")
Back to Journals » International Journal of Nanomedicine » Volume 13
A facile doxorubicin-dichloroacetate conjugate nanomedicine with high drug loading for safe drug delivery
Authors Yang CL, Wu TT, Qin YT, Qi Y, Sun Y, Kong M, Jiang X, Qin XY, Shen YQ , Zhang ZP
Received 17 October 2017
Accepted for publication 18 January 2018
Published 6 March 2018 Volume 2018:13 Pages 1281—1293
DOI https://doi.org/10.2147/IJN.S154361
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lei Yang
Conglian Yang,1 Tingting Wu,1 Yuting Qin,1 Yan Qi,1 Yu Sun,1 Miao Kong,1 Xue Jiang,1 Xianya Qin,1 Yaqi Shen,2 Zhiping Zhang1,3,4
1Tongji School of Pharmacy, 2Department of Radiology, Tongji Hospital, Tongji Medical College, 3National Engineering Research Center for Nanomedicine, 4Hubei Engineering Research Center for Novel Drug Delivery System, Huazhong University of Science and Technology, Wuhan, People’s Republic of China
Background: Doxorubicin (DOX) is an effective chemotherapeutic agent but severe side effects limit its clinical application. Nanoformulations can reduce the toxicity while still have various limitations, such as complexity, low drug loading capability and excipient related concerns.
Methods: An amphiphilic conjugate, doxorubicin-dichloroacetate, was synthesized and the corresponding nanoparticles were prepared. The in vitro cytotoxicity and intracellular uptake, in vivo imaging, antitumor effects and systemic toxicities of nanoparticles were carried out to evaluate the therapeutic efficiency of tumor.
Results: Doxorubicin-dichloroacetate conjugate can self-assemble into nanoparticles with small amount of DSPE-PEG2000, leading to high drug loading (71.8%, w/w) and diminished excipient associated concerns. The nanoparticles exhibited invisible systemic toxicity and high maximum tolerated dose of 75 mg DOX equiv./kg, which was 15-fold higher than that of free DOX. It also showed good tumor targeting capability and enhanced antitumor efficacy in murine melanoma model.
Conclusion: This work provides a promising strategy to simplify the drug preparation process, increase drug loading content, reduce systemic toxicity as well as enhance antitumor efficiency.
Keywords: self-assembly, doxorubicin, drug delivery, chemotherapy, nanomedicine
Introduction
Doxorubicin (DOX) is an effective chemotherapeutic drug for many cancers, such as breast cancer, bladder cancer, Kaposi’s sarcoma, lymphoma and acute lymphocytic leukemia.1 However, its clinical application is limited by the acute and chronic toxicities, such as cardiotoxicity, hepatotoxicity, vomiting and nausea, phlebosclerosis and myelosuppression.2 The dose-dependent and cumulative cardiotoxicity, including cardiomyopathy and congestive heart failure, is grave for the life-threatening characteristic and prevalent for the organs’ greater sensitivity to free radicals induced by DOX.3,4 Another common damage is hepatotoxicity, which was found in 40% patients during the treatment. The serious side effects of DOX lead to a quite narrow therapeutic window and limited clinical application. To diminish or even abolish the DOX-induced organic dysfunctions, numerous work has been done to develop nanodrug delivery systems, such as micelles, nanoparticles (NPs), hydrogels, liposomes and so on.5 Nanotechnology has gained profound benefit of reducing side effects and improving the therapeutic efficacy for chemotherapeutics.6 MacKay et al7 developed polypeptide-DOX NPs with 4-fold higher maximum tolerated dose (MTD) and impressive therapeutic efficacy against tumor. Some other DOX-loaded NPs were also constructed to realize reduced adverse effect and enhanced therapeutic efficiency.8–12 These nanomedicines demonstrated diverse advantages, such as enhanced drug stability and intracellular uptake, prolonged circulation time, decreased systemic toxicity and improved antitumor efficacy, by virtue of the multifunctional materials as well as the enhanced permeability and retention (EPR) effect. However, the content of excipient in most nanomedicines can reach as high as 90%, resulting in low drug loading content (DLC) (usually less than 10%, w/w).13 Such a large amount of excipient can bring extra burden to patients, including high cost, biodegradability concern, systemic toxicity and metabolism/excretion problem.14–16 Despite the fact that thousands of nanomedicines have been developed, only a few of them can be translated into the clinic. It could be attributed to the complicated formulation design and construction, high cost and excipient content and potential side effects of nanoformulations.17–19 As the most successful nanoformulations, Doxil® and Abraxane® are prosperous owing to their simplicity.20 Therefore, it is highly desired to develop a simple nanomedicine with facile operability, high DLC, low side effect and high therapeutic efficacy.
In the past few years, self-assembled nanomedicine by exploiting the unique π–π stacking and hydrophobic interactions has attracted attentions from researchers.21–24 It was prepared through a simple nanoprecipitation method by the use of amphiphilic drug, which was synthesized by conjugating hydrophobic drug with small molecule or hydrophilic drug. This kind of nanomedicine has exhibited many advantages.25 First, the simple preparation procedure of nanomedicine makes it easy to translate into large-scale manufacture. Second, the self-assembly ability can greatly decrease the usage of excipient, resulting in high DLC and diminished excipient associated concerns. Furthermore, it also shows good stability under different conditions. DOX, a hydrophobic drug with rich aromatic structure, can facilitate weak intermolecular interactions, including π–π stacking and hydrophobic interactions, which is the fundamental for self-assembled nanomedicine. However, DOX cannot self-assemble in water due to the strong lipophilicity. We herein try to conjugate DOX with a hydrophilic drug to promote the capability of self-assembly. Dichloroacetate (DCA) is a small hydrophilic drug that is used to treat congenital lactic acidosis in children.26 It can also reverse the Warburg effect by inhibiting the activity of pyruvate dehydrogenase kinase in many cancer cells.27 However, the dosage of DCA used to realize tumor inhibition is as high as 100 mg/kg,28 which is much higher than that of DOX (5 mg/kg). Herein, we constructed an amphiphilic DOX-DCA conjugate via the reaction of amino and carboxyl group in DOX and DCA, respectively. DOX-DCA can self-assemble into NPs with small amount of PEGylated lipid DSPE-PEG2000. This kind of NPs exhibited many advantages: 1) facile fabrication; 2) high DLC; 3) reduced excipients related concerns; 4) low adverse effect and 5) enhanced therapeutic response. These findings could give insights in promoting large-scale manufacture and clinical translation of nanomedicines.
Material and methods
Materials
Doxorubicin hydrochloride (DOX·HCl) was purchased from Beijing Huafeng United Technology Co. (Beijing, People’s Republic of China). DCA was obtained from Aladdin (Shanghai, People’s Republic of China). Dichloroacetic acid anhydride was purchased from J&K Scientific, Ltd (Beijing, People’s Republic of China). 1,1′-Dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine iodide (DIR) was obtained from AAT Bioquest Inc. (Sunnyvale, CA, USA). 3-(4, 5-dimethyl-thiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) and 4′,6-diamidino-2-phenylindole (DAPI) were purchased from BioSharp (Seoul, South Korea). Radioimmunoprecipitation assay buffer and bicinchoninic acid (BCA) protein quantitation kit were purchased from Beyotime Institute of Biotechnology (Haimen, People’s Republic of China). N,N-dimethylformamide (DMF) was treated with CaH2 (Aladdin) before use. Other regents and solvents were used directly without further treatment.
Murine hepatic carcinoma cells H22 and murine melanoma cells B16F10 were purchased from Chinese Academy of Sciences Cells Bank (Shanghai, People’s Republic of China). B16F10 were cultured in DMEM at 37°C in a humidified atmosphere of 5% CO2. The medium contained 10% FBS, 100 U/mL penicillin and streptomycin. C57/BL6 and Kunming mice were purchased from the Experiment Animal Center of Wuhan University (Wuhan, People’s Republic of China). All the animal experiments were performed in the Animal Center of Huazhong University of Science and Technology in accordance with National Institutes of Health Animal Care guidelines. The animals were fed in sterile atmosphere with enough food and water. The in vivo studies were approved by the Ethical Committee of Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology Institutional.
1H-NMR was performed on a Bruker BioSpin GmbH 400 MHz spectrometer. The chemical shift was referenced relative to the residual solvent peaks. Electrospray ionization mass spectrum (ESI-MS) was carried out on an API 4000 QTrap® triple quadrupole mass spectrometer (AB SCIEX, Foster City, CA, USA). Fourier transform infrared spectroscopy (FTIR) was performed on a Bruker VERTEX 70 spectrophotometer (Ettlingen, Germany). The HPLC spectrogram was recorded on UltiMate 3000 Thermo Fisher Scientific machine (Waltham, MA, USA) with fluorescence detector and Restek Viva C18 column (150 × 4.6 mm, 5 μm) at a flow rate of 1 mL/min. The injection volume was 10 μL, and the temperature of column oven was set at 30°C. The identification and purity measurement of DOX and DOX-DCA were accomplished using a gradient elution of 25%–60% solvent B for 10 min where solvent A was water with 0.1% trifluoroacetic acid and solvent B was acetonitrile.
Synthesis of DOX-DCA
To synthesize DOX-DCA, DOX·HCl (50 mg, 0.086 mmol) and triethylamine (17.5 μL, 0.129 mmol) were mixed and stirred in anhydrous DMF (5 mL) at room temperature for 3 h. Then dichloroacetic acid anhydride (19.7 μL, 0.129 mmol) was added. After stirring for 24 h, DMF was removed by rotary evaporation under vacuum condition to yield a red residue. Afterward, dichloromethane was added and unreacted DOX was removed by the repetitive washing with water (3 × 10 mL). The final product was isolated and obtained using the silica gel column chromatography (dichloromethane:methanol, 20:1). The whole experimental process was performed in dark under normal atmospheric conditions. Yield: 33.2 mg (59%). 1H-NMR (ppm, 400 MHz, DMSO-d6): δ 1.14 (d, 3H), 1.49 (dd, 1H), 1.86 (td, 1H), 2.14 (m, 2H), 2.93 (m, 2H), 3.4 (s, 1H), 3.97 (s, 3H), 4.2 (q, 1H), 4.58 (s, 2H), 4.87 (s, 1H), 4.92 (m, 1H), 5.05 (d, 1H), 5.25 (d, 1H), 5.45 (s, 1H), 6.50 (s, 1H), 7.63 (m, 1H), 7.88 (m, 2H), 8.34 (d, 1H), 13.25 (s, 1H), 14.02 (s, 1H). ESI-MS (negative mode) C29H29Cl2NO12: [M-H]−, cald, (m/z) 652.1; found, 652.3. Purity (HPLC): 98.5%.
Preparation and characterization of DOX-DCA nanoparticles (DOX-DCA NPs)
DOX-DCA NPs were prepared via a simple nanoprecipitation method. Briefly, 1 mg DOX-DCA and 0.3 mg DSPE-PEG2000 (w/w, 1/0.3) were mixed together in 50 μL of dimethyl sulfoxide (DMSO) and added dropwise into 1 mL of distilled water with magnetic stirring at 100 rpm for 5 min. Thereafter, the solution was dialyzed against water to remove DMSO (MWCO 1 kDa). For animal administration, the NPs were concentrated with centrifugal filter (Ultracel YM-50, MWCO 50kDa, Millipore, Ireland). DOX NPs were constructed by the same nanoprecipitation method. In all, 1 mg desalinated DOX and 0.3 mg DSPE-PEG2000 (w/w, 1/0.3) were mixed for further NP preparation. For the preparation of DIR-labeled NPs, DIR and DOX-DCA were mixed with a ratio of 1:20 (w/w) and performed by the similar method.
The morphology of DOX-DCA NPs was observed by transmission electron microscopy (TEM, JEM-1230; JEOL, Tokyo, Japan) via negative staining method. The particle size, distribution and zeta potential were measured by dynamic light scattering (DLS; Zeta Plus, Brookhaven, USA). The stability of DOX-DCA NPs was monitored for 7 days by DLS in phosphate-buffered saline (PBS) at room temperature. The drug encapsulation efficiency (EE) and DLC were calculated according to the following formulas: EE (%) = drug mass in NPs/drug feeding amount × 100%; DLC (%) = drug mass in NPs/total mass of NPs × 100%.
Intracellular uptake and in vitro cellular pharmacokinetics
Intracellular uptake was carried out on B16F10 melanoma cells using confocal microscopy (710META, Zeiss, Oberkochen, Germany). B16F10 cells were harvested and seeded into 24-well plates (1 × 105 per well), which were pretiled with a round glass. After overnight incubation, the medium was changed into DOX, free DOX-DCA or DOX-DCA NPs with an equivalent (equiv.) concentration of DOX (3 μg/mL). After 4 h and 24 h, B16F10 cells were washed with precooling PBS (1 mL) for three times and then fixed with 4% paraformaldehyde at room temperature for 15 min. Followed by another repeated washing with PBS, 5 μg/mL DAPI was added and the cells were incubated at room temperature for 8 min. Finally, the glass was turned over on a glass slide for confocal microscopy imaging. For in vitro cellular pharmacokinetics, B16F10 cells were seeded into 6-well plates (2 × 105 per well) and incubated with DOX-DCA NPs for 4 h, 8 h, 12 h and 24 h, respectively. The cells were lysed with lysis buffer containing phenylmethanesulfonyl fluoride. DOX and DOX-DCA concentrations were determined by HPLC and the relative cellular protein contents were measured by a BCA assay kit.
Evaluation of cell viability by MTT
The in vitro antitumor activity of DOX-DCA NPs was measured using the MTT test. Briefly, B16F10 cells were harvested and seeded into 96-well plates (5 × 103 per well). After overnight incubation, the medium was replaced with different concentrations of DOX, free DOX-DCA or DOX-DCA NPs, respectively. After 24 h, 10 μL of MTT (5 mg/mL) was added in each well followed by 4 h of incubation. The supernatant was replaced by 150 μL of DMSO. The absorbance at 490 nm was measured by a microplate reader (Multiskan MK3, Thermo Fisher Scientific).
In vivo imaging and distribution study
The in vivo distribution of DOX-DCA NPs was investigated on B16F10 tumor-bearing mice by noninvasive optical imaging system (Pearl Trilogy, LI-COR Biosciences, Lincoln, NE, USA). The mice were administrated free DIR or DIR-labeled DOX-DCA NPs via tail vein at the dose of 15 μg DIR per mouse. The real-time fluorescence images of DIR or DIR-labeled DOX-DCA NP-treated mice were recorded at 4 h, 8 h, 12 h, 24 h and 48 h postadministration. The main organs (heart, liver, spleen, lung and kidney) and tumors were harvested and their fluorescence intensities were recorded at 8 h, 24 h and 48 h. The fluorescence intensities of each organ were calculated for semi-quantitative biodistribution analysis (n = 3).
In vivo antitumor effect
The in vivo therapeutic effect was detected on two tumor models, H22 sarcoma and B16F10 melanoma. In total, 5 × 106 of H22 cells were inoculated subcutaneously at the right flank of Kunming mice (20–22 g). Tumor volume was calculated by the use of following equation: Tumor volume = length × width2/2. The mice were randomly divided into five groups: PBS, DOX, DOX + DCA, DOX-DCA and DOX-DCA NPs. These mice were injected every 2 days for four times via the tail vein at an equiv. dosage of 5 mg/kg DOX. Tumor volume and body weight were monitored every day. For B16F10 melanoma model, 200 μL of 5 × 104 B16F10 cells were subcutaneously injected at the right flank of C57/BL6 mice (16–18 g). When the tumor size was about 30–50 mm3, the mice were randomly divided into four groups. PBS, DOX 5 mg/kg, 5 mg DOX-DCA NPs equiv./kg or 15 mg DOX-DCA NPs equiv./kg was intravenously administrated every 2 days for three times. The tumor size and body weight change were recorded every other day. After 11 days, all mice were scarified. And all tumors were harvested for imaging and weighting.
MTD and systemic toxicity study of DOX-DCA NPs
The systemic toxicity was carried out by determining MTD in tumor-free C57/BL6 mice. The mice were intravenously administrated a single injection with PBS, 5, 10 or 15 mg/kg DOX, 25, 50 or 75 mg DOX-DCA NPs equiv./kg on day 0, respectively. The body weight, survival conditions and physical states of each mouse were monitored every day for the following 10 days. MTD was defined as the maximum dosage of DOX or DOX-DCA NPs that lead to less than 15% body weight loss and no other obvious toxicities during 10 days.8 At day 10, all mice were scarified. The serum and main organs were harvested for further toxicity studies. The activities of lactate dehydrogenase (LDH), aspartate transaminase (AST), alanine transaminase (ALT), blood urea nitrogen (BUN) and creatinine were assayed as indicators of cardiac, hepatic and renal functions. Pathological studies were carried on the H&E staining analysis of major organs.
Statistical analysis
The statistical analyses were conducted using Graphpad Prism 7.00 software. The significance level among two groups was calculated using two-tailed unpaired t-test. The significance level among multiple groups was identified by one-way ANOVA with Tukey post hoc test. p-value < 0.05 was considered to show a significant difference.
Results and discussion
Synthesis and characterization of DOX-DCA
The synthesis route of DOX-DCA is shown in Figure 1A. Briefly, DOX-DCA was synthesized by directly conjugating DOX with dichloroacetic acid anhydride. The chemical structure of DOX-DCA was characterized by 1H-NMR, HPLC, ESI-MS and FTIR (Figure 1B–D). As shown in Figure 1B, the successful synthesis of DOX-DCA was verified by the generation of amide bond (a) and the signal from DCA (b), which were demonstrated by the proton signal at 8.2–8.4 ppm and 6.4–6.6 ppm, respectively. All the proton signal of DOX was remained in the spectrum of DOX-DCA with little chemical shift. Besides, the proton signal at 5.7–5.8 ppm came from the trace amount of dichloromethane. HPLC chromatograms exhibited that DOX-DCA showed absolutely different retention time (9.4 min) compared to DOX (5.4 min) (Figure 1C), suggesting the successful synthesis of DOX-DCA. As identified by HPLC analysis, the purity of DOX-DCA was 98.5%. Moreover, the m/z of DOX-DCA was 652.3 Da in negative mode, which was in accordance with the calculated value of 652.1 Da (Figure 1D). In the FTIR spectrum of DOX-DCA, the peak at 1,687 cm−1 further confirmed the generation of amide bound (Figure 1E). All results demonstrated the successful synthesis of DOX-DCA with high purity.
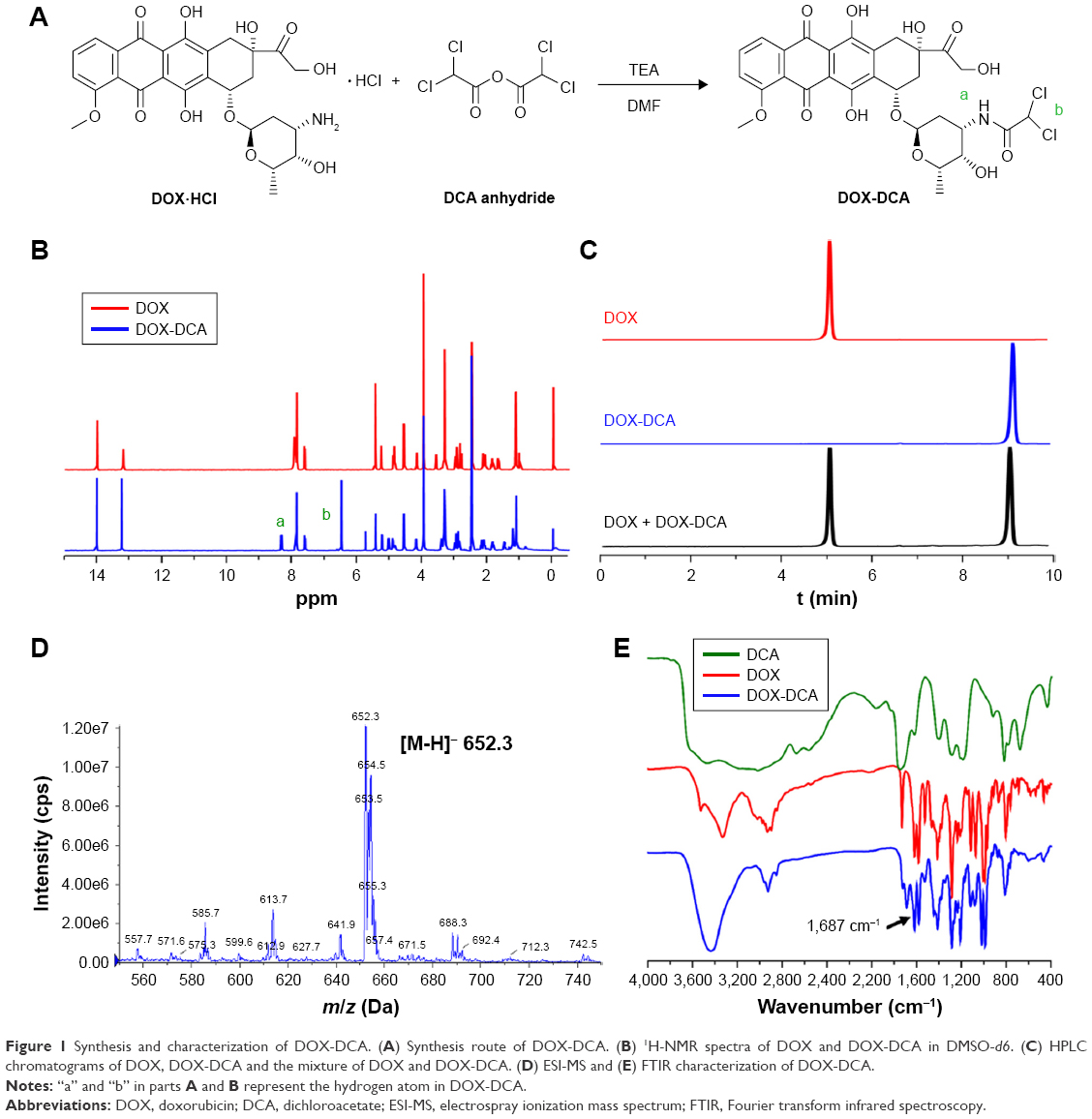 | Figure 1 Synthesis and characterization of DOX-DCA. (A) Synthesis route of DOX-DCA. (B) 1H-NMR spectra of DOX and DOX-DCA in DMSO-d6. (C) HPLC chromatograms of DOX, DOX-DCA and the mixture of DOX and DOX-DCA. (D) ESI-MS and (E) FTIR characterization of DOX-DCA. |
Preparation and characterization of DOX-DCA NPs
DOX-DCA NPs were prepared via a facile nanoprecipitation method. Interestingly, DOX-DCA can spontaneously assemble into stable and uniform NPs with small amount of DSPE-PEG2000. In a sharp contrast, when the same method was performed using free DOX, large amount of precipitate appeared. This phenomenon indicated that DCA played an important role in the process of NP formation. It may be attributed to the amphiphilic structure of DOX-DCA, which promoted the assembly of DOX in NPs. Moreover, the introduced PEGylated lipid in the formulation would facilitate the stability and in vivo circulation of NPs but had minor influence on the DLC. As shown in Figure 2A and B, the NPs possessed near-spherical morphology with an average particle size of approximate 56 nm, which was close to the hydrodynamic size (55.8 nm). DLS analysis indicated that the surface charge of NPs was −28.6 mV. The appropriate size and negative surface charge of NP make it suitable for long blood circulation and tumor accumulation via EPR effect.6 Furthermore, the NPs were homogeneous with a narrow distribution, which was evidenced from the low polydispersity index (0.198). Moreover, the preparation method of DOX-DCA NPs is simple with good reproducibility, suggesting the possibility of large-scale manufacture. The drug concentration can be as high as 15.3 mg/mL after the NPs were concentrated. More importantly, DOX-DCA NPs possessed a quite high EE (76.5% ± 6.5%) and DLC (71.8% ± 1.7%), whereas the NPs constructed with DOX demonstrated a relatively low EE (6.4% ± 1.1%) as well as DLC (17.6% ± 2.4%) (Figure 2C). The self-assembly ability of DOX-DCA and small amount of DSPE-PEG2000 resulted in the high drug/carrier ratio and DLC, which showed superiority against most of conventional nanoformulations (usually less than 10%).13 High DLC is essential for low excipient associated toxicity and biodegradability concerns as well as high therapeutic response. What is more, the NPs showed very high stability in PBS with small average size change over a period of 7 days (Figure 2D). TEM analysis was also used to testify the stability. Figure 2E represents the TEM images of DOX-DCA NPs after 7 days at room temperature, 24 h at 37°C and 24 h in acetate buffer (pH 5.0). DOX-DCA NPs demonstrated no significant difference in morphology and good stability against time, temperature and acidic condition. Compared with conventional nanomedicines, DOX-DCA NPs would attract much attention for the simplicity, reproducibility, high DLC, reduced excipient-related toxicity and good stability.
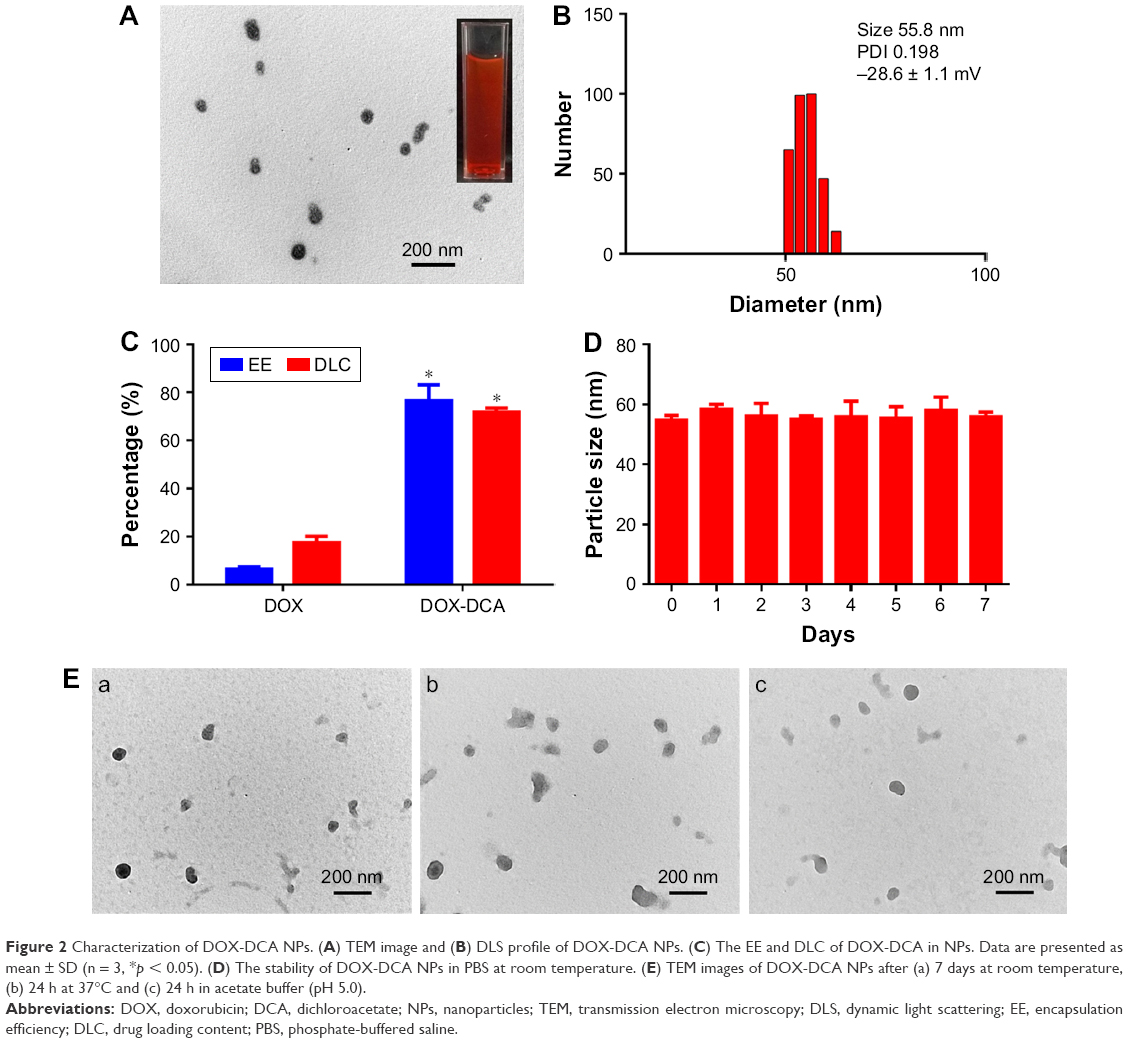 | Figure 2 Characterization of DOX-DCA NPs. (A) TEM image and (B) DLS profile of DOX-DCA NPs. (C) The EE and DLC of DOX-DCA in NPs. Data are presented as mean ± SD (n = 3, *p < 0.05). (D) The stability of DOX-DCA NPs in PBS at room temperature. (E) TEM images of DOX-DCA NPs after (a) 7 days at room temperature, (b) 24 h at 37°C and (c) 24 h in acetate buffer (pH 5.0). |
In vitro cellular uptake and cytotoxicity assay
The cellular uptake and localization of free DOX, DOX-DCA and DOX-DCA NPs were investigated in B16F10 tumor cells at 4 h and 24 h using confocal microscopy. As seen from Figure 3A, the red fluorescence of DOX and blue fluorescence of DAPI overlapped well in the group of free DOX after 4 h of incubation, which was attributed to the high affinity of DOX with nucleic acids. On the contrary, most of free DOX-DCA localized into the cell cytoplasm at 4 h and subsequently diffused into cell nucleus at 24 h. DOX-DCA NPs exhibited similar and enhanced intracellular uptake compared with free DOX-DCA both at 4 h and 24 h. After DOX-DCA NPs were internalized into cells by the endocytosis pathway and escaped to the cytoplasm, DOX-DCA was released and showed gradual accumulation in nucleus to exert the cell cytotoxicity effect. Similar results can also be found in the internalization and intracellular drug delivery of other DOX-loaded NPs.7,16
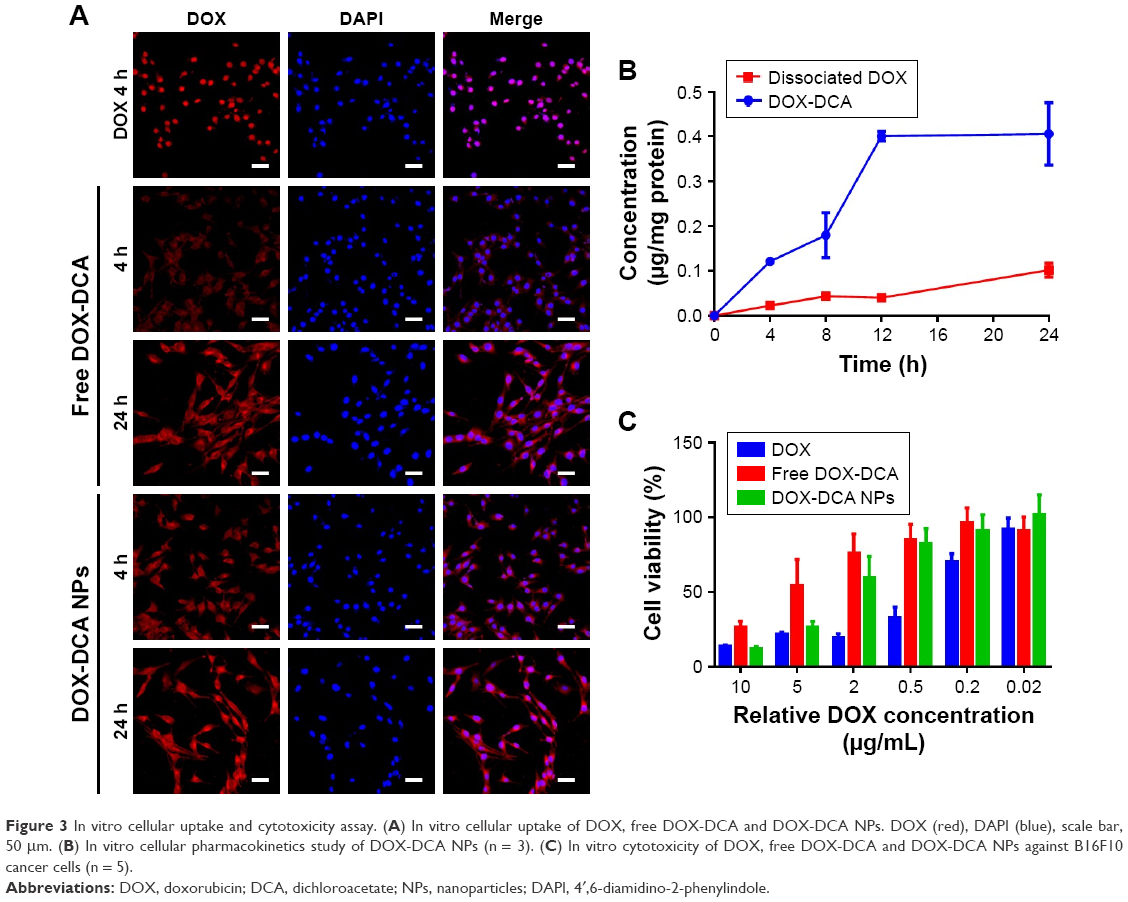 | Figure 3 In vitro cellular uptake and cytotoxicity assay. (A) In vitro cellular uptake of DOX, free DOX-DCA and DOX-DCA NPs. DOX (red), DAPI (blue), scale bar, 50 μm. (B) In vitro cellular pharmacokinetics study of DOX-DCA NPs (n = 3). (C) In vitro cytotoxicity of DOX, free DOX-DCA and DOX-DCA NPs against B16F10 cancer cells (n = 5). |
In order to investigate the fate of DOX-DCA NPs after uptake, the cellular pharmacokinetics study was performed. As shown in Figure 3B, the concentration of DOX-DCA and dissociated DOX increased with time going on. DOX can be released from DOX-DCA after cell uptake under acidic and esterase conditions. We further examined the in vitro antitumor effect of DOX, free DOX-DCA and DOX-DCA NPs by MTT assay. Free DOX showed the best antitumor efficiency against cancer cells, followed by DOX-DCA NPs and then free DOX-DCA (Figure 3C). The results were consistent with the tendency of intracellular uptake characteristics.
In vivo imaging and biodistribution study
To study the in vivo tumor targeting effect and biodistribution of NPs, B16F10 tumor-bearing mice were intravenously injected with free DIR or DIR-labeled DOX-DCA NPs (DIR@DOX-DCA NPs). As shown in Figure 4A, free DIR was quickly eliminated from body at 4 h and invisible DIR signal was found in tumor. On the contrary, strong and durable signal in tumor was found in DIR@DOX-DCA NP-treated mice up to 48 h. At 8 h, the fluorescence signal reached the peak. The prolonged circulation may be attributed to small particle size, PEGylation and negative charge of DOX-DCA NPs. It seemed that DOX-DCA NPs showed significant tumor targeting and retention capabilities.
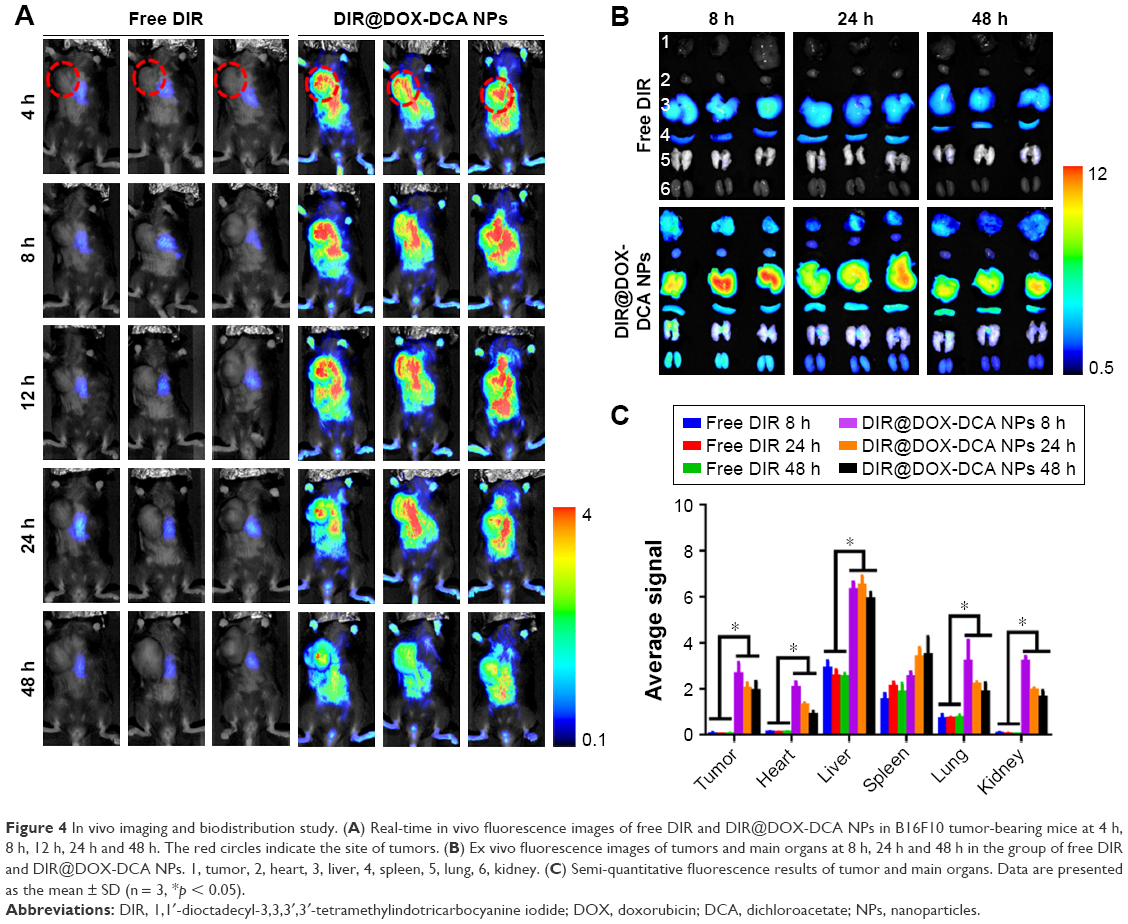 | Figure 4 In vivo imaging and biodistribution study. (A) Real-time in vivo fluorescence images of free DIR and DIR@DOX-DCA NPs in B16F10 tumor-bearing mice at 4 h, 8 h, 12 h, 24 h and 48 h. The red circles indicate the site of tumors. (B) Ex vivo fluorescence images of tumors and main organs at 8 h, 24 h and 48 h in the group of free DIR and DIR@DOX-DCA NPs. 1, tumor, 2, heart, 3, liver, 4, spleen, 5, lung, 6, kidney. (C) Semi-quantitative fluorescence results of tumor and main organs. Data are presented as the mean ± SD (n = 3, *p < 0.05). |
We further investigated ex vivo biodistribution of free DIR and DIR@DOX-DCA NPs after harvesting the main organs and tumor. DIR@DOX-DCA NPs showed absolute advantage against free DIR in the battle of tumor targeting, which was evidenced by the fluorescence image (Figure 4B) and semi-quantitative results (Figure 4C). Similar results can also be found in the investigation of DIR-labeled PTX-S-S-OA/TPGS2k NPs.15 The tumor fluorescence signal of DIR@DOX-DCA NPs was 47.2-, 48.2- and 39.4-fold compared with that of free DIR at 8 h, 24 h and 48 h, respectively. More importantly, the fluorescence in excised tumor can last as long as 48 h. The higher drug targeting and retention abilities can result in better therapeutic response. These findings suggested that DOX-DCA NPs showed good system circulation, tumor targeting and retention abilities.
In vivo antitumor efficiency
We explored the antitumor efficiency of DOX-DCA NPs on two tumor models, H22 sarcoma and B16F10 melanoma. Figure 5A represents the tumor volume growth with the treatment of PBS, DOX, DOX + DCA, free DOX-DCA and DOX-DCA NPs, respectively. The treatment with the admixture of free DOX and DCA (DOX + DCA), free DOX-DCA or DOX-DCA NPs showed comparable tumor inhibition capability in comparison to DOX (Figure 5A–C). The tumor inhibitory rate of DOX, DOX + DCA, free DOX-DCA and DOX-DCA NPs was 63.2%, 51.5%, 62.4% and 51.0%, respectively. Notably, compared with the stably increased body weight in DOX-DCA NPs, there was obvious body weight loss in mice receiving DOX or DOX + DCA (Figure 5D). It may be resulted from the systemic toxicity of free DOX. It seemed that the conjugation of DOX with DCA can reduce the systemic toxicity of free DOX.
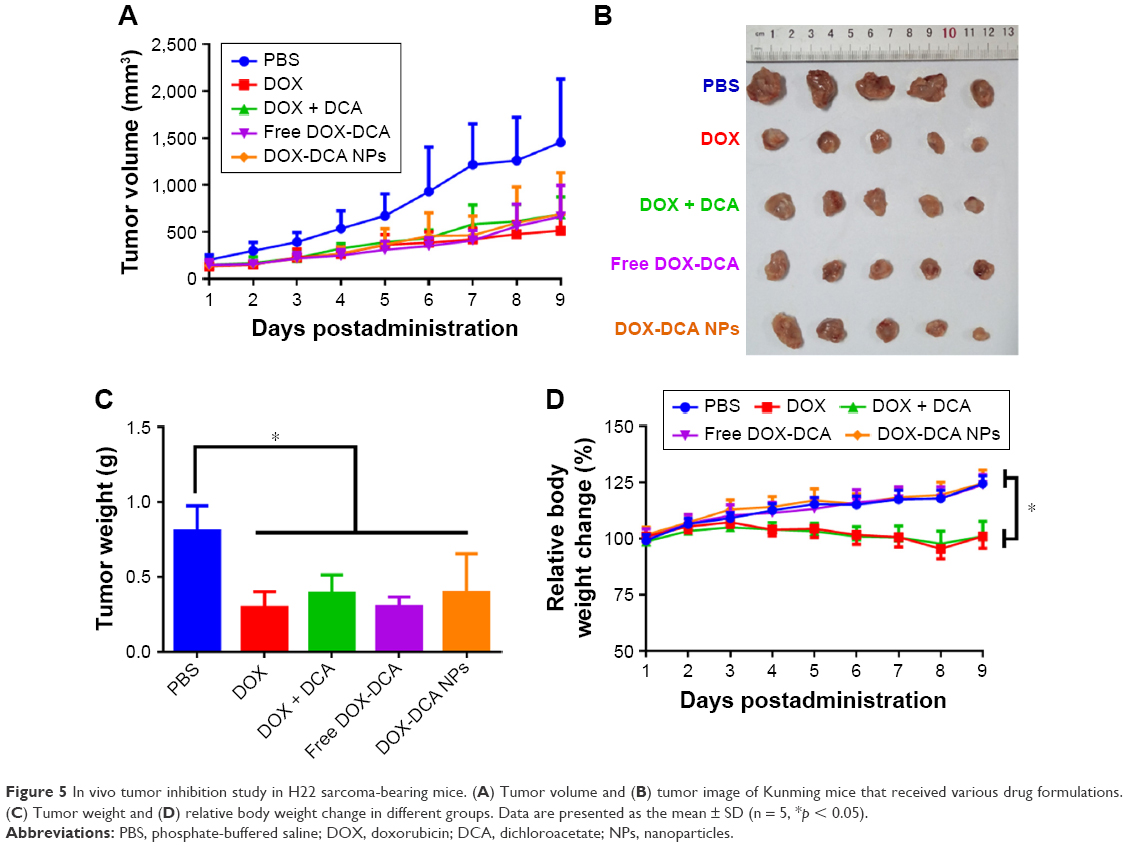 | Figure 5 In vivo tumor inhibition study in H22 sarcoma-bearing mice. (A) Tumor volume and (B) tumor image of Kunming mice that received various drug formulations. (C) Tumor weight and (D) relative body weight change in different groups. Data are presented as the mean ± SD (n = 5, *p < 0.05). |
We further investigated the antitumor efficiency on aggressive murine melanoma B16F10 model. To improve the therapeutic effect, we intensified the dosage of DOX-DCA NPs to 15 mg DOX/kg. However, the dosage of free DOX was restricted to 5 mg/kg due to the systemic toxicity mentioned earlier. As shown in Figure 6A and B, the tumor- bearing mice administrated with PBS exhibited a rapid tumor volume growth. By contrast, other groups showed delayed tumor growth. Notably, DOX-DCA NPs 15 mg DOX/kg demonstrated the best tumor inhibition. The tendency of tumor weight was consistent with that of tumor volume (Figure 6C). The tumor inhibitory rate of DOX, DOX-DCA NPs 5 mg DOX/kg and DOX-DCA NPs 15 mg DOX/kg was 72.7%, 62.3% and 83.0%, respectively. The decreased therapeutic response of DOX-DCA NPs with equiv. DOX could be attributed to the inferior cellular uptake and restricted cytotoxicity. As an indicator of systemic toxicity, the body weight was also monitored every day. Compared with PBS group, mice receiving DOX suffered from significant body weight loss during the treatment, which was similar to the result in H22 sarcoma (Figure 6D). However, the body weight of mice treated with PBS, DOX-DCA NPs 5 mg DOX/kg or even DOX-DCA NPs 15 mg DOX/kg exhibited an increase tendency. It further confirmed the reduced systemic toxicity of DOX-DCA NPs in comparison to free DOX. These results suggested that DOX-DCA NPs demonstrated enhanced antitumor efficiency and reduced toxicity profile, which can be a safe and effective nanomedicine.
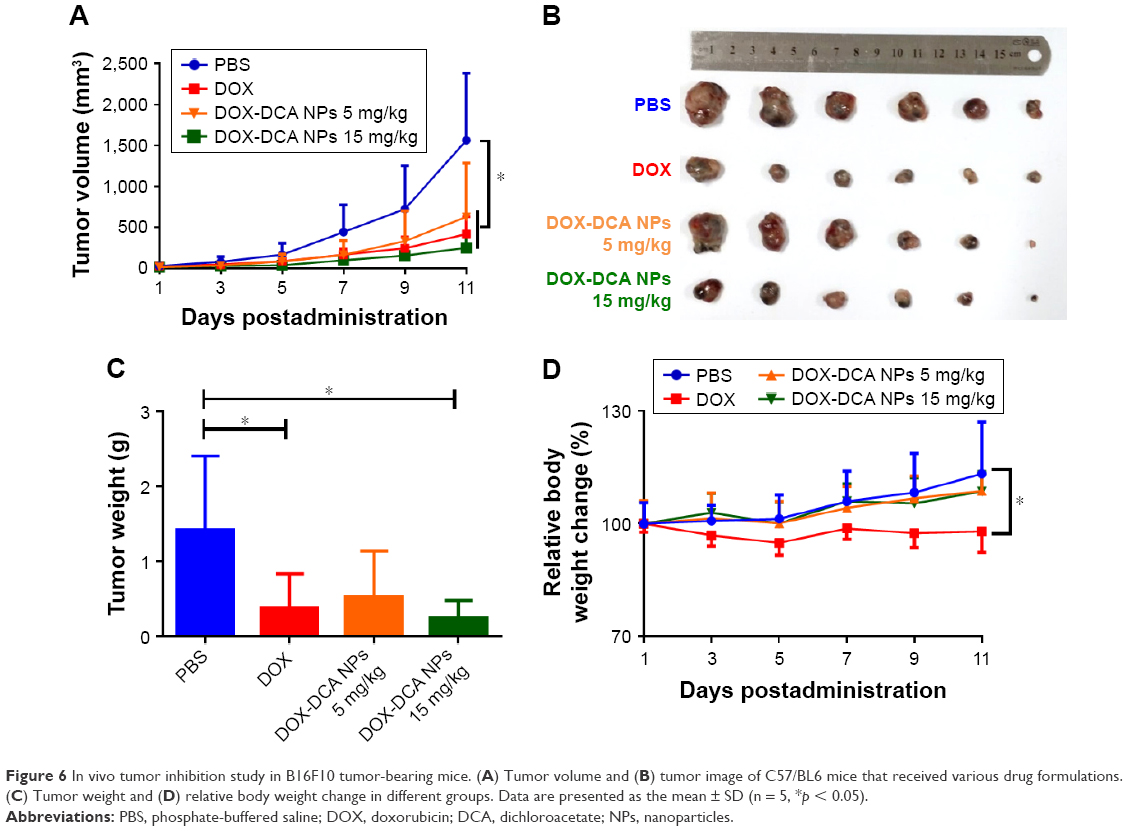 | Figure 6 In vivo tumor inhibition study in B16F10 tumor-bearing mice. (A) Tumor volume and (B) tumor image of C57/BL6 mice that received various drug formulations. (C) Tumor weight and (D) relative body weight change in different groups. Data are presented as the mean ± SD (n = 5, *p < 0.05). |
MTD and systemic toxicity study of DOX-DCA NPs
DOX has been reported to cause serious cardiotoxicity and hepatotoxicity, leading to a narrow therapeutic window.2,3 To explore the nanoformulation, it is essential to study the systemic toxicity of DOX-DCA NPs. Therefore, MTD study was first performed to investigate the systemic toxicity and safety concern of the developed DOX-DCA NPs. MTD was determined on the basis that all animals survived with a body weight loss less than 15% as well as no other obvious toxicities during 10 days.8 As evidenced from the aforementioned animal tests, DOX-DCA NPs have exhibited significant safety in comparison to free DOX and DOX + DCA at the same dosage of 5 mg/kg (Figure 5D). Moreover, even at DOX equiv. dosage of 15 mg/kg, the nanoformulation still demonstrated improved safety (Figure 6D). Therefore, the dosage of free DOX was selected as 5 mg/kg, 10 mg/kg and 15 mg/kg for MTD study, whereas the dosage of DOX-DCA NPs was set as 25 mg, 50 mg and 75 mg DOX equiv./kg. As expected, DOX-DCA NPs demonstrated extremely low systemic toxicity. Compared with free DOX, DOX-DCA NPs caused negligible body weight loss (Figure 7A). The maximum body weight loss for DOX 5 mg/kg-, DOX 10 mg/kg- and DOX 15 mg/kg-treated mice was 4.2%, 17.0% and 24.0% respectively, whereas only 1.8% for DOX-DCA NPs-treated mice with the dosage of 75 mg DOX equiv./kg. No body weight loss was found in the groups of control, 25 mg DOX equiv./kg and 50 mg DOX equiv./kg. Interestingly, the body weight change were almost the same in the groups of control and 25 mg DOX equiv./kg, indicating the safe drug delivery of DOX-DCA NPs. Induced by the severe systemic toxicity, one out of six mice died in the group of 15 mg/kg DOX-treated mice (Figure 7B). The relative body weight of remaining five mice ranged from 71.1% to 74.5% and showed no sign of increasing during the following days. Moreover, their physiological activity were sluggish, and the body temperature decreased. The MTD value of DOX-DCA NPs was 75 mg DOX equiv./kg, which was 15-fold higher compared with free DOX (5 mg/kg). The MTD of DOX-DCA NPs was higher than most of the reported nanomedicines. For example, the MTD of PolyMPC-DOX Prodrugs,29 SQ-DOX NAs30 and DOX-loaded HSA NPs31 was 10 mg/kg, 20 mg/kg and 30 mg/kg, respectively. The significantly enhanced MTD indicated that DOX-DCA NPs can immensely minimize the systemic toxicity of DOX, and thus widen the therapeutic window for promising cancer treatment.
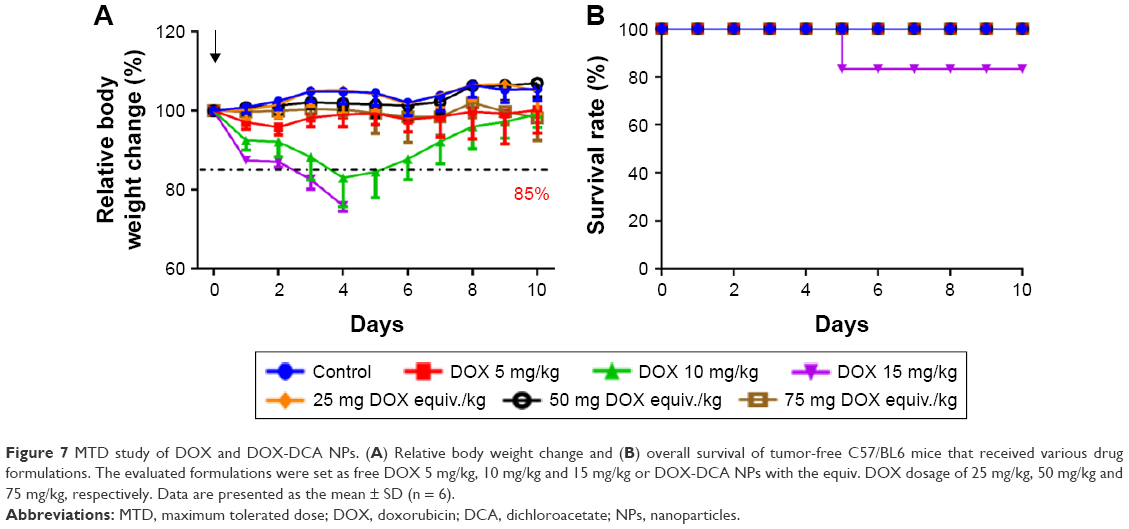 | Figure 7 MTD study of DOX and DOX-DCA NPs. (A) Relative body weight change and (B) overall survival of tumor-free C57/BL6 mice that received various drug formulations. The evaluated formulations were set as free DOX 5 mg/kg, 10 mg/kg and 15 mg/kg or DOX-DCA NPs with the equiv. DOX dosage of 25 mg/kg, 50 mg/kg and 75 mg/kg, respectively. Data are presented as the mean ± SD (n = 6). |
At day 10, all the survived mice were scarified and major organs were excised. Blood biochemical parameter analysis (Figure 8A–C) and pathological studies (Figure 8D) were carried out to further investigate the systemic toxicity. Cardiotoxicity could be life threatening during the use of DOX for chemotherapy and should be the foremost investigation indicator. As the biggest site for drug accumulation and metabolism, liver can also reflect the pathological state. Administration of DOX at a dose of 15 mg/kg did affect the blood level of LDH, ALT and AST (Figure 8A–C), indicating the occurrence of cardiotoxicity and hepatotoxicity. Minor increases in LDH, ALT and AST were also found in DOX 10 mg/kg-treated mice compared with the control. On the contrary, the levels of LDH, ALT and AST were within normal range in DOX-DCA NP-treated mice even at the highest dose, 75 mg DOX equiv./kg, suggesting the negligible cardiotoxicity and hepatotoxicity. Likewise, H&E staining analysis revealed that cardiac injury (vacuolar degeneration in cardiomyocytes, blue arrow) and hepatic damage (large amount of inflammatory cell infiltration and hepatocyte necrosis, yellow arrow) were caused by 10 mg/kg and 15 mg/kg DOX treatment (Figure 8D). No obvious damage in heart and liver was found in the DOX-DCA NP-treated mice. It was worth noting that DOX-DCA NPs can immensely mitigate the hepatotoxicity and cardiotoxicity compared with DOX. Moreover, there are invisible toxicities in kidney, lung and spleen in any groups, which were evidenced from the results of BUN, creatinine level (data not shown) and H&E staining analysis (Figure 8D). These results suggested that systemic toxicity can be greatly reduced by the use of DOX-DCA NPs, indicating the feasibility of this kind of self-assembled nanoformulation for clinic translation.
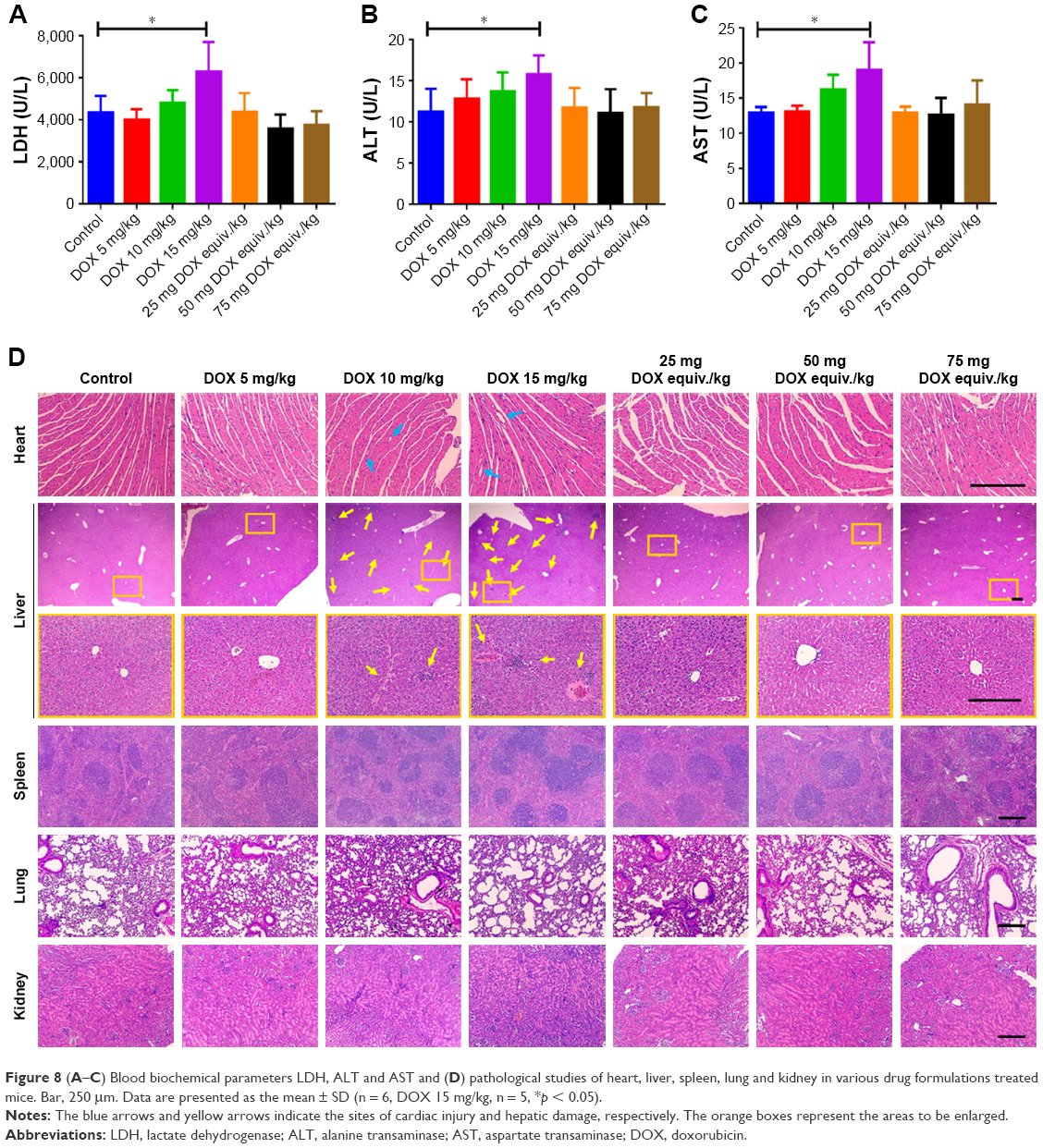 | Figure 8 (A–C) Blood biochemical parameters LDH, ALT and AST and (D) pathological studies of heart, liver, spleen, lung and kidney in various drug formulations treated mice. Bar, 250 μm. Data are presented as the mean ± SD (n = 6, DOX 15 mg/kg, n = 5, *p < 0.05). |
Conclusion
In summary, we developed a new DOX derivative, DOX-DCA, by directly conjugating DCA with DOX. It has high purity and can be easily self-assembled into NPs with small amount of PEGylated lipid DSPE-PEG2000. The DOX-DCA nanoformulation exhibited high DLC as 71.8%, which can greatly decrease the side effects caused by excipient. After systemic administration, the NPs demonstrated good tumor targeting and retention capability as well as good antitumor efficacy with high tumor inhibitory rate in murine melanoma model. It is noteworthy that the MTD of DOX-DCA NPs was 15 times higher than that of free DOX. No obvious cardiotoxicity and hepatotoxicity were found in DOX-DCA NP-treated mice. DOX-DCA nanoformulation can reduce the systemic toxicity and widen the therapeutic window. These results suggest the clinical applicability of DOX-DCA nanoformulation for the simplicity, reproducibility, high drug loading capability, reduced side effect and enhanced therapeutic effect.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81673374, 81373360 and 81701657) and Fundamental Research Funds for the Central Universities (2015ZDTD048). The authors thank the Analytical and Testing Center of Huazhong University of Science and Technology for performing the TEM and FTIR.
Disclosure
The author reports no conflicts of interest in this work.
References
Tacar O, Sriamornsak P, Dass CR. Doxorubicin: an update on anticancer molecular action, toxicity and novel drug delivery systems. J Pharm Pharmacol. 2013;65(2):157–170. | ||
De Beer EL, Bottone AE, Voest EE. Doxorubicin and mechanical performance of cardiac trabeculae after acute and chronic treatment: a review. Eur J Pharmacol. 2001;415(1):1–11. | ||
Doroshow JH. Doxorubicin-induced cardiac toxicity. N Engl J Med. 1991;324(12):843–845. | ||
Granados-Principal S, Quiles JL, Ramirez-Tortosa CL, Sanchez-Rovira P, Ramirez-Tortosa MC. New advances in molecular mechanisms and the prevention of adriamycin toxicity by antioxidant nutrients. Food Chem Toxicol. 2010;48(6):1425–1438. | ||
Fojtu M, Gumulec J, Stracina T, et al. Reduction of doxorubicin-induced cardiotoxicity using nanocarriers: a review. Curr Drug Metab. 2017;18(3):237–263. | ||
Brannon-Peppas L, Blanchette JO. Nanoparticle and targeted systems for cancer therapy. Adv Drug Deliver Rev. 2012;64:206–212. | ||
MacKay JA, Chen MN, McDaniel JR, Liu WG, Simnick AJ, Chilkoti A. Self-assembling chimeric polypeptide-doxorubicin conjugate nanoparticles that abolish tumours after a single injection. Nat Mater. 2009;8(12):993–999. | ||
Zou Y, Fang Y, Meng H, et al. Self-crosslinkable and intracellularly decrosslinkable biodegradable micellar nanoparticles: a robust, simple and multifunctional nanoplatform for high-efficiency targeted cancer chemotherapy. J Control Release. 2016;244(Pt B):326–335. | ||
Tian XQ, Ni XW, Xu HL, et al. Prevention of doxorubicin-induced cardiomyopathy using targeted MaFGF mediated by nanoparticles combined with ultrasound-targeted MB destruction. Int J Nanomedicine. 2017;12:7103–7119. | ||
Pistone A, Iannazzo D, Ansari S, et al. Tunable doxorubicin release from polymer-gated multiwalled carbon nanotubes. Int J Pharm. 2016;515(1–2):30–36. | ||
Mashat A, Deng L, Altawashi A, Sougrat R, Wang GC, Khashab NM. Zippered release from polymer-gated carbon nanotubes. J Mater Chem. 2012;22(23):11503–11508. | ||
Iannazzo D, Pistone A, Salamò M, et al. Graphene quantum dots for cancer targeted drug delivery. Int J Pharm. 2017;518(1–2):185–192. | ||
Yadav AK, Mishra P, Jain S, Mishra P, Mishra AK, Agrawal GP. Preparation and characterization of HA-PEG-PCL intelligent core-corona nanoparticles for delivery of doxorubicin. J Drug Target. 2008;16(6):464–478. | ||
Choi SJ, Choy JH. Layered double hydroxide nanoparticles as target-specific delivery carriers: uptake mechanism and toxicity. Nanomedicine (Lond). 2011;6(5):803–814. | ||
Luo C, Sun J, Sun B, et al. Facile fabrication of tumor redox-sensitive nanoassemblies of small-molecule oleate prodrug as potent chemotherapeutic nanomedicine. Small. 2016;12(46):6353–6362. | ||
Yu CT, Zhou MJ, Zhang XJ, Wei WJ, Chen XF, Zhang XH. Smart doxorubicin nanoparticles with high drug payload for enhanced chemotherapy against drug resistance and cancer diagnosis. Nanoscale. 2015;7(13):5683–5690. | ||
Venditto VJ, Szoka FC Jr. Cancer nanomedicines: so many papers and so few drugs! Adv Drug Deliver Rev. 2013;65(1):80–88. | ||
Hoffman AS. Stimuli-responsive polymers: biomedical applications and challenges for clinical translation. Adv Drug Deliv Rev. 2013;65(1):10–16. | ||
Dawidczyk CM, Kim C, Park JH, et al. State-of-the-art in design rules for drug delivery platforms: lessons learned from FDA-approved nanomedicines. J Control Release. 2014;187:133–144. | ||
Danhier F. To exploit the tumor microenvironment: since the EPR effect fails in the clinic, what is the future of nanomedicine? J Control Release. 2016;244(Pt A):108–121. | ||
Mou Q, Ma Y, Zhu X, Yan D. A small molecule nanodrug consisting of amphiphilic targeting ligand-chemotherapy drug conjugate for targeted cancer therapy. J Control Release. 2016;230:34–44. | ||
Zhang T, Huang P, Shi LL, et al. Self-assembled nanoparticles of amphiphilic twin drug from floxuridine and bendamustine for cancer therapy. Mol Pharm. 2015;12(7):2328–2336. | ||
Huang P, Wang DL, Su Y, et al. Combination of small molecule prodrug and nanodrug delivery: amphiphilic drug-drug conjugate for cancer therapy. J Am Chem Soc. 2014;136(33):11748–11756. | ||
Cheetham AG, Zhang PC, Lin YA, Lock LL, Cui HG. Supramolecular nanostructures formed by anticancer drug assembly. J Am Chem Soc. 2013;135(8):2907–2910. | ||
Jiang Y, Wang X, Liu X, et al. Enhanced antiglioma efficacy of ultrahigh loading capacity paclitaxel prodrug conjugate self-assembled targeted nanoparticles. ACS Appl Mater Interfaces. 2017;9(1):211–217. | ||
Stacpoole PW, Kerr DS, Barnes C, et al. Controlled clinical trial of dichloroacetate for treatment of congenital lactic acidosis in children. Pediatrics. 2006;117(5):1519–1531. | ||
Bonnet S, Archer SL, Allalunis-Turner J, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007;11(1):37–51. | ||
Su L, Zhang H, Yan C, et al. Superior anti-tumor efficacy of diisopropylamine dichloroacetate compared with dichloroacetate in a subcutaneous transplantation breast tumor model. Oncotarget. 2016;7(40):65721–65731. | ||
Wong KE, Mora MC, Skinner M, et al. Evaluation of polyMPC-Dox prodrugs in a human ovarian tumor model. Mol Pharm. 2016;13(5):1679–1687. | ||
Maksimenko A, Dosio F, Mougin J, et al. A unique squalenoylated and nonpegylated doxorubicin nanomedicine with systemic long-circulating properties and anticancer activity. Proc Natl Acad Sci U S A. 2014;111(2):E217–E226. | ||
Yuan A, Wu JH, Song CC, et al. A novel self-assembly albumin nanocarrier for reducing doxorubicin-mediated cardiotoxicity. J Pharm Sci. 2013;102(5):1626–1635. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.