")
Back to Journals » International Journal of Nanomedicine » Volume 13
A flow cytometric analysis of macrophage–nanoparticle interactions in vitro: induction of altered Toll-like receptor expression
Authors Njoroge JM, Yourick JJ, Principato MA
Received 15 May 2018
Accepted for publication 3 October 2018
Published 12 December 2018 Volume 2018:13 Pages 8365—8378
DOI https://doi.org/10.2147/IJN.S174184
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Joyce M Njoroge, Jeffrey J Yourick, Mary Ann Principato
Division of Toxicology, Office of Applied Research and Safety Assessment, Center for Food Safety and Applied Nutrition, US Food and Drug Administration, Laurel, MD 20708, USA
Background: Nanoparticles exhibit unique physiochemical characteristics that provide the basis for their utilization. The diversity of potential and actual applications compels a thorough understanding regarding the consequences of their containment within the cellular environment.
Purpose: This paper presents a flow cytometric examination of the biologic effects associated with the internalization of citrate-buffered silver (Ag) nanoparticles (NP) by the murine macrophage cell line, RAW264.7.
Materials and methods: Cells were cultured with varying concentrations of citrate-buffered Ag nanoparticle and analyzed for changes in cellular volume, fluorescence emissions, and surface receptor expression.
Results: Notable changes in side scatter (SSC) signal occurred following the phagocytosis of citrate-buffered Ag NP representative of the 10 nm, 50 nm, and 100 nm particle size by cultured RAW 264.7 cells. A characteristic associated with the internalization of all the citrated Ag NP sizes tested, was the detection of emitted infra-red and near-infrared wavelength emissions. This characteristic consistently permitted the detection of 10 nm, 50 nm, and 100 nm Ag NP particles internalized within the RAW cells by flow cytometry. A functional distinction between monocyte subsets within the RAW 264.7 cell line was noted as Ag NP are taken up by the F4/80+ subset of cells within the culture. Further, the internalization of Ag NP by the cells resulted in an increased cell surface expression of the Toll-like Receptor (TLR) 3, but not TLR4.
Conclusion: Taken together, these results implicate the more mature macrophage in the ingestion of Ag NP; and an influence upon at least one of the Toll receptors present in macrophages following exposure to Ag NP. Further, our flow cytometric approach presents a potentially viable detection method for the identification of occult Ag NP material using an indicator cell line.
Keywords: silver nanoparticle, flow cytometry, innate immunity, macrophage, Toll-like receptor
Introduction
Nanotechnology has presented an astounding array of multidisciplinary applications in applied technology, medicine, and consumer products due to the unique physiochemical properties attributable to the size and formulation of nanoparticles (NPs). Because of their size, NPs exhibit unique cellular interactions at the subcellular level, and have provided wide-ranging effects that are dependent upon the route of administration, composition, size, and shape of the nanomaterial.1,2 While their potential or actual application presents strong advantages, there is little understanding regarding the physiology of NP absorption, and clarification must be made of the toxicological consequences of their containment within the cellular environment.
The recent increased use of silver (Ag) NP in a wide variety of consumer products, including food packaging,1 has been driven mainly by interest in its antimicrobial properties.3–5 Prior to the advent of nanotechnology, Ag was long recognized for its antimicrobial characteristics6 and has been used as an ingredient within consumer products, such as dental composite material7 or in wound salves, or integrated into wound dressings.8–10 Exposure of microbial organisms to Ag results in the retraction of the cytoplasm from the cell wall, condensation of the DNA into electron-dense granules, and an accumulation of Ag ions into the cytoplasm. The damage, as inferred in these studies, is due to the inability to replicate at the DNA level.11
Nanoparticles, including Ag NP, have been shown to be taken up by dendritic cells and macrophages, either derived from the peripheral lymph nodes or as cell lines,12–15 and recent experimental evidence has shown that the endocytosis of Ag NP is dependent upon scavenger receptors.16 Research using the J774A.1 mouse macrophage cell line has demonstrated that the endocytosis of 57 nm PEG-treated Ag NP is dependent upon actin and clathrin-mediated endocytosis.16 Potential toxicities associated with murine and human cellular exposure to Ag NP in vitro have been identified that is related to the ratio of Ag ion vs nanoparticulate Ag in the preparation,17 raising concerns regarding their potential for adverse health effects. Research has shown that a 24-hour exposure to Ag NP will induce elevated oxidative stress as demonstrated by ROS formation in Jurkat T cells.18 In this instance, signaling cascades involved in the regulation of oxidative stress have demonstrated increases in p38 MAP kinase protein, expression of the redox-sensitive nuclear factor-kappa B, and nuclear factor-E2–related factor 2. Cell cycle analysis demonstrated an increased number of cells within the G2/M and S phase populations, indicative of DNA accumulation, while Annexin V staining has shown increased apoptosis and necrosis in Ag NP-exposed cells. In other research, U937 human monocytic cells exposed to polyvinylpyrrolidone (PVP)-treated Ag NP demonstrated apoptotic cell death and ROS formation; however, the cellular effects were dependent upon the size of the particle, as these events were induced by the smaller 4 nm np, but not by the 40 or 70 nm NP.15 Other studies using primary human monocytes have similarly demonstrated a particle size-dependent effect. In this research, the smaller 5 and 28 nm Ag NP induced greater levels of the pro-inflammatory cytokine IL-1β and led to formation of regulatory nucleotide binding domain and leucine –rich repeat with pyrin domain-containing 3 (NLRP3) inflammasomes with the subsequent activation of caspase-1, two events that are critical for the cleavage of pro-IL-1β to produce mature IL-1β.19 Together, these observations suggest a significant immunologic impact by the Ag NP, as IL-1β is involved in both adaptive and innate immune responses.
Given the multiple observations attesting to NP’s effect upon the cells of the immune system, and the observations implicating macrophage in the uptake of NPs, we conducted a flow cytometric analysis of the effects of Ag NP uptake upon the macrophage using the murine RAW264.7 macrophage cell line. Relatively, few studies have addressed the changes in cell surface marker expression that occur when macrophages come into contact with a non-targeted Ag NP. Using a gating strategy that eliminated dead cells, and smaller-sized cellular debris and particulates, we used the gated viable cells to generate histograms for forward scatter (FSC), side scatter (SSC), and fluorescence. SSC signals are attributed to cellular internal structure and organelles, and are sensitive to changes within the internal structures.20 Thus, SSC signals were used in the present experiments to demonstrate the phagocytosis of citrated Ag NP. Furthermore, we sought to define the cellular subpopulations responsible for Ag NP uptake, and assess the possibility of determining biomarkers expressed during the phagocytosis of Ag NP. Our results implicate the more mature macrophage in the ingestion of Ag NP, detection of emitted wavelengths indicative of the presence of Ag NP within the cells, and increased expression of Toll-like receptor (TLR) in macrophages following exposure to Ag NP. We also demonstrate the amenability of our approach to being adapted for the detection of Ag NP in a food matrix, ie, milk.
Materials and methods
Cell lines
The murine monocyte/macrophage cell line, RAW264.7 (TIB71), was obtained from ATCC (Manassas, VA) and maintained as per the vendor’s recommendations. Briefly, cells were cultured in T75 culture flasks containing DMEM media supplemented with 10% (v/v) fetal calf serum, 1% (v/v) glutamine, and 1% (v/v) penicillin–streptomycin. Cultures were maintained at 37°C with 5% CO2 and used prior to passage 15 for all experiments. Confirmation of the cell line’s monocytic lineage was done by flow cytometric examination of the line’s cell surface phenotype using monoclonal antibodies of defined specificity. This cell line is not identified within the International Cell Line Authentication Committee (ICLAC) list of misidentified or cross-contaminated cell lines.21
Latex microbeads
Fluorescently labeled polystyrene latex YG microbeads (Fluoresbrite® by Polysciences, Inc., Warrington, PA) were obtained in sizes including 0.05, 0.1, 0.2, 0.5, and 1.0 μm. The particles are reported to have an excitation wavelength of 441 nm with emission at 486 nm. Unlabeled beads of equivalent sizes (Polybead® by Polysciences, Inc.) were used as controls in these experiments to assure that the detected emitted fluorescence was due to the presence of Ag NP, and not due to the phagocytosis of any other particulates. RAW264.7 cells were first incubated with fluorescent beads to ascertain that the phagocytic events could be detected by flow without extraneous stimulation.22
Silver nanoparticles
Biopure Ag NP (1 mg/mL) in citrate buffer were obtained from NanoComposix Inc. (San Diego, CA). Ag NP characterization was provided by the company. Three sizes of Ag NP were used in this research: 10, 50, and 100 nm. In the absence of any referenced data regarding the concentration of Ag in consumer products, arbitrary concentrations of 1, 5, and 10 μg/mL were used with cultures containing 5×105 cells.
Ag NP sizing by dynamic light scattering (DLS)
A Malvern Zetasizer Nano ZS DLS with a backscattering detector (173°) was used for measuring the hydrodynamic size of Ag NP at 25°C. Ag NP were pre-diluted 100-fold in either milli-Q water or DMEM cell-cultured medium prepared as above. A total of six measurements were performed and each measurement consisted of 12 subruns with 10-second duration each.
Ag NP characterization by transmission electron microscopy (TEM)
The size of Ag NPs was determined by using the Jeol JEM 1400 TEM operated at 80 kV. A 10 μL aliquot of colloidal Ag NP sample (10× diluted in Milli-Q water) was deposited on a clean piece of parafilm. A NanoPlus grid (Dune Sciences) was placed upside down onto the sample for 5 minutes. The grids were then rinsed with 10 μL Milli-Q water and excessive solution was blotted off with a piece of filter paper. The grids were dried at room temperature and subsequently imaged in TEM.
Inductively coupled plasma mass spectrometry (ICP-MS)
A Thermo X-Series II quadruple ICP-MS was used to determine the total concentration of Ag in the stock solution and the concentration of Ag ions after 24 hours of incubation with DMEM cell culture media. For ICP-MS analysis of Ag in the stock solution, samples were diluted with 2% HNO3. To compare the release of Ag ions before and after the incubation, samples were pre-diluted 100-fold in either milli-Q water or DMEM cell culture media (24 hours of incubation) and then centrifuged for 2 hours at 25,500 ×g (Thermo Fisher Scientific, Waltham, MA). The supernatant was collected and then further diluted with 2% HNO3 prior to ICP-MS analysis.
The ICP-MS was tuned using 1ppb Tune A solution (Thermo Fisher Scientific) to meet the required performance and NIST SRM 3151 Ag standard solution was used as a full-quant calibration standard. An internal standard solution (Perkin Elmer Inc., Waltham, MA, containing 10ppb Bi Ge In Li Sc Tb Y) was introduced along with samples through a T-connector to correct signal drift and matrix effects.
Phagocytosis of Ag NP
The 5×105 cells were plated in T25 flasks with 10 mL DMEM media containing 10% fetal calf serum and supplements. The cultures were grown to 60% confluence before treatment with the appropriate particle. Negative control cultures consisted of naive cells devoid of any particles, and separate cultures given unlabeled latex beads. Fluorescently labeled polystyrene latex beads of 50 nm size were used as a fluorescent control to detect and confirm the phagocytosis of NPs by the cells. Ag NP (10, 50, and 100 nm; NanoComposix.com) were added to each T25 flask to produce cultures of 1, 5, and 10 μg/mL. Following 24 hours of incubation, the growth media was aspirated and the cells were flushed with 1 mL cold media, scraped lightly, and spun at 300 ×g for 5 minutes. The pellet was suspended in 1 mL cold Hanks Balanced Salt Solution (HBSS) with 1% FBS and 0.1% sodium azide and the cells were counted using Trypan blue exclusion. The cells were washed once more and suspended in fresh media prior to flow analysis.
Phagocytosis of Ag NP diluted in milk
Briefly, 10, 50, or 100 nm Ag NP were diluted in 2% milk and added in a 100 μL volume to 9.9 mL of cell culture media to produce a final concentration of 10 μg/mL. Control cultures did not receive Ag NP. Latex beads were added to some cultures as a control against the induction of red fluorescence by the phagocytosis of particulate by the cells. The RAW264.7 cells were incubated for 24 hours and then assayed. For assay, the growth media was aspirated and the cells were flushed with 1 mL cold media, scraped lightly, and spun at 300 ×g for 5 minutes. The pellet was suspended in 1 mL cold HBSS with 1% FBS and 0.1% sodium azide and the cells were counted using Trypan blue exclusion. The cells were washed once more and suspended in fresh media prior to flow analysis. Emissions from the Ag NP were detected in the infrared region, ie, PE-Cy7.
Flow cytometry
All flow data were acquired using a BD FACS Aria II (BD Biosciences, San Jose, CA) equipped with a Violet 405, Blue 488, Red 633 nm and operated using the BD FACSDiva 6.1.1 software. The wavelengths detectable with the 405 nm laser standard filters include 430–470 nm (Pacific Blue, DAPI, or Hoechst). The wavelengths detected with the 488 nm standard filters included the ranges of 515–545 nm (FITC), 563–606 nm (phycoerythrin [PE]), 600–620 nm (PE-Texas-Red), 665–715 nm (PI, PerCP-Cy-5.5, PECy5.5), and 750–780 nm (PE-Cy7). The wavelengths detected from the 633 nm laser filters included 650–670 nm (Allophycocyanin [APC]) and 750–800 nm (APC-Cy7). The instrument’s day to day performance was checked according to manufacturer’s specifications, using Cytometer Setup and Tracking within the FACSDiva software (BD Biosciences). Briefly, a solution of multiple fluorochrome-labeled (Cytometer Setup and Tracking beads [CST]) beads (BD Biosciences) was diluted as per instructions and run on the cytometer. The settings were checked to ensure consistency in instrument performance.
Flow assay for the detection of phagocytized particles
Data were initially collected and gated using the BD DIVA 6.1.1 software. All viable, cultured cells were gated using a linear FSC vs log SSC, eliminating the smaller debris particles and dead cells from the histograms that were generated from within the gated region. A comparative quantitation of the level of phagocytosis by the RAW cells was determined by SSC and fluorescence intensity emitted by the phagocytized NP or labeled bead within the gated region. The log median fluorescence intensity of FITC within the gated populations was used to identify cells that had phagocytized FITC-labeled latex beads. Phagocytized Ag NP emission was detectable by filters in the red and far red spectra, with the strongest signals observed within the far red 750–800 nm (PE-Cy7) region. Thus, the log median SSC and PE-Cy7 fluorescence of the gated population was used to analyze Ag NP ingestion. Data analysis was done using DIVA 6.1.1 and FlowJo V10 software (Tree star, Inc.).
Cell surface staining and flow cytometry
Native RAW cells were used in the initial phenotyping. The following antibodies were obtained from BD Biosciences Pharmingen and used for staining: Phycoerythrin (PE)-labeled CD11b Mac1 Integrin (cat. #552850), PECy5-labeled rat anti-mouse B220 monoclonal (CD45R/B220 glycoprotein) (cat. #553085), F4/80-FITC (cat. #552958), rat anti-mouse CD32 FcR FITC (cat. #553144), and unlabeled anti-Fc receptor monoclonal (Mouse Fc Block; cat. #553141). Unstained cells that were treated with unlabeled FcR block were used as negative controls. The optimal concentration of each antibody used in the staining experiments was determined via titration prior to use. Staining was performed with 50 μL cell suspension containing 1×106 cells in staining buffer (HBSS containing 1% fetal calf serum and 0.1% sodium azide), blocked with unlabeled anti-Fc receptor monoclonal (Mouse Fc Block; cat. #553141), stained with the selected monoclonal at 4°C for 30 minutes. The samples were washed twice prior to addition of fixative (BD Cytofix). After fixation, the cells were washed once and resuspended in staining buffer.
Toll receptor staining and flow cytometry
FITC-rat anti mouse CD283 (clone 11F8, MCA5891F, Bio-Rad AbD Serotec) and FITC anti-CD284 were obtained from AbD Serotec and were used to stain RAW274.7 cells that were cultured in the presence or absence of added Ag NP. Intracellular staining for CD283 was performed23 using cell suspension containing 1×106 cells in staining buffer (HBSS containing 1% fetal calf serum and 0.1% sodium azide). The cells were blocked with unlabeled anti-Fc receptor monoclonal, and washed twice with BD Fix wash as per manufacturer’s instructions (BD Cytofix/Cytoperm kit; cat. #554714). Staining of the cells was done using the optimal CD283 FITC dilution as determined by titration, and incubated at 4°C for 30 minutes. The samples were washed twice with BD Fix wash and resuspended in staining buffer.
Statistical analyses
All data points were included in the analyses, and no outliers were excluded in calculations of means or statistical significance. All error bars are reported from the standard error of the means. Two-way ANOVA was used to determine the statistical significance differences between resultant means. For all comparisons, a P-value of <0.05 was considered statistically significant. Statistical differences between two populations was determined using Tukey’s Honest Significant Difference test Kaldeidagraph 4.1.1 statistical software (Synergy Software; Reading, PA), or Excel was used for calculations and graphs.
Results
Characterization of Ag NPs
The 10, 50, and 100 nm Ag NP used in this study were characterized by DLS, TEM, and ICP-MS. Their Z-average hydrodynamic size in water was determined by DLS and were found to be 27.5, 53.4, and 99.3 nm, respectively (Figure 1), consistent with the manufacturer’s-provided values. However, after 24 hours of incubation in DMEM cell culture media, the Z-average hydrodynamic size for all three Ag NP increased even though the value of viscosity had been adjusted in the software for size calculation. The average core size of the 10, 50, and 100 nm Ag NPs determined by TEM analysis were 11.1, 46.3, and 95.6 nm, respectively. The TEM images showed minimal aggregation or agglomeration of these Ag NP when diluted with water. However, the NPs exhibited variable degrees of agglomeration following 24 hours of incubation in DMEM culture media as shown in Figure 2.
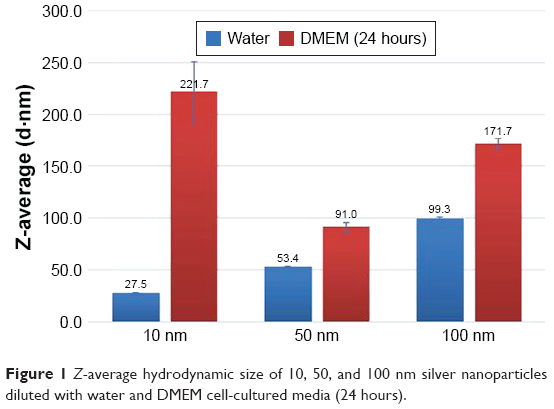 | Figure 1 Z-average hydrodynamic size of 10, 50, and 100 nm silver nanoparticles diluted with water and DMEM cell-cultured media (24 hours). |
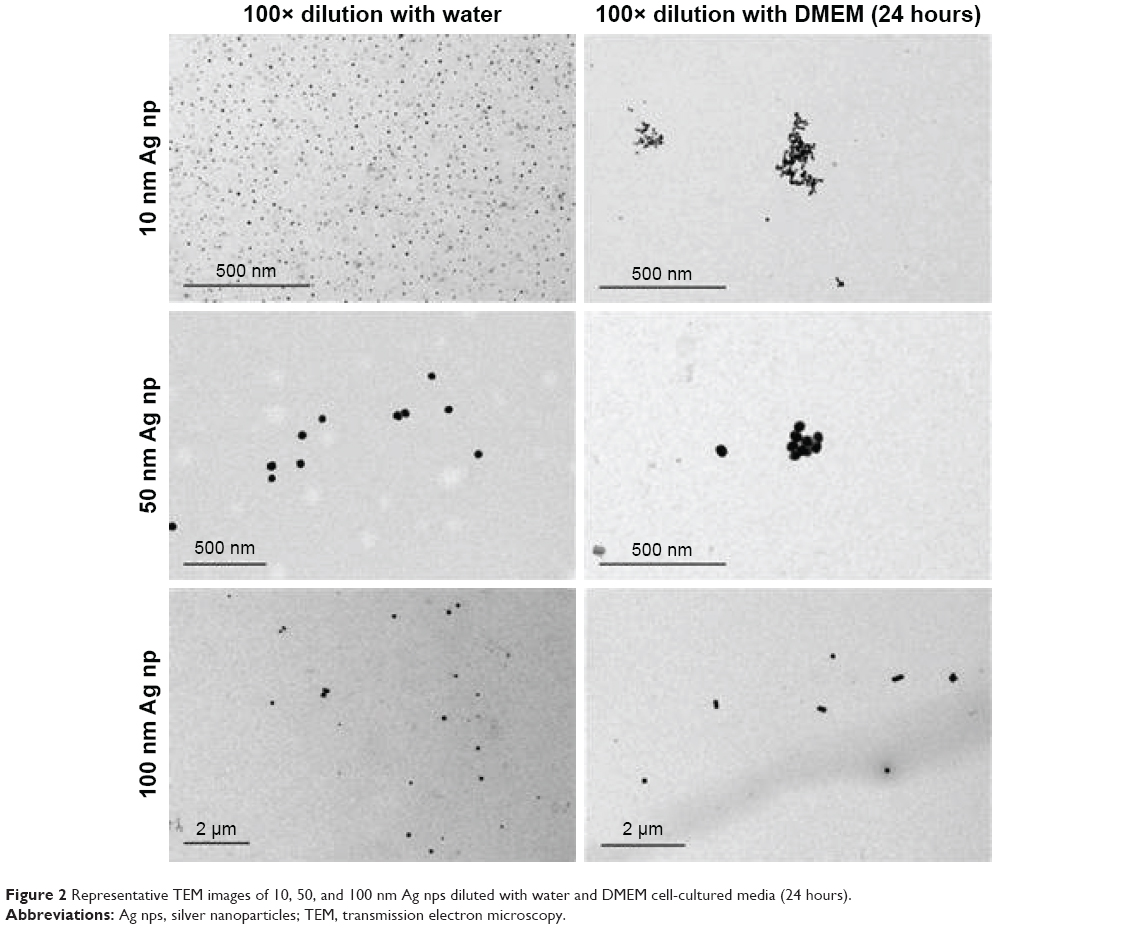 | Figure 2 Representative TEM images of 10, 50, and 100 nm Ag nps diluted with water and DMEM cell-cultured media (24 hours). |
The concentration of the 10, 50, and 100 nm Ag NP solution determined by ICP-MS analysis were 1.03±0.09, 1.03±0.02, and 1.05±0.04 mg/mL, respectively, compared with the manufacturer-provided concentration of 1.0 mg/mL. Ag ion content was determined before and after 24 hours of incubation of the NPs in DMEM media (Figure S1). The concentration of Ag ions after centrifugation increased except that for 10 nm. This is due to insufficient centrifugation force to allow 10 nm Ag NPs to precipitate. (Table S1)
Detection of Ag NP within RAW264.7
For these experiments, RAW264.7 cells were incubated with three different sizes of Ag-citrate NP, 10, 50, and 100 nm, at concentrations between 0 and 10 μg/mL for 24 hours. It has been reported that Ag NP presents some toxicity to the cells. The cultures generated in this study demonstrated a median averaged viability of ~50% or greater. For flow cytometric evaluation, we used a gating strategy that eliminated smaller-sized cellular debris dead cells, and particulates, and used the gated viable cells to obtain FSC, SSC, and emitted fluorescence data.
The cellular uptake of Ag-citrate NP by the RAW264.7 cell line was detected by flow cytometry, due to the observed alterations in SSC in cultures receiving Ag NP. As shown in the histograms depicted in Figure 3A, the SSC parameter increased with each increasing size of Ag-citrate NP. Data acquired by the cytometer software were transferred into FlowJo to obtain the histogram overlay depicted in Figure 3B. The histogram provides a clear depiction of the differences in SSC changes attributable to the size of the phagocytized NP. Interestingly, RAW cells did not demonstrate SSC changes when incubated with 50 nm latex particles, yet exhibited a notable SSC shift following coculture with the smaller 10 nm Ag NP. SSC changes were easily noted with all of the next larger-sized Ag NP. The observed SSC changes occurred reliably throughout all experiments described in this report.
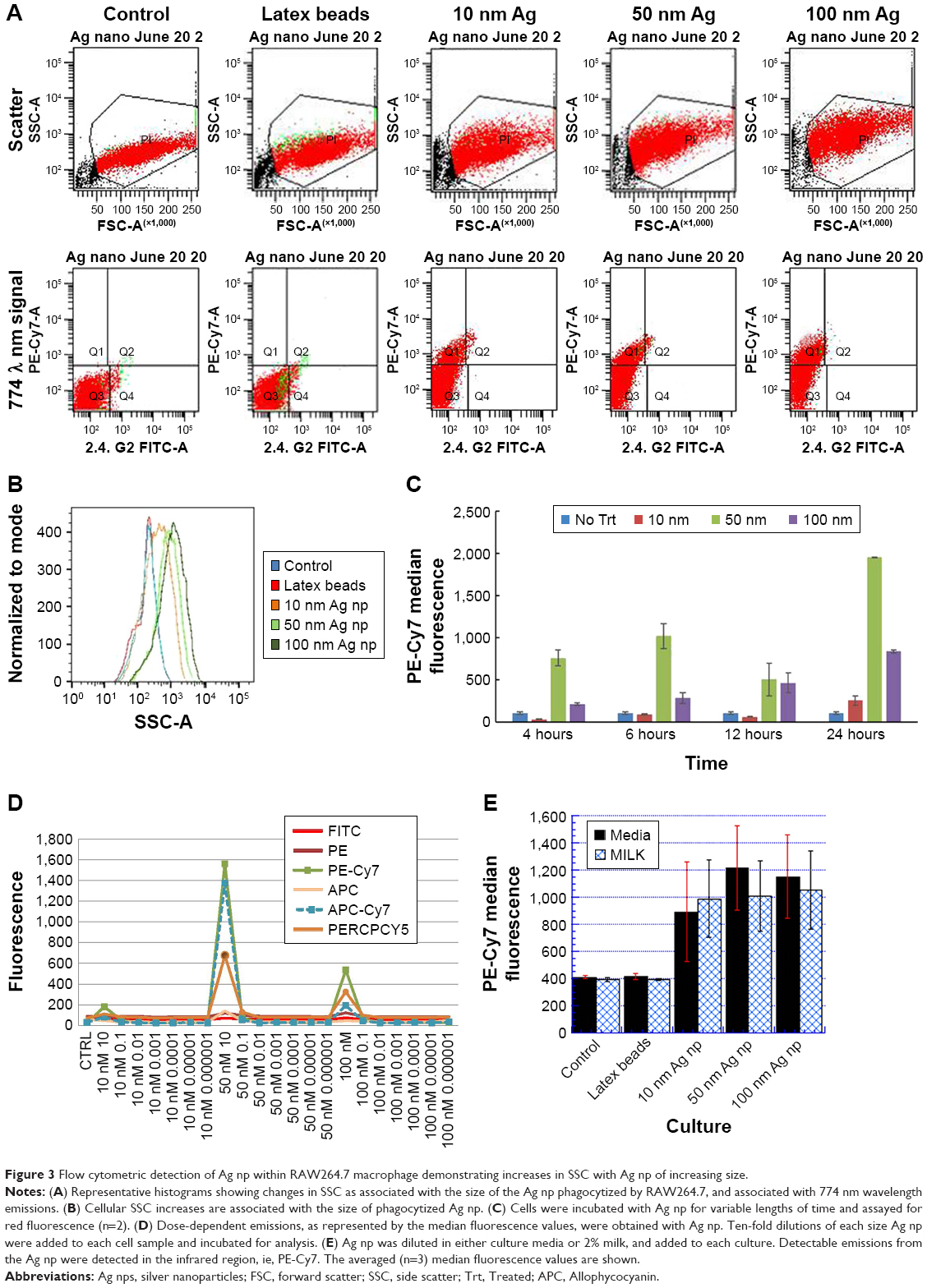 | Figure 3 Flow cytometric detection of Ag np within RAW264.7 macrophage demonstrating increases in SSC with Ag np of increasing size. |
Cultures that had been incubated with Ag NP for 24 hours and examined with laser wavelengths of 488 and 640 nm emitted detectable infrared and near-infrared signals that were used as an indication of the presence of Ag NP within the cell (Figure 3A). Cytometric analysis revealed two populations: cells that had ingested particles and demonstrated far-red fluorescence (quadrant Q1), and cells that did not appear to internalize NP that lacked observable far-red emissions (Q3). A timecourse study was performed to ascertain the cells’ activity with respect to the phagocytosis of Ag NP. The intake of Ag NP was evident as early as 4 hours of culture, and was more apparent at 24 hours for all three sizes of NP tested (Figure 3C). We, therefore, selected the 24-hour timepoint for the remainder of the project.
The averaged PE-Cy7 fluorescence data obtained from six representative experiments are shown in Table 1. Titered amounts of each of the three sizes of Ag NP were added to the cells, and the PE-Cy7 median fluorescence values were collected. The size of the Ag NP, its concentration (Table 2), and the interaction of the two factors had a statistically significant contribution (P<0.001) to the median PE-Cy7 fluorescence detected in the RAW macrophage cell line. Tukey’s test post ad hoc analysis showed that the RAW macrophage cells treated with 100 nm Ag NPs emitted statistically significant higher (P<0.05) PE-Cy7 fluorescence than the those treated with the 10 nm Ag NP. However, there were no statistically significant differences in the PE-Cy7 fluorescence emitted from RAW cells treated with 50 nm Ag NPs compared with cultures receiving 10 or 100 nm NPs. The Tukey’s test pairwise comparison also showed that the PE-Cy7 signal emitted by cells treated with Ag NP at 10 μg/mL was significantly higher than the flouresence obtained at 0, 1, or 5 μg/mL. In contrast, no statistically significant differences were observed in PE-Cy7 emissions from untreated (0 μg/mL) cells compared with those treated with 1 or 5 μg/mL of NP. Similarly, there were no significant differences in PE-Cy7 emission between RAW cells exposed to concentrations of 1 μg/mL compared with 5 μg/mL of Ag NP in culture media.
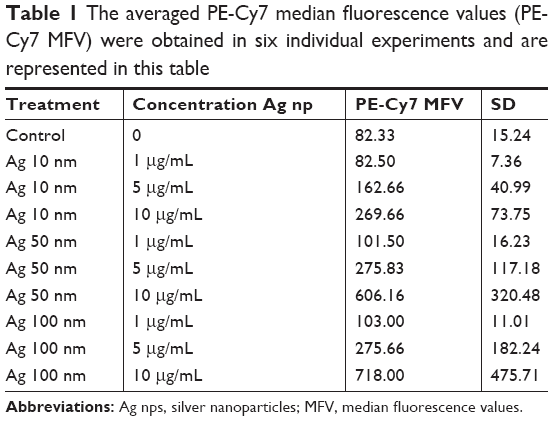 | Table 1 The averaged PE-Cy7 median fluorescence values (PE-Cy7 MFV) were obtained in six individual experiments and are represented in this table |
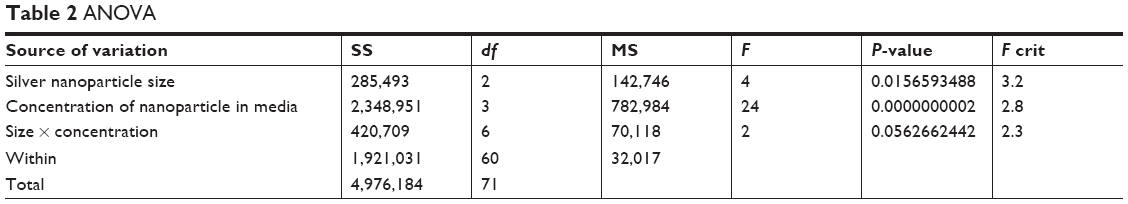 | Table 2 ANOVA |
Cells that had phagocytized Ag NP demonstrated a dose-dependent increase in the infra-red and far-red fluorescence intensity. While the lowest level of red emissions were routinely obtained with 10 nm Ag NP, greater red emissions were routinely obtained with both 50 and 100 nm Ag NP at the highest concentrations used in this study. However, the largest red emission was not always attributed to the largest 100 nm particle, but could also be detected following coculture with the highest concentration of 50 nm Ag NP. In contrast, SSC changes always occurred in accordance to the size of the particle and in a dose-dependent manner. The optimal emitted fluorescence intensity occurred at wavelengths between 690 and 774 nm for 50 and 100 nm Ag NP. The smaller 10 nm Ag NP was best detected in the infrared at 774 nm (Figure 3D).
Since NP can be used in a wide variety of consumer products and applications, we next examined whether our flow cytometric approach would detect Ag NP diluted within a food matrix such as milk. For this determination, the Ag NP were diluted in milk and added to the cultures. Cells were incubated with each of the test concentrations of Ag NP for 24 hours and analyzed. Our flow analysis detected the SSC changes observed in our previous experiments. While the assay also detected emitted far red fluorescence signals in a dose-dependent manner, there was an overall reduction with respect to signal strength (Figure 3E). Cultures receiving Ag NP spiked in milk demonstrated lower PE-Cy7 emissions as compared to cultures that received the citrated Ag NP in media. A greater variability was observed in the strength of the signal emission, but these reproducible observations showed that the detected signal followed the relationship with administered dose.
Flow cytometric examination of RAW cells following ingestion of Ag NP
Exposure of RAW264.7 cells to Ag NP resulted in the ingestion of the NP by a portion of the cellular populations as evidenced by our gating strategy. We next sought to address the observed functional differences within the subsets of the cell line by immunophenotyping. We selected FITC fluorochrome conjugated to monoclonals of defined specificity toward cell surface antigens, in order to minimize the overlap of emission spectra from the far-red (>660 nm) detection regions. In addition, two excitation sources (488 and 633 nm) were used.
The cell line demonstrated partial staining with monoclonal antibodies associated with macrophages including the transmembrane alpha M glycoprotein CD11b, a component of the Mac-1 integrin, and CD14, a glycophosphatidylinositol-linked glycoprotein known to be a receptor for LPS and is involved in TLR2-mediated macrophage activation,24 indicating that these populations were represented as subsets within the cell line. Staining was also done for the F4/80 glycoprotein, a member of the epidermal growth factor-seven transmembrane family of receptors, which are expressed on murine tissue macrophages and can be increased in inflammatory responses. The native RAW264.7 line expresses a predominant (70.47%±2.6233% SD, n=3), brightly staining F4/80-positive (F4/80+) population and a smaller F4/80-negative (F4/80−) subset of cells. Control RAW cells cultured in the presence of 50 nm latex NP retain this expression. Given the significant proportion of F4/80+ cells, we next asked whether Ag NP would be phagocytized by this subset. We next used RAW cultures that had received each of the three sizes of Ag NP. Our results demonstrate the characteristic increase in emitted red fluorescence associated with increasing amounts of Ag NP (PE-Cy7), and an association of this emitted red fluorescence with the expression of FITC-labeled F4/80 (Figure 4). The F4/80+ subset phagocytized Ag NP of all three sizes, in a dose-dependent manner, with the greatest intake observed at the highest dose of the 100 nm Ag NP. Interestingly, with each concentration of Ag NP, the F4/80+ subset was further subdivided into two cohorts; those that had phagocytized a lot of Ag NP and are detected as emitting greater levels of PE-Cy7 fluorescence vs those that did not ingest as much and are lower along the Y axis.
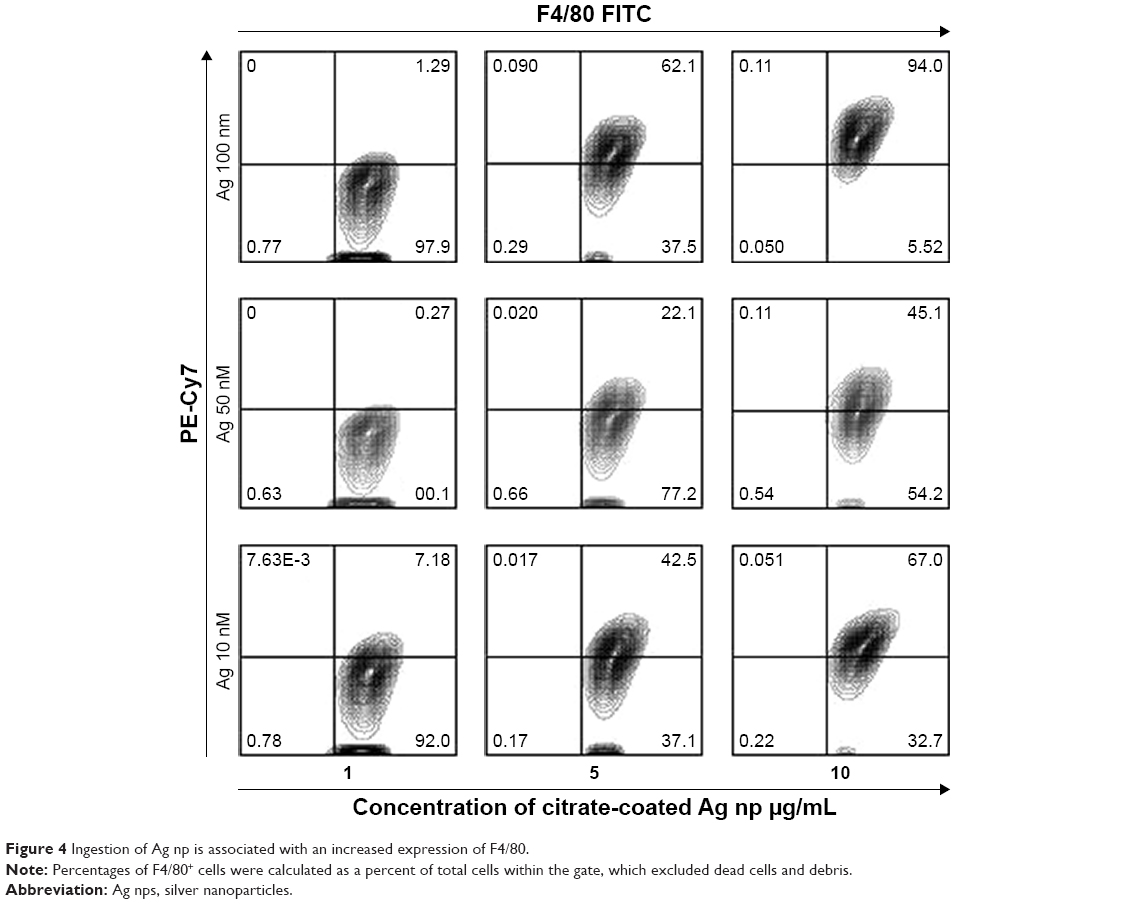 | Figure 4 Ingestion of Ag np is associated with an increased expression of F4/80. |
We next examined the expression of TLR of innate immunity relative to the presence of Ag NP. We used two methods for a comparison of the staining obtained using an anti-CD283 monoclonal. To this end, RAW cells were incubated with three distinctly sized Ag NP at increasing concentrations as previously described, collected, washed, and then stained using a cross-reactive monoclonal generated to a peptide sequence within human TLR3. Surface staining of RAW264.7 cells showed that cultures that had not received either latex beads or Ag NP demonstrated detectable levels of TLR3 (Figure 5A). A definite subset of TLR3+ cells that demonstrated the characteristic PE-Cy7 emission associated with Ag NP was also evident in these experiments. Interestingly, the staining levels of TLR3 demonstrated variations that were associated with the presence of Ag NP and the levels increased with increasing concentration of Ag NP. While the percentages of cells staining with the monoclonal showed variation, the greatest variability was seen with the 10 nm Ag NP, and in particular with the higher concentrations of Ag NP, which showed statistical significance when compared to cultures receiving no treatment or latex beads.
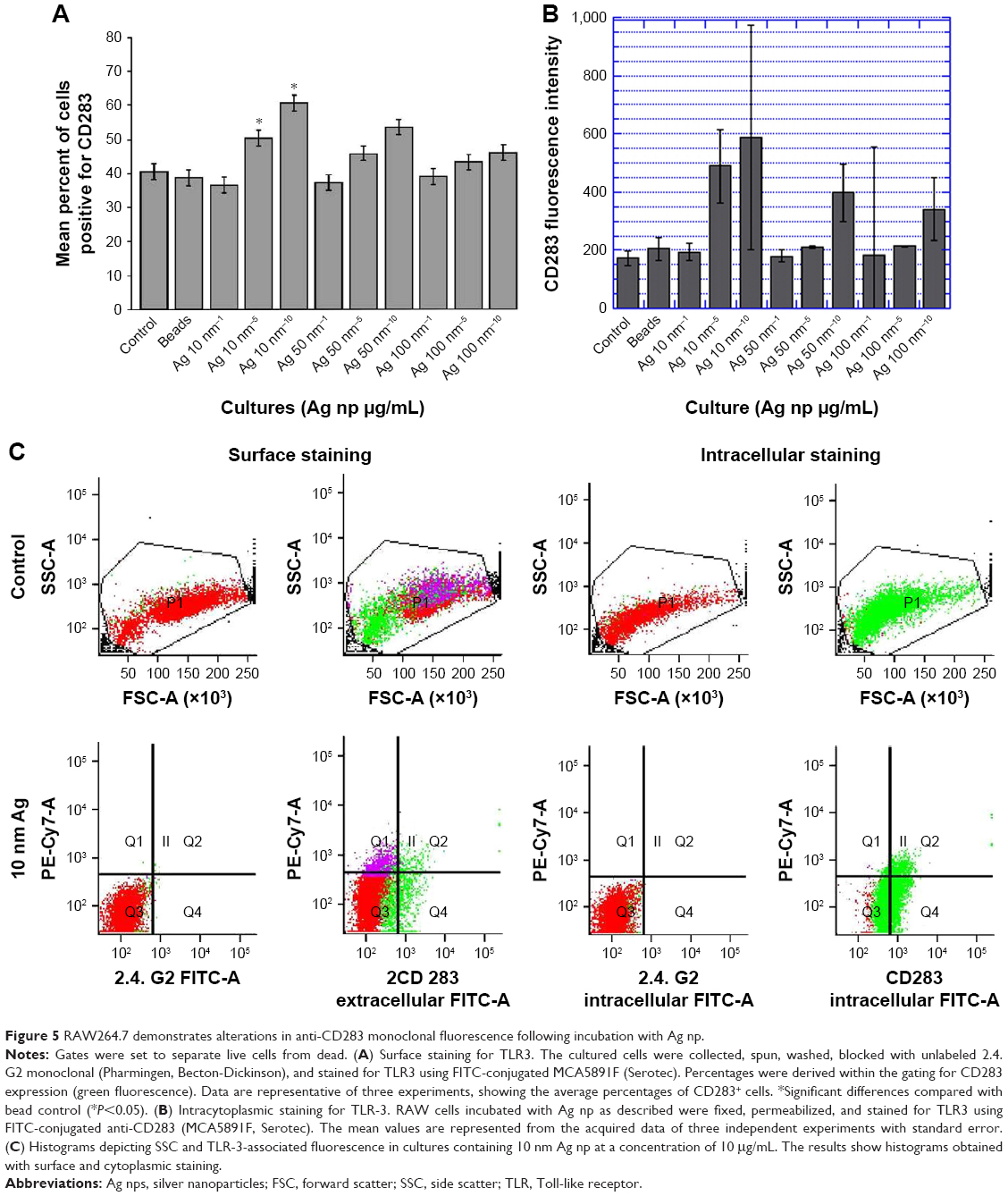 | Figure 5 RAW264.7 demonstrates alterations in anti-CD283 monoclonal fluorescence following incubation with Ag np. |
Our staining also assessed TLR3 by intracytoplasmic staining. To this end, cells that had been cultured with Ag NP were collected, fixed, and then stained with the monoclonal of interest. This method of staining showed a greater variability in the levels of staining obtained with the anti-TLR3 monoclonal (Figure 5B). However, the greatest variability was seen with the 10 nm Ag NP at the highest concentration. In both types of staining, Ag NP retained the characteristics observed in all previous experiments, by showing the smallest changes in SSC. However, there was evidence that the red emission characteristic of Ag NP coexisted with the FITC signal elicited by the labeled monoclonal (Figure 5C).
We next asked whether there would be other TLR that showed changes in expression as a result of incubation with Ag NP. Low, but measurable quantities of CD284 were detected in native RAW cells by flow (Figure 4). In the presence of Ag NP, our measurements demonstrated the existence of a weakly staining CD284+ population and a CD284− population within the culture. With the culture of either 10 nm Ag NP or 100 nm Ag NP, very little CD284 staining was found to coincide with the red fluorescence associated with the NP. A small subpopulation of CD284+ cells demonstrated red fluorescence. Thus, while there was some association between cells that stained for CD284 and had been cultured with Ag NP, there had been no increase in the level of stain obtained with the monoclonal. By contrast, there were no changes in the levels of this receptor following ingestion of Ag NP (Figure 6). Taken together, these results implicate the more mature macrophage in the ingestion of Ag NP, and an increase in TLR3 expression present on the surface of macrophages following exposure to Ag NP.
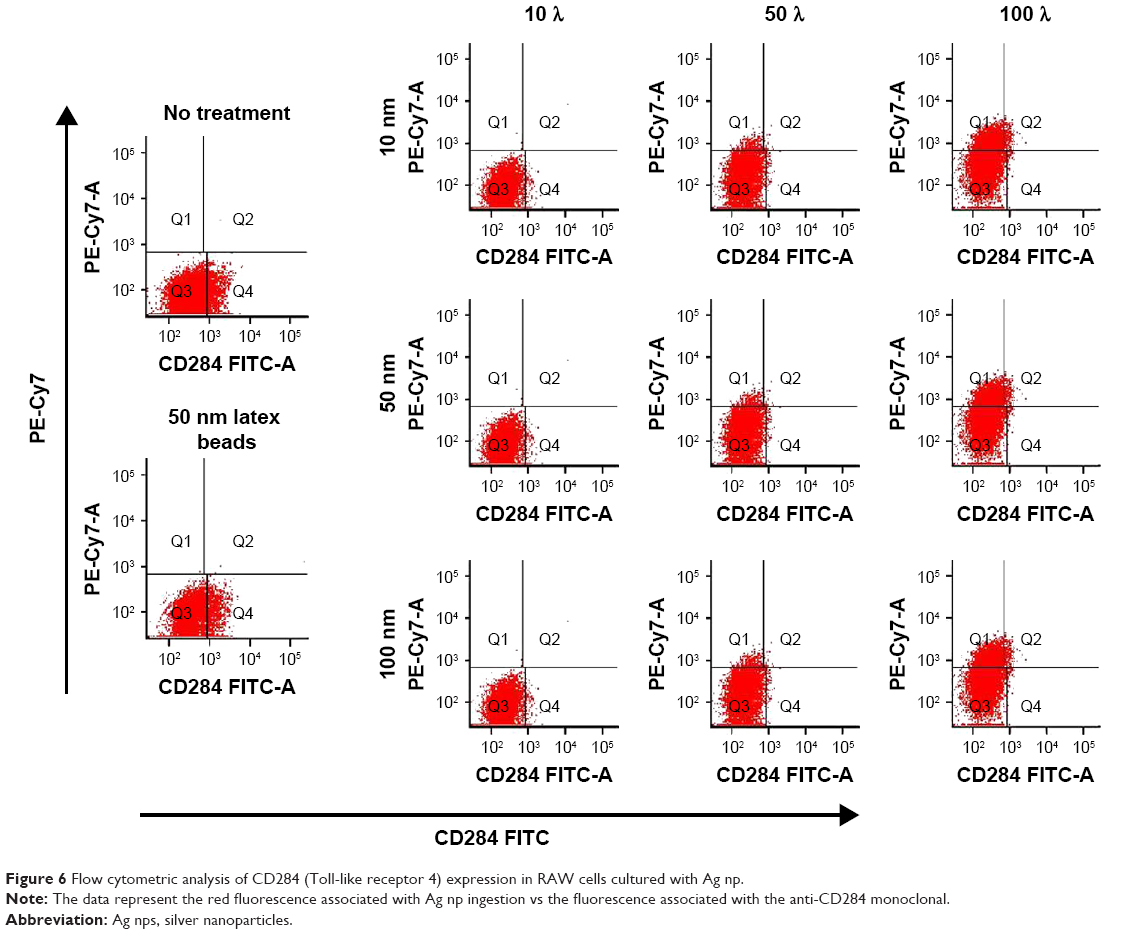 | Figure 6 Flow cytometric analysis of CD284 (Toll-like receptor 4) expression in RAW cells cultured with Ag np. |
Discussion
This report presents a flow cytometric assay for the detection of internalized Ag NP within the murine macrophage cell line RAW264.7. Specifically, the assay permits the detection of internalized citrated Ag NP using the combined flow cytometric parameters of SSC and red fluorescence. The changes in SSC occurred in a dose-dependent manner, and the emitted infrared and near-infrared signals were used as an indication of the presence of Ag NP within the cell.
The size and shape of the NP is known to affect the optical properties of NPs, and the absorption of UV-visible light energy is related both to the diameter of the particle and the oscillations inherent to its electron cloud. This is known as the surface plasmon resonance (SPR) or plasmon absorbance of NPs, and the optical absorption spectrum of metal NPs is dominated by the SPR. Absorption bands in the visible light region have been reported to be typical for Ag NP. Smitha et al reported an SPR at 454 nm for citrate colloids of Ag NP,25 while Zhao et al have reported that the plasmon absorption of Ag NP prepared by the citrate reduction method is 440 nm.26 The SPR or plasmon absorbance of NPs is also influenced by external factors such as solvents and pH. Thus, Smitha et al demonstrated that increasing the citrate concentration of a colloidal Ag solution resulted in the decrease of particle size, and a lowering of the SPR band from 454 to 423 nm. While both studies utilized particles in colloidal solutions, our study utilized particles that had been internalized by the cell line. In our study, the phagocytosed Ag NP were stimulated by laser wavelengths of either 488 and 640 nm wavelengths, resulting in detectable emissions that were in the infrared and the near-infrared regions. We propose that these emissions were a function of the plasmon resonance of the internalized NP. The results of our study are in agreement with other published reports demonstrating the flow cytometric detection of Ag NP, or other NPs, within other indicator cell lines by SSC27–29 and red fluorescence.30 In studies comparing a number of different cytometers, excitation of Ag NP was achieved using a red laser of 633 nm wavelength, a blue laser at 488 nm, and a 405 nm laser.30 The excitation by lasers of different wavelengths in our studies and others may represent an alteration of the Ag NP within the cell, resulting in ionic interactions that give rise to new combinations of Ag NP and cellular metabolic products, or Ag NP aggregates that are subject to excitation with the laser beams of varied wavelengths. Our TEM analysis showed that the citrated Ag NP used in these studies was able to form aggregates under the conditions used in these studies. Interestingly, it is the 10 nm NP that showed the greater tendency to agglomerate; the 100 nm NP did not show the same level of agglomeration. In contrast, the increased SSC size and the output of PE-Cy7 signal were associated with the larger Ag NPs, suggesting that there are other additional factors contributing to these observed changes. The formation of new Ag NP combinations with cellular products within the cells may be determinable by the metabolic activities of the cell. Such new combinations may be specific to the cellular population used in the study and influenced by the local environment of the cell. This further implies that a variance in conditions would alter our ability to detect Ag NP within the cells, and that reported detection values may vary depending on the culture conditions being used. Indeed, this was the case when we tested the ability of this technique to detect Ag NP that had been diluted in 2% milk prior to phagocytosis.
An interesting observation to arise from these studies was the change in TLR expression following ingestion of Ag NP by the cells. Ag NP have been shown to enter cells via receptor-mediated endocytosis, specifically scavenger-type receptors.16 The intracellular localization of TLR ligands has been correlated with their ability to induce cellular activation through the TLRs and their associated signaling pathways,31 and we propose that compartmentalization of the Ag NP in the present study supported downstream cellular events that resulted in the observed increase of TLR3. TLR3 is recognized to be a transmembrane receptor with an N-terminal extracellular domain (ECD), a transmembrane helix, and a C-terminal cytoplasmic signaling domain of the Toll/IL-1 receptor family. The extracellular domain is located within endosomes where it can encounter and form dimeric complexes with 45 bp segments of ds RNA, a viral replication intermediate, in a sequence-independent manner. Whether the Ag NP acts as a direct molecular trigger for the upregulation of TLR3 expression is not known, but the conditions used in our study supports the suggestion that the internalization of Ag NP by the cells during culture with Ag NP is prerequisite, and may point to a role of endosomal localization with respect to the triggering of a signaling pathway that supports the increase of TLR3 expression. The increased effect was limited to the expression of TLR3, and not exhibited by other cellular biomarkers such as F4/80 and TLR4. Other studies have demonstrated a relationship between the introduced Ag NP and a biological effect mediated by a TLR. In a comparison of two cell types, tongue squamous cell carcinoma cells SCC-9 and PDL (periodontal ligament) cells, SCC-9 cells were observed to be less sensitive to the effects of Ag NP than PDL cells using an MTT assay for viability. The difference in sensitivity was correlated to a difference in the level of TLR2 as measured by RT-PCR; SCC-9 cells contained less TLR2 than the PDL cells.32 Furthermore, a reduction of TLR2 expression by the addition of TLR2-specific Si-RNA or a TLR-2-directed antibody, resulted in a reduced Ag NP–induced cytotoxicity. The apoptosis of Ag NP–treated cells was dependent upon the phosphorylation of c-Jun, which was shown to be increased by Ag NP addition to the cells. Thus, Ag NP-mediated toxicity, in this instance, is attributable to the TLR2 pathway.
While our observations raise the question as to how does the increase in TLR3 come about, it also presents the additional question of what are the selective consequences of this increased expression. The interaction of these receptors with their ligand will result in the signaling cascades involving proteins identified as TRIF, TRAM, or MyD88 that can interact with the Toll-IL-1 receptor. Innate immunity, through the evolutionarily conserved, germline-encoded TLRs, depends upon the recognition of a broad range of pathogen-associated molecular patterns, or PAMPs that are found on a wide array of pathogens. The TLR detect conserved microbial products such as bacterial lipopolysaccharides lipoproteins and flagella, and double- and single-stranded RNA.33–35 The interactions will result in a series of downstream signaling cascades, ultimately resulting in the production of proinflammatory cytokines that engage host immune defenses.36–38
Our results implicate the mature macrophage in the ingestion of Ag NP and an influence upon the TLR3 Toll receptor present on the surface of macrophages following exposure to Ag. These results point a possible effect on toll receptor–mediated innate response pathways that are critical for the activation of regulatory T cells and their function, possibly leading to further consequences in the host’s immune status. The assay presented in this report provides a valuable approach to assess the relationship between NP ingestion, compartmentalization, and cellular downstream events. The identification of foreign particulate matter in tissues is central to discussions pertaining to exposure. This assay also presents an approach for the development of a flow method by which Ag NP could be identified in either pathological or toxicological examination of tissues exposed to Ag NP.
Conclusion
We have developed a method for the detection of Ag NP in cells. The size-dependent uptake of Ag NP by the RAW264.7 cell line was measurable using both SSC and red fluorescence intensity. The optimal emitted fluorescence intensity occurred at wavelengths between 690 and 774 nm for 50 and 100 nm Ag NP. Smaller 10 nm Ag NP was best detected in the infrared at 774 nm. There is a linear correlation with increased fluorescence intensity associated with the ingestion of increased number of NP. Our results implicate the more mature F4/80+ macrophage in the ingestion of Ag NP and an influence upon the subpopulation of macrophages expressing TLR3 following exposure to Ag NP.
Acknowledgments
We gratefully thank Dr Kristina Williams for use of the flow cytometer used in these studies. We recognize and thank Drs Yong Wu, Jiwen Zheng and Ms Lynn Chen from the FDA Advanced Characterization Facility (ACF) for the characterization of the nanoparticles used in this study.
Disclosure
The findings and views presented in this article are those of the authors and do not reflect the views or policies of the US Food and Drug Administration. The authors report no conflicts of interest in this work.
References
Principato M. Gastrointestinal immunoregulation and the challenges of nanotechnology in foods. In: Muzzalupo I, editor. Food Industry. Croatia: InTech; 2013:491–517. | ||
Dunkle A, Blanchette C, Boone T, et al. Co-delivery of adjuvant and subunit antigens via a nanoparticle platform induces tissue-associated and systemic adaptive immune responses (P4409). J Immunol. 2013;190(Meeting Abstracts 1):205–214. | ||
Shameli K, Ahmad MB, Zargar M, Yunus WM, Rustaiyan A, Ibrahim NA. Synthesis of silver nanoparticles in montmorillonite and their antibacterial behavior. Int J Nanomedicine. 2011;6:581–590. | ||
Pal S, Tak YK, Song JM. Does the antibacterial activity of silver nanoparticles depend on the shape of the nanoparticle? A study of the Gram-negative bacterium Escherichia coli. Appl Environ Microbiol. 2007;73(6):1712–1720. | ||
Anh-Tuan L, Thi Tam L, Van Quy N, et al. Powerful colloidal silver nanoparticles for the prevention of gastrointestinal bacterial infections. Adv Nat Sci Nanosci Nanotechnol. 2012;3(4):045007. | ||
Russell AD, Hugo WB. Antimicrobial activity and action of silver. Prog Med Chem. 1994;31:351–370. | ||
Brune D. Metal release from dental biomaterials. Biomaterials. 1986;7(3):163–175. | ||
Parsons D, Bowler PG, Myles V, Jones S. Silver antimicrobial dressings in wound management: comparison of antibacterial, physical, and chemical characteristics. Wound. 2005;7(8):222–232. | ||
Illingworth B, Bianco RW, Weisberg S. In vivo efficacy of silver coated fabric against fungal burn wound pathogens. 2000;27:344–350. | ||
Klasen HJ. Historical review of the use of silver in the treatment of burns. I. Early uses. Burns. 2000;26(2):117–130. | ||
Feng QL, J W, Chen GQ, Cui FZ, Kim TN, Kim OJ. A mechanistic study of the antibacterial effect of silver ions in Escherichia coli and Staphylococcus aureus. Appl Environ Microbiol. 2008;74:2171–2178. | ||
Sadauskas E, Wallin H, Stoltenberg M, et al. Kupffer cells are central in the removal of nanoparticles from the organism. Part Fibre Toxicol. 2007;4(10):10. | ||
Illum L, Jacobsen LO, Müller RH, Mak E, Davis SS. Surface characteristics and the interaction of colloidal particles with mouse peritoneal macrophages. Biomaterials. 1987;8(2):113–117. | ||
Loeschner K, Hadrup N, Qvortrup K, et al. Distribution of silver in rats following 28 days of repeated oral exposure to silver nanoparticles or silver acetate. Part Fibre Toxicol. 2011;8:18. | ||
Park J, Lim DH, Lim HJ, et al. Size dependent macrophage responses and toxicological effects of Ag nanoparticles. Chem Commun. 2011;47(15):4382–4384. | ||
Wang H, Wu L, Reinhard BM, Brm R. Scavenger receptor mediated endocytosis of silver nanoparticles into J774A.1 macrophages is heterogeneous. ACS Nano. 2012;6(8):7122–7132. | ||
Hadrup N, Lam HR. Oral toxicity of silver ions, silver nanoparticles and colloidal silver – a review. Regul Toxicol Pharmacol. 2014;68(1):1–7. | ||
Eom HJ, Choi J. p38 MAPK activation, DNA damage, cell cycle arrest and apoptosis as mechanisms of toxicity of silver nanoparticles in Jurkat T cells. Environ Sci Technol. 2010;44(21):8337–8342. | ||
Yang EJ, Kim S, Kim JS, Choi IH. Inflammasome formation and IL-1β release by human blood monocytes in response to silver nanoparticles. Biomaterials. 2012;33(28):6858–6867. | ||
Kerker M, Chew H, Mcnulty PJ, et al. Light scattering and fluorescence by small particles having internal structure. J Histochem Cytochem. 1979;27(1):250–263. | ||
Masters JR. Cell-line authentication: end the scandal of false cell lines. Nature. 2012;492(7428):186. | ||
Steinberg BE, Scott CC, Grinstein S. High-throughput assays of phagocytosis, phagosome maturation, and bacterial invasion. Am J Physiol Cell Physiol. 2007;292(2):C945–C952. | ||
Jelinek I, Leonard JN, Price GE, et al. TLR3-specific double-stranded RNA oligonucleotide adjuvants induce dendritic cell cross-presentation, CTL responses, and antiviral protection. J Immunol. 2011;186(4):2422–2429. | ||
da Silva TA, Zorzetto-Fernandes ALV, Cecílio NT, Sardinha-Silva A, Fernandes FF, Roque-Barreira MC. CD14 is critical for TLR2-mediated M1 macrophage activation triggered by N-glycan recognition. Sci Rep. 2017;7(1):7083. | ||
Smitha SL, Nissamudeen KM, Philip D, Gopchandran KG. Studies on surface plasmon resonance and photoluminescence of silver nanoparticles. Spectrochim Acta A Mol Biomol Spectrosc. 2008;71(1):186–190. | ||
Zhao Y, Jiang Y, Fang Y. Spectroscopy property of Ag nanoparticles. Spectrochim Acta A Mol Biomol Spectrosc. 2006;65(5):1003–1006. | ||
Suzuki H, Toyooka T, Ibuki Y. Simple and easy method to evaluate uptake potential of nanoparticles in mammalian cells using a flow cytometric light scatter analysis. Environ Sci Technol. 2007;41(8):3018–3024. | ||
Toduka Y, Toyooka T, Ibuki Y. Flow cytometric evaluation of nanoparticles using side-scattered light and reactive oxygen species-mediated fluorescence-correlation with genotoxicity. Environ Sci Technol. 2012;46(14):7629–7636. | ||
Zucker RM, Massaro EJ, Sanders KM, Degn LL, Boyes WK. Detection of TiO2 nanoparticles in cells by flow cytometry. Cytometry A. 2010;77(7):677–685. | ||
Zucker RM, Daniel KM, Massaro EJ, Karafas SJ, Degn LL, Boyes WK. Detection of silver nanoparticles in cells by flow cytometry using light scatter and far-red fluorescence. Cytometry Part A. 2013;83AA:962–972. | ||
Guiducci C, Ott G, Chan JH, et al. Properties regulating the nature of the plasmacytoid dendritic cell response to Toll-like receptor 9 activation. J Exp Med. 2006;203(8):1999–2008. | ||
Kim AS, Chae CH, Kim J, Choi JY, Kim SG, Băciut G. Silver nanoparticles induce apoptosis through the Toll-like receptor 2 pathway. Oral Surg Oral Med Oral Pathol Oral Radiol. 2012;113(6):789–798. | ||
Takeuchi O, Hoshino K, Kawai T, et al. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. 1999;11(4):443–451. | ||
Medzhitov R, Janeway C. Innate immune recognition: mechanisms and pathways. Immunol Rev. 2000;173(1):89–97. | ||
Kawai T, Akira S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int Immunol. 2009;21(4):317–337. | ||
Yamamoto M, Sato S, Hemmi H, et al. Role of adaptor TRIF in the MyD88-independent Toll-like receptor signaling pathway. Science. 2003;301(5633):640–643. | ||
Yamamoto M, Sato S, Mori K, et al. Cutting edge: a novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-beta promoter in the Toll-like receptor signaling. J Immunol. 2002;169(12):6668–6672. | ||
Sato S, Sugiyama M, Yamamoto M, et al. Toll/IL-1 receptor domain-containing adaptor inducing IFN-beta (TRIF) associates with TNF receptor-associated factor 6 and TANK-binding kinase 1, and activates two distinct transcription factors, NF-kappa B and IFN-regulatory factor-3, in the Toll-like receptor signaling. J Immunol. 2003;171(8):4304–4310. |
Supplementary materials
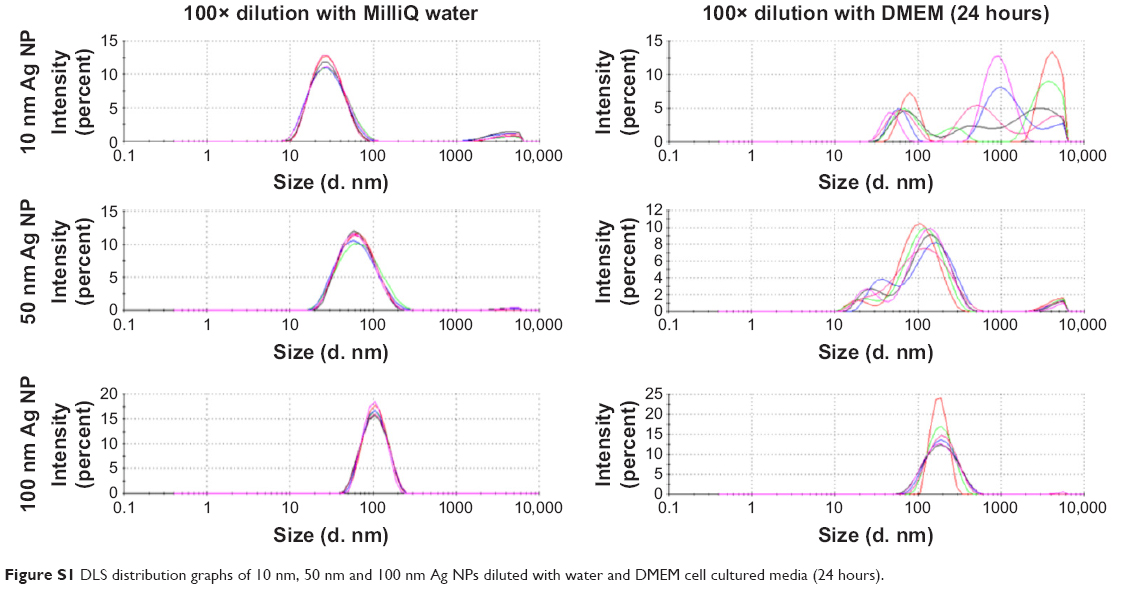 | Figure S1 DLS distribution graphs of 10 nm, 50 nm and 100 nm Ag NPs diluted with water and DMEM cell cultured media (24 hours). |
 | Table S1 ICP-MS analysis of supernatants of 10 nm, 50 nm and 100 nm Ag NPs diluted to 10 μg/mL in water and DMEM cell cultured media (24 hours) showing the concentration of Ag ions increased upon incubation with DMEM cell media except that for 10 nm. The higher concentration of Ag ions was detected in the water supernatant because the centrifugal force used, 25,500 g, was not high enough to completely precipitate 10 nm Ag NPs |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.