2020;156(1):140–146. doi:10.1016/j.ygyno.2019.11.005
17. Menko FH, Ter Stege JA, van der Kolk LE, et al. The uptake of presymptomatic genetic testing in hereditary breast-ovarian cancer and Lynch syndrome: a systematic review of the literature and implications for clinical practice. Fam Cancer. 2019;18(1):127–135. doi:10.1007/s10689-018-0089-z
18. Henrikson NB, Blasi P, Figueroa Gray M, et al. Patient and family preferences on health system-led direct contact for cascade screening. J Pers Med. 2021;11(6):538. doi:10.3390/jpm11060538
19. Voorwinden JS, Jaspers JPC. Prognostic factors for distress after genetic testing for hereditary cancer. J Genet Couns. 2016;25(3):495–503. doi:10.1007/s10897-015-9894-9
20. DudokdeWit AC, Tibben A, Duivenvoorden HJ, et al. Psychological distress in applicants for predictive DNA testing for autosomal dominant, heritable, late onset disorders. The Rotterdam/leiden genetics workgroup. J Med Genet. 1997;34(5):382–390. doi:10.1136/jmg.34.5.382
21. Godino L, Turchetti D, Jackson L, Hennessy C, Skirton H. Presymptomatic genetic testing for hereditary cancer in young adults: a survey of young adults and parents. Eur J Hum Genet. 2019;27(2):291–299. doi:10.1038/s41431-018-0262-8
22. Julian-Reynier C, Chabal F, Frebourg T, et al. Professionals assess the acceptability of preimplantation genetic diagnosis and prenatal diagnosis for managing inherited predisposition to cancer. J Clin Oncol. 2009;27(27):4475–4480. doi:10.1200/JCO.2008.21.2712
23. Patel AP, Wang M, Fahed AC, et al. Association of rare pathogenic DNA variants for familial hypercholesterolemia, hereditary breast and ovarian cancer syndrome, and lynch syndrome with disease risk in adults according to family history. JAMA Netw Open. 2020;3(4):e203959–e. doi:10.1001/jamanetworkopen.2020.3959
24. Hampel H, de la Chapelle A. The search for unaffected individuals with Lynch syndrome: do the ends justify the means? Cancer Prev Res. 2011;4(1):1–5. doi:10.1158/1940-6207.CAPR-10-0345
25. Hampel H, de la Chapelle A. How do we approach the goal of identifying everybody with Lynch syndrome? Fam Cancer. 2013;12(2):313–317. doi:10.1007/s10689-013-9611-5
26. Downing M, Pollitt R. Newborn bloodspot screening in the UK–past, present and future. Ann Clin Biochem. 2008;45(1):11–17. doi:10.1258/acb.2007.007127
27. Manchanda R, Patel S, Gordeev VS, et al. Cost-effectiveness of population-based BRCA1, BRCA2, RAD51C, RAD51D, BRIP1, PALB2 mutation testing in unselected general population women. J Natl Cancer Inst. 2018;110(7):714–725. doi:10.1093/jnci/djx265
28. Guzauskas GF, Jiang S, Garbett S, et al. Cost-effectiveness of population-wide genomic screening for Lynch syndrome in the United States. Genet Med. 2022;24(5):1017–1026. doi:10.1016/j.gim.2022.01.017
29. Manchanda R, Legood R. Population based germline testing for primary cancer prevention. Oncotarget. 2018;9(69):33062–33063. doi:10.18632/oncotarget.25995
30. De Simone LM, Arjunan A, Vogel Postula KJ, Maga T, Bucheit LA. Genetic counselors’ perspectives on population-based screening for BRCA-related hereditary breast and ovarian cancer and Lynch syndrome. J Genet Couns. 2021;30(1):158–169. doi:10.1002/jgc4.1305
31. Offit K, Tkachuk KA, Stadler ZK, et al. Cascading after peridiagnostic cancer genetic testing: an alternative to population-based screening. J Clin Oncol. 2020;38(13):1398–1408. doi:10.1200/JCO.19.02010
32. Dragojlovic N, Borle K, Kopac N, et al. The composition and capacity of the clinical genetics workforce in high-income countries: a scoping review. Genet Med. 2020;22(9):1437–1449. doi:10.1038/s41436-020-0825-2
33. Ryan NAJ, Morris J, Green K, et al. Association of mismatch repair mutation with age at cancer onset in Lynch syndrome: implications for stratified surveillance strategies. JAMA Oncol. 2017;3(12):1702–1706. doi:10.1001/jamaoncol.2017.0619
34. Hampel H, Frankel W, Panescu J, et al. Screening for Lynch syndrome (hereditary nonpolyposis colorectal cancer) among endometrial cancer patients. Cancer Res. 2006;66(15):7810–7817. doi:10.1158/0008-5472.CAN-06-1114
35. Snowsill T, Huxley N, Hoyle M, et al. A systematic review and economic evaluation of diagnostic strategies for Lynch syndrome. Health Technol Assess. 2014;18(58):1–406. doi:10.3310/hta18580
36. Snowsill TM, Ryan NAJ, Crosbie EJ. Cost-effectiveness of the Manchester approach to identifying Lynch syndrome in women with endometrial cancer. J Clin Med. 2020;9:6. doi:10.3390/jcm9061664
37. Snowsill TM, Ryan NAJ, Crosbie EJ, Frayling IM, Evans DG, Hyde CJ. Cost-effectiveness analysis of reflex testing for Lynch syndrome in women with endometrial cancer in the UK setting. PLoS One. 2019;14(8):e0221419. doi:10.1371/journal.pone.0221419
38. Atwal A, Snowsill T, Dandy MC, et al. The prevalence of mismatch repair deficiency in ovarian cancer: a systematic review and meta-analysis. Int J Cancer. 2022;151(9):1626–1639. doi:10.1002/ijc.34165
39. Crosbie EJ, Kitson SJ, McAlpine JN, Mukhopadhyay A, Powell ME, Singh N. Endometrial cancer. Lancet. 2022;399(10333):1412–1428. doi:10.1016/S0140-6736(22)00323-3
40. Lu KH, Dinh M, Kohlmann W, et al. Gynecologic cancer as a “sentinel cancer” for women with hereditary nonpolyposis colorectal cancer syndrome. Obstet Gynecol. 2005;105(3):569–574. doi:10.1097/01.AOG.0000154885.44002.ae
41. Lynch HT, Shaw MW, Magnuson CW, Larsen AL, Krush AJ. Hereditary factors in cancer. Study of two large midwestern kindreds. Arch Intern Med. 1966;117(2):206–212. doi:10.1001/archinte.1966.03870080050009
42. Rodriguez-Bigas MA, Boland CR, Hamilton SR, et al. A national cancer institute workshop on hereditary nonpolyposis colorectal cancer syndrome: meeting highlights and Bethesda guidelines. J Natl Cancer Inst. 1997;89(23):1758–1762. doi:10.1093/jnci/89.23.1758
43. Vasen HF, Watson P, Mecklin JP, Lynch HT. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the international collaborative group on HNPCC. Gastroenterology. 1999;116(6):1453–1456. doi:10.1016/S0016-5085(99)70510-X
44. Sjursen W, Haukanes BI, Grindedal EM, et al. Current clinical criteria for Lynch syndrome are not sensitive enough to identify MSH6 mutation carriers. J Med Genet. 2010;47(9):579–585. doi:10.1136/jmg.2010.077677
45. Kastrinos F, Uno H, Ukaegbu C, et al. Development and validation of the PREMM(5) model for comprehensive risk assessment of lynch syndrome. J Clin Oncol. 2017;35(19):2165–2172. doi:10.1200/JCO.2016.69.6120
46. Barnetson RA, Tenesa A, Farrington SM, et al. Identification and survival of carriers of mutations in DNA mismatch-repair genes in colon cancer. N Engl J Med. 2006;354(26):2751–2763. doi:10.1056/NEJMoa053493
47. Chen S, Wang W, Lee S, et al. Prediction of germline mutations and cancer risk in the Lynch syndrome. JAMA. 2006;296(12):1479–1487. doi:10.1001/jama.296.12.1479
48. George A, Riddell D, Seal S, et al. Implementing rapid, robust, cost-effective, patient-centred, routine genetic testing in ovarian cancer patients. Sci Rep. 2016;6(1):29506. doi:10.1038/srep29506
49. Percival N, George A, Gyertson J, et al. The integration of BRCA testing into oncology clinics. Br J Nurs. 2016;25(12):690–694. doi:10.12968/bjon.2016.25.12.690
50. Scott N, O’Sullivan J, Asgeirsson K, Macmillan D, Wilson E. Changing practice: moving to a specialist nurse-led service for BRCA gene testing. Br J Nurs. 2020;29(10):S6–S13. doi:10.12968/bjon.2020.29.10.S6
51. Bokkers K, Vlaming M, Engelhardt EG, et al. The feasibility of implementing mainstream germline genetic testing in routine cancer care-a systematic review. Cancers. 2022;14:4. doi:10.3390/cancers14041059
52. NHS England. Implementing Lynch syndrome testing and surveillance pathways; 2021. Available from: https://www.england.nhs.uk/wp-content/uploads/2021/07/B0622-implementing-lynch-syndrome-testing-and-surveillance-pathways.pdf.
53. Monahan KJ, Bradshaw N, Dolwani S, et al. Guidelines for the management of hereditary colorectal cancer from the British Society of Gastroenterology (BSG)/Association of Coloproctology of Great Britain and Ireland (ACPGBI)/United Kingdom Cancer Genetics Group (UKCGG). Gut. 2020;69(3):411–444. doi:10.1136/gutjnl-2019-319915
54. Thompson BA, Spurdle AB, Plazzer JP, et al. Application of a 5-tiered scheme for standardized classification of 2360 unique mismatch repair gene variants in the InSiGHT locus-specific database. Nat Genet. 2014;46(2):107–115. doi:10.1038/ng.2854
55. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. 2015;17(5):405–424. doi:10.1038/gim.2015.30
56. Graves KD, Sinicrope PS, Esplen MJ, et al. Communication of genetic test results to family and health-care providers following disclosure of research results. Genet Med. 2014;16(4):294–301. doi:10.1038/gim.2013.137
57. Vears DF, Minion JT, Roberts SJ, et al. Return of individual research results from genomic research: a systematic review of stakeholder perspectives. PLoS One. 2021;16(11):e0258646. doi:10.1371/journal.pone.0258646
58. Bradbury AR, Patrick-Miller L, Egleston BL, et al. Returning individual genetic research results to research participants: uptake and outcomes among patients with breast cancer. JCO Precis Oncol. 2018;2:1–24.
59. Keogh L, McClaren B, Maskiell J, et al. How do individuals decide whether to accept or decline an offer of genetic testing for colorectal cancer? Hered Cancer Clin Pract. 2011;9(Suppl 1):P17. doi:10.1186/1897-4287-9-S1-P17
60. Campbell-Salome G, Buchanan AH, Hallquist MLG, Rahm AK, Rocha H, Sturm AC. Uncertainty management for individuals with Lynch syndrome: identifying and responding to healthcare barriers. Patient Educ Couns. 2021;104(2):403–412. doi:10.1016/j.pec.2020.07.017
61. Van Leerdam ME, Roos VH, van Hooft JE, et al. Endoscopic management of Lynch syndrome and of familial risk of colorectal cancer: European Society of Gastrointestinal Endoscopy (ESGE) guideline. Endoscopy. 2019;51(11):1082–1093. doi:10.1055/a-1016-4977
62. Ahadova A, Gallon R, Gebert J, et al. Three molecular pathways model colorectal carcinogenesis in Lynch syndrome. Int J Cancer. 2018;143(1):139–150. doi:10.1002/ijc.31300
63. Haanstra JF, Vasen HF, Sanduleanu S, et al. Quality colonoscopy and risk of interval cancer in Lynch syndrome. Int J Colorectal Dis. 2013;28(12):1643–1649. doi:10.1007/s00384-013-1745-2
64. Sánchez A, Roos VH, Navarro M, et al. Quality of colonoscopy is associated with adenoma detection and postcolonoscopy colorectal cancer prevention in Lynch syndrome. Clin Gastroenterol Hepatol. 2022;20(3):611–21.e9. doi:10.1016/j.cgh.2020.11.002
65. Newton K, Green K, Lalloo F, Evans DG, Hill J. Colonoscopy screening compliance and outcomes in patients with Lynch syndrome. Colorectal Dis. 2015;17(1):38–46. doi:10.1111/codi.12778
66. Monahan KJ, Alsina D, Bach S, et al. Urgent improvements needed to diagnose and manage Lynch syndrome. BMJ. 2017;356:j1388. doi:10.1136/bmj.j1388
67. Edwards P, Monahan KJ. Diagnosis and management of Lynch syndrome. Frontline Gastroenterol. 2022;13(e1):e80. doi:10.1136/flgastro-2022-102123
68. McNeil JJ, Gibbs P, Orchard SG, et al. Effect of aspirin on cancer incidence and mortality in older adults. J Natl Cancer Inst. 2021;113(3):258–265. doi:10.1093/jnci/djaa114
69. Schmeler KM, Lynch HT, Chen L-M, et al. Prophylactic surgery to reduce the risk of gynecologic cancers in the Lynch syndrome. N Engl J Med. 2006;354(3):261–269. doi:10.1056/NEJMoa052627
70. Dominguez-Valentin M, Crosbie EJ, Engel C, et al. Risk-reducing hysterectomy and bilateral salpingo-oophorectomy in female heterozygotes of pathogenic mismatch repair variants: a prospective Lynch syndrome database report. Genet Med. 2021;23(4):705–712. doi:10.1038/s41436-020-01029-1
71. Garry R, Fountain J, Mason S, et al. The eVALuate study: two parallel randomised trials, one comparing laparoscopic with abdominal hysterectomy, the other comparing laparoscopic with vaginal hysterectomy. BMJ. 2004;328(7432):129. doi:10.1136/bmj.37984.623889.F6
72. Ryan N, Snowsill T, McKenzie E, Monahan K, Nebgen D. Should women with Lynch syndrome be offered gynaecological cancer surveillance? BMJ. 2021;374:n2020. doi:10.1136/bmj.n2020
73. Ryan N, Nobes M, Sedgewick D, Teoh SN, Evans DG, Crosbie EJ. A mismatch in care: results of a United Kingdom-wide patient and clinician survey of gynaecological services for women with Lynch syndrome. BJOG. 2021;128(4):728–736. doi:10.1111/1471-0528.16432
74. Funston G, O’Flynn H, Ryan NAJ, Hamilton W, Crosbie EJ. Recognizing gynecological cancer in primary care: risk factors, red flags, and referrals. Adv Ther. 2018;35(4):577–589. doi:10.1007/s12325-018-0683-3
75. Burn J, Sheth H, Elliott F, et al. Cancer prevention with aspirin in hereditary colorectal cancer (Lynch syndrome), 10-year follow-up and registry-based 20-year data in the CAPP2 study: a double-blind, randomised, placebo-controlled trial. Lancet. 2020;395(10240):1855–1863. doi:10.1016/S0140-6736(20)30366-4
76. Havrilesky LJ, Moorman PG, Lowery WJ, et al. Oral contraceptive pills as primary prevention for ovarian cancer: a systematic review and meta-analysis. Obstet Gynecol. 2013;122:1. doi:10.1097/AOG.0b013e318291c235
77. MacKintosh ML, Crosbie EJ. Prevention strategies in endometrial carcinoma. Curr Oncol Rep. 2018;20(12):101. doi:10.1007/s11912-018-0747-1
78. Lu KH, Loose DS, Yates MS, et al. Prospective multicenter randomized intermediate biomarker study of oral contraceptive versus depo-provera for prevention of endometrial cancer in women with Lynch syndrome. Cancer Prev Res. 2013;6(8):774–781. doi:10.1158/1940-6207.CAPR-13-0020
79. Bancroft EK, Page EC, Brook MN, et al. A prospective prostate cancer screening programme for men with pathogenic variants in mismatch repair genes (IMPACT): initial results from an international prospective study. Lancet Oncol. 2021;22(11):1618–1631. doi:10.1016/S1470-2045(21)00522-2
80. Narine N, Rana D, Shelton D, et al. Synchronous uterine and bladder cancers detected in urine and vaginal samples by cytology. Diagn Cytopathol. 2022;50(3):E86–E91. doi:10.1002/dc.24906
81. Myrhøj T, And Ersen MB, Bernstein I. Screening for urinary tract cancer with urine cytology in Lynch syndrome and familial colorectal cancer. Fam Cancer. 2008;7(4):303–307. doi:10.1007/s10689-008-9193-9
82. Coletta AM, Peterson SK, Gatus LA, et al. Energy balance related lifestyle factors and risk of endometrial and colorectal cancer among individuals with lynch syndrome: a systematic review. Fam Cancer. 2019;18(4):399–420. doi:10.1007/s10689-019-00135-7
83. Lindor NM, Petersen GM, Hadley DW, et al. Recommendations for the care of individuals with an inherited predisposition to lynch syndrome a systematic review. JAMA. 2006;296(12):1507–1517. doi:10.1001/jama.296.12.1507
84. Fogleman AJ, Zahnd WE, Lipka AE, et al. Knowledge, attitudes, and perceived barriers towards genetic testing across three rural Illinois communities. J Community Genet. 2019;10(3):417–423. doi:10.1007/s12687-019-00407-w
85. Ryan N, Wall J, Crosbie EJ, et al. Lynch syndrome screening in gynaecological cancers: results of an international survey with recommendations for uniform reporting terminology for mismatch repair immunohistochemistry results. Histopathology. 2019;75(6):813–824. doi:10.1111/his.13925
86. Eriksson J, Amonkar M, Al-Jassar G, et al. Experience of mismatch repair/microsatellite instability (MMR/MSI) testing among patients with advanced/metastatic colorectal cancer in the US. Curr Med Res Opin. 2020;36(8):1355–1361. doi:10.1080/03007995.2020.1776235
87. Kang SY, Kim DG, Ahn S, Ha SY, Jang K-T, Kim K-M. Comparative analysis of microsatellite instability by next-generation sequencing, MSI PCR and MMR immunohistochemistry in 1942 solid cancers. Pathol Res Pract. 2022;233:153874. doi:10.1016/j.prp.2022.153874
88. Stoffel EM, Mangu PB, Gruber SB, et al. Hereditary colorectal cancer syndromes: American society of clinical oncology clinical practice guideline endorsement of the familial risk-colorectal cancer: European society for medical oncology clinical practice guidelines. J Clin Oncol. 2015;33(2):209–217. doi:10.1200/JCO.2014.58.1322
89. Gupta S, Provenzale D, Llor X, et al. NCCN guidelines insights: genetic/familial high-risk assessment: colorectal, version 2.2019: featured updates to the NCCN guidelines. J Natl Compr Canc Netw. 2019;17(9):1032–1041. doi:10.6004/jnccn.2019.0044
90. Stjepanovic N, Moreira L, Carneiro F, et al. Hereditary gastrointestinal cancers: ESMO clinical practice guidelines for diagnosis, treatment and follow-up†. Ann Oncol. 2019;30(10):1558–1571. doi:10.1093/annonc/mdz233
91. Jenkins MA, Ait Ouakrim D, Boussioutas A, et al. Revised Australian national guidelines for colorectal cancer screening: family history. Med J Aust. 2018;209(10):455–460. doi:10.5694/mja18.00142
92. Boland CR, Shike M. Report from the Jerusalem workshop on Lynch syndrome-hereditary nonpolyposis colorectal cancer. Gastroenterology. 2010;138(7):2197.e1–e21977. doi:10.1053/j.gastro.2010.04.024
93. Ghorbanoghli Z, Jabari C, Sweidan W, et al. A new hereditary colorectal cancer network in the Middle East and eastern Mediterranean countries to improve care for high-risk families. Fam Cancer. 2018;17(2):209–212. doi:10.1007/s10689-017-0018-6
94. Thibodeau SN, Bren G, Schaid D. Microsatellite instability in cancer of the proximal colon. Science. 1993;260(5109):816–819. doi:10.1126/science.8484122
95. Ramchander NC, Ryan NAJ, Walker TDJ, et al. Distinct immunological landscapes characterize inherited and sporadic mismatch repair deficient endometrial cancer. Front Immunol. 2020;10:3023. doi:10.3389/fimmu.2019.03023
96. von Knebel Doeberitz M, Kloor M. Towards a vaccine to prevent cancer in Lynch syndrome patients. Fam Cancer. 2013;12(2):307–312. doi:10.1007/s10689-013-9662-7
97. Gebert J, Gelincik O, Oezcan-Wahlbrink M, et al. Recurrent frameshift neoantigen vaccine elicits protective immunity with reduced tumor burden and improved overall survival in a lynch syndrome mouse model. Gastroenterology. 2021;161(4):1288–302.e13. doi:10.1053/j.gastro.2021.06.073
98. Sahin U, Türeci Ö. Personalized vaccines for cancer immunotherapy. Science. 2018;359(6382):1355–1360. doi:10.1126/science.aar7112
99. Hernandez-Sanchez A, Grossman M, Yeung K, Sei SS, Lipkin S, Kloor M. Vaccines for immunoprevention of DNA mismatch repair deficient cancers. J Immunother Cancer. 2022;10(6):e004416. doi:10.1136/jitc-2021-004416
100. Zwink N, Holleczek B, Stegmaier C, Hoffmeister M, Brenner H. Complication rates in colonoscopy screening for cancer. Dtsch Arztebl Int. 2017;114(18):321–327. doi:10.3238/arztebl.2017.0321
101. Morris EJ, Rutter MD, Finan PJ, Thomas JD, Valori R. Post-colonoscopy colorectal cancer (PCCRC) rates vary considerably depending on the method used to calculate them: a retrospective observational population-based study of PCCRC in the English national health service. Gut. 2015;64(8):1248–1256. doi:10.1136/gutjnl-2014-308362
102. Schreuders EH, Grobbee EJ, Spaander MCW, Kuipers EJ. Advances in fecal tests for colorectal cancer screening. Curr Treat Options Gastroenterol. 2016;14(1):152–162. doi:10.1007/s11938-016-0076-0
103. Lincoln A, Lincoln A, Benton S, Sasieni P. PTH-27 Risk-stratified FIT for urgent colonoscopy in Lynch syndrome: a clinical service throughout the COVID-19 pandemic. Gut. 2021;70(Suppl 4):A184.
104. Jones ER, Carter S, Flynn H, et al. DEveloping Tests for Endometrial Cancer deTection (DETECT): protocol for a diagnostic accuracy study of urine and vaginal samples for the detection of endometrial cancer by cytology in women with postmenopausal bleeding. BMJ Open. 2021;11(7):e050755. doi:10.1136/bmjopen-2021-050755
105. O’Flynn H, Ryan NAJ, Narine N, Shelton D, Rana D, Crosbie EJ. Diagnostic accuracy of cytology for the detection of endometrial cancer in urine and vaginal samples. Nat Commun. 2021;12(1):952. doi:10.1038/s41467-021-21257-6
106. Phelps R, Gallon R, Hayes C, et al. Detection of microsatellite instability in colonoscopic biopsies and postal urine samples from Lynch syndrome cancer patients using a multiplex PCR assay. Cancers. 2022;14(15). doi:10.3390/cancers14153838
107. Liu MC, Oxnard GR, Klein EA, et al. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann Oncol. 2020;31(6):745–759. doi:10.1016/j.annonc.2020.02.011
108. Chen X, Dong Z, Hubbell E, et al. Prognostic significance of blood-based multi-cancer detection in plasma cell-free DNA. Clin Cancer Res. 2021;27(15):4221–4229. doi:10.1158/1078-0432.CCR-21-0417
109. Ryan NAJ, Walker TDJ, Bolton J, et al. Histological and somatic mutational profiles of mismatch repair deficient endometrial tumours of different aetiologies. Cancers. 2021;13(18):4538. doi:10.3390/cancers13184538
110. Glaire MA, Ryan NAJ, Ijsselsteijn ME, et al. Discordant prognosis of mismatch repair deficiency in colorectal and endometrial cancer reflects variation in antitumour immune response and immune escape. J Pathol. 2022;257(3):340–351. doi:10.1002/path.5894
111. Kim JM, Chen DS. Immune escape to PD-L1/PD-1 blockade: seven steps to success (or failure). Ann Oncol. 2016;27(8):1492–1504. doi:10.1093/annonc/mdw217
112. Kaushik I, Ramachandran S, Zabel C, Gaikwad S, Srivastava SK. The evolutionary legacy of immune checkpoint inhibitors. Semin Cancer Biol. 2022;86:491–498. doi:10.1016/j.semcancer.2022.03.020
113. Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372(26):2509–2520. doi:10.1056/NEJMoa1500596
114. Evans B, Evans S. Immune checkpoint inhibitors in cancer: pharmacology and toxicities. Pharm J. 2018;10:1.
115. Grivas P, Monk BJ, Petrylak D, et al. Immune checkpoint inhibitors as switch or continuation maintenance therapy in solid tumors: rationale and current state. Target Oncol. 2019;14(5):505–525. doi:10.1007/s11523-019-00665-1
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.