")
Back to Journals » International Journal of Nanomedicine » Volume 12
A dual-mediated liposomal drug delivery system targeting the brain: rational construction, integrity evaluation across the blood–brain barrier, and the transporting mechanism to glioma cells
Authors Liu C, Liu X, Wang G, Hei Y , Meng S, Yang L , Yuan L, Xie Y
Received 30 December 2016
Accepted for publication 1 March 2017
Published 28 March 2017 Volume 2017:12 Pages 2407—2425
DOI https://doi.org/10.2147/IJN.S131367
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Linlin Sun
Chang Liu,1,* Xiao-Na Liu,1,* Gui-Ling Wang,1 Yu Hei,1 Shuai Meng,1 Ling-Fei Yang,1 Lan Yuan,2 Ying Xie1
1Beijing Key Laboratory of Molecular Pharmaceutics and New Drug Delivery Systems, Department of Pharmaceutics, School of Pharmaceutical Sciences, 2Medical and Healthy Analysis Center, Peking University, Beijing, People’s Republic of China
*These authors contributed equally to this work
Abstract: As the global population ages, cancer rates increase worldwide, and degenerative diseases of the central nervous system (CNS), brain tumors, and inflammation threaten human health more frequently. We designed a dual-mediated (receptor-mediated and adsorption-mediated) liposome, named transferrin–cell penetrating peptide–sterically stabilized liposome (TF-CPP-SSL), to improve therapy for gliomas through combining molecular recognition of transferrin receptors (TF-Rs) on the blood–brain barrier (BBB) and glioma cells with the internalization and lysosomal escaping ability of CPP. Based on the systematic investigation of structure–activity relations on the cellular level, we constructed TF-CPP-SSL rationally by conjugating TF and CPP moieties to the liposomes via PEG3.4K and PEG2.0K, respectively, and found the optimum densities of TF and CPP were 1.8% and 4%, respectively. These liposomes had the highest targeting efficacy for brain microvascular endothelial cell and C6 cell uptake but avoided capture by normal cells. Fluorescence resonance energy transfer technology and coculture models of BBB and glioma C6 cells indicated that TF-CPP-SSL was transported across the BBB without drug leakage, liposome breakup, or cleavage of ligand. TF-CPP-SSL offered advantages for crossing the BBB and entering into glioma C6 cells. Real-time confocal viewing revealed that TF-CPP-SSL was entrapped in endosomes of glioma C6 cells and then escaped from lysosomes successfully to release the liposomal contents into the cytosol. Entrapped contents, such as doxorubicin, could then enter the nucleus to exert pharmacological effects.
Keywords: dual ligand-modified liposomes, transferrin, cell-penetrating peptide, real-time confocal, fluorescence resonance energy transfer, tumor targeting
Introduction
With aging of populations and rising morbidity rates of cancer worldwide, degenerative diseases of the central nervous system (CNS), brain tumors, and inflammation are threats to human health.1 Over the past decade, liposomes have been developed as brain-targeted drug delivery systems (DDSs).2 Based on the abundant receptor expression on the blood–brain barrier (BBB), studies have mainly focused on receptor-mediated active transport of DDS,3–5 which can achieve high drug levels at the target site. However, the effect of the treatment is limited by receptor saturation and lysosomal digestion.6
In fact, beyond expressing various receptors, the BBB also possesses abundant negative surface charge along the tight junctions between cells. Therefore, this provides an effective mechanism for positively charged peptides to be transported across the BBB by adsorptive processes. Cell-penetrating peptides (CPPs),7 discovered in the late 20th century, can maintain electropositivity under physiological conditions because of their arginine-rich residues interacting with anionic materials on the cell surface and further activating cellular uptake. By coupling polyarginine to the surface of liposomes or micelles with polyethylene glycol (PEG)-phosphoethanolamine, DDSs accumulate in tumor cells through the enhanced permeability and retention (EPR) effect,8 thus reaching subcellular organelles, including the nucleus, mitochondria, and lysosomes.9 When the liposomes modified with CPP are trapped in endosomes after uptake, CPP adsorbs more H+ and then promotes the lipid fusion between cationic CPP-H+ liposomes and anionic components in the endosomal membrane via electronic attraction. Fusion pore is subsequently formed to release the liposomal cargoes into the cytosol.10 Unfortunately, CPPs still lack cell specificity in cargo delivery, even though they have been a known technology for considerable time. In addition, they accumulate in normal as well as tumor cells, which causes unintended damage to normal tissues.11
In order to improve the brain-targeting selectivity of CPP, we designed a dual-mediated (receptor-mediated and adsorption-mediated) liposome, named transferrin–cell penetrating peptide–sterically stabilized liposome (TF-CPP-SSL), in anticipation that it could provide effective therapy for gliomas through combining molecular recognition of TF by the TF receptor (TF-R) both on the BBB and on tumor cells with the internalization and lysosomal escaping ability of CPP. In the construction of these liposomes, PEG is an important component, which is used as a coating to protect the liposomes from binding with opsonin in the bloodstream and also provides linkage to conjugate ligands on the liposomes that provide molecular recognition of targeted cells.12 However, the molecular weight and modification density of the PEG determine the thickness of the hydration layer, which may hamper the interaction of the ligands with the receptors on cells. In addition, the modified density of the ligands determines its numbers and position on the liposomes, which relate to the possible combination of ligands with receptors on the cell surfaces to exert the optimum targeting effects.13 Therefore, it is necessary to screen and optimize the chain length/molecular weight of PEG linked to the ligands and the modified densities of the ligands to construct a rational TF-CPP-SSL in order to maximize the targeting efficiency for gliomas.
In contrast to conventional solid tumors, gliomas are located deep in the brain parenchyma.14 Encapsulated DDS must cross the BBB in order to be delivered to a glioma.15 Although there are many strategies to improve the targeting of DDS to brain tumors, how DDS will penetrate the BBB is still unclear. It is doubtful whether liposomes maintain their integrity and the ligands persist in the liposome after crossing the BBB. Fluorescence resonance energy transfer (FRET) techniques have recently helped us to understand the process of liposomal transit across the BBB. 1,1′-Dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine iodide (DiI) and 3,3′-dioctadecyloxacarbocyanine perchlorate (DiO) are common fluorescent molecular pairs for FRET.16 The principle of FRET is that the fluorescent intensity of DiI increases when the distance between two fluorescent molecules becomes <10 nm. That is, FRET occurs when DiI and DiO are coencapsulated into the same liposome, which means that the liposome has maintained its integrity. Furthermore, the coculture model, combining an in vitro BBB model and C6, can also reveal the mechanisms by which dual-mediated liposomes penetrate the BBB.
In the present study, we screened and optimized the molecular weight of PEG linked to TF and CPP with liposomes and the modification densities of ligands through cellular uptake, for the rational construction of dual-mediated liposomes (TF-CPP-SSL). In addition, we demonstrated the transport processes and mechanisms by which TF-CPP-SSL crosses the BBB, thus providing an explicit basis for designing liposomal formulations for entering glioma cells. We expect this novel and effective TF-CPP-SSL DDS to perform well in maintaining prolonged circulation times, brain targeting, and effective intracellular penetration, as well as efficient lysosomal escape, to accomplish nucleus targeting of anticancer drugs in the treatment of gliomas.
Materials and methods
Materials
Soybean phosphatidylcholine (SPC, 99% purity) was kindly donated by Shanghai Taiwei Chemical Company (Shanghai, China). Cholesteryl (Chol) was purchased from Alfa Aesar Co. Ltd. (Shanghai, China). 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)2000] (DSPE-PEG-2K) was purchased from Shanghai Advanced Vehicle Technology Pharmaceutical Ltd. (Shanghai, China). 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[poly(ethylene glycol)-X]-maleimide (DSPE-PEG(X)-MAL, X =2K, 3.4K) were obtained from NOF Corp. (Tokyo, Japan). 1,2-Distearoyl-sn-glycerol-3-phosphoethanolamine-N-[poly(ethyleneglycol)-Y]-hydroxy succinamide (DSPE-PEG(Y)-NHS, Y =1K, 2K, 3.4K) was obtained from Shearwater Polymers Inc. (Huntsville, AL, USA). CPP (GGRRRRRRRRR-amide) was provided by KTG Pharmaceuticals, Inc. (Wuhan, Hubei Province, China). Transferrin, coumarin-6, triethylamine, 4-dimethylaminopyridine (DMAP), and dimethylformamide (DMF) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Doxorubicin (DOX) was purchased from Melunbio. Co., Ltd. (Dalian, Liaoning Province, China). 1,10-Dioctadecyl-3,3,30,30-tetramethylindotricarbocyanine iodide (DiR) was purchased from Fanbo Biochemicals (Beijing, China). DiI and DiO were purchased from Anaspec Inc. (Fremont, CA, USA).
Cell culture
The glioma C6 cell line was purchased from the Institute of Basic Medical Science, Chinese Academy of Medical Science (Beijing, China). Brain microvascular endothelial cells (BMVECs) from BALB/c mice were generously gifted by Prof Jin-Ning Lou from China–Japan Friendship Hospital, China. The glioma C6 cells and BMVECs were cultured in HAM’S F10 containing 10% (v/v) heat-inactivated fetal bovine serum (FBS) and endothelial cell culture medium (ECCM) (dulbecco’s modified eagle medium [DMEM], 20% FBS, 100 μg/mL endothelial cell growth factor [ECGF], 20 μg/mL heparin), respectively, supplemented with 100 U/mL penicillin, 100 μg/mL streptomycin, and 2 mmol/L L-glutamine. Cultures were maintained at 37°C in a humidified 5% CO2 incubator.
Animals
BALB/c nude mice (20±2 g) were provided by the Animal Center of Peking University Health Science Center. All the animal experiments were conducted strictly according to the Guiding Principles for the Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of Peking University.
Preparation of TF-SSL, CPP-SSL, and TF-CPP-SSL
TF-SSL was prepared by a postreaction method.17 Briefly, a thin film containing SPC/cholesterol/DSPE-PEG(X)-MAL (X =2K, 3.4K)/DSPE-PEG2K was hydrated with phosphate-buffered saline (PBS, pH 7.4) under sonication at 37°C for 20 min to form SSL with an active maleimide functional group (SSL-MAL). Sulfhydryl transferrin (TF-SH; the synthesis method and the determination of thiol groups’ amount are depicted in the “Supplementary materials” section) was incubated with SSL-MAL (TF:DSPE-PEG(X)-MAL =1:4, mol/mol) for 12 h at 37°C to form TFX-SSL, in which X represents the molecular weight of PEG linked to TF. TFX-SSL was separated from free TF by a Sepharose CL-4B column, and the coupling efficiency of TF with SSL was measured to be ~75% by a Bicinchoninic Acid (BCA) protein quantitation kit.
CPP-SSL was prepared by a thin-film hydration method.18 First, the succinamide group in DSPE-PEG(Y)-NHS (Y =1K, 2K, 3.4K) reacted with the amino group in CPP to produce DSPE-PEG(Y)-CPP (Y =1K, 2K, 3.4K), and the product was identified by MALDI-TOF-MS (AB SCIEX TOF/TOF 5800, Singapore) after being filtered and freeze-dried (Figure S3). Then, DSPE-PEG(Y)-CPP was mixed with SPC/cholesterol/DSPE-PEG2K in a solution of chloroform. After evaporation to remove the organic solvent, the thin film was hydrated with PBS (pH 7.4) and CPP-modified liposomes (CPPY-SSL) with a high CPP binding ratio (95%) were prepared: in this expression, Y represents the molecular weight of PEG linked to CPP.
Dual-mediated liposomes, TF-CPP-SSL, were prepared by a combination method of thin-film hydration and postreaction technique. Briefly, the thin film containing SPC/cholesterol/DSPE-PEG2K/DSPE-PEG(X)-MAL (X =2K, 3.4K)/DSPE-PEG(Y)-CPP (Y =1K, 2K, 3.4K) was hydrated with PBS, and then TF-SH was added to react with the MAL group to produce TFX-CPPY-SSL.
For optimizing the molecular weight of PEG linked to ligands on liposomes and the ligands’ modified density to construct TF-CPP-SSL rationally, via detecting the cellular uptake ratio by flow cytometry (FCM) and investigating the transporting mechanism of TF-CPP-SSL across the BBB, coumarin-6-loaded liposomes were prepared (SPC: coumarin-6 =2,000:1, w/w). In the FRET evaluation, single fluorescent probe (DiI and DiO)-loaded liposomes were prepared at the ratio of 200:1 (mol/mol) for SPC:DiI(DiO), respectively. In dual fluorescent probe (DiI-DiO)-loaded liposomes, DiI and DiO were coencapsulated into liposomes at the molar ratio of 200:1:1 for SPC:DiI:DiO. All the abovementioned liposomes that encapsulated hydrophobic drugs were prepared using the lipid film hydration method as described previously.19 For confocal images, DOX was loaded into liposomes by a pH-gradient method.20
Optimization of molecular weight of PEG linked to ligands in TF-CPP-SSL
The ligand moiety was usually conjugated to the surface of the liposomes via PEG as a linker during the construction of the active-targeting liposomes.21 However, PEG might either enhance or hamper the cellular uptake of liposomes because of its molecular weight and distribution morphology around the liposomes.22 TFX-SSL with different molecular weights of PEG was prepared by the postreaction method as described earlier, and the molar ratio of SPC/cholesterol/DSPE-PEG(X)-MAL (X =2K, 3.4K)/DSPE-PEG2K/TF-SH was 100:50:2:6:0.5.
BMVECs were seeded in six-well plates precoated with 2% gelatin at the density of 1×105 cells/well and cultured at 37°C in a humidified 5% CO2 incubator. After 24 h in culture, the cells were rinsed with PBS and incubated with free serum DMEM containing TFX-SSL (coumarin-6) solutions (0.2 mg/mL SPC) for 2 h. Cells were then trypsinized, washed thrice with cold PBS, resuspended in 0.5 mL PBS, and passed through a 300-mesh cell strainer prior to fluorescence-activated cell sorting (FACS) using a FACS Calibur flow cytometer (BD Biosciences, San Jose, CA, USA). Cells were enclosed in a forward versus side scatter gate to exclude debris and dead cells prior to analysis of 10,000 cell counts. Fluorescent intensity was measured at λex =467 nm, λem =505 nm. Data were analyzed via BD Cell Quest Pro software. SSL served as the control.
The molecular weight of PEG linked to CPP in TF-CPP-SSL was optimized on the BMVECs. TF-Rs expressed on BMVECs were presaturated with free TF, which would prevent the TF moiety of the TF-CPP-SSL from exerting molecular recognition. The optimum PEG linked to CPP would be the one that can maximally increase BMVEC uptake but cannot promote CPP penetration of the cells that expressed little or no TF-R. TF3.4K-CPPY-SSL (coumarin-6) was prepared with the thin film hydration method as described earlier. The molar ratio of SPC/cholesterol/DSPE-PEG (3.4K)-MAL/DSPE-PEG(Y)-CPP (Y =0, 1K, 2K, 3.4K)/DSPE-PEG2K/TF-SH was 100:50:2:1:5:0.5. BMVECs were seeded in six-well plates at the density of 1×105 cells/well and cultured at 37°C in a humidified 5% CO2 incubator. After 24 h of culture, the cells were rinsed with PBS and incubated with free serum DMEM containing 1 mg/mL TF for 30 min, and the TF3.4K-CPPY-SSL (coumarin-6; Y =0, 1K, 2K, 3.4K; 0.2 mg/mL SPC) was then added. After 2 h incubation, the fluorescence intensity (FI) of each sample was measured by a flow cytometer based on the regular protocol of FCM described earlier.
Optimization of modified density of TF and CPP in TF-CPP-SSL
The density of ligands on the liposomes was closely related to the receptor recognition efficiency of the target cells.23 TF3.4K-SSL with 0, 0.25%, 0.5%, 1.5%, 1.8%, and 2% of TF densities were each prepared by regulating the molar ratio of SPC/cholesterol/DSPE-PEG3.4K-MAL/DSPE-PEG2K/TF-SH =100:50:4n:(8-4n):n (n=0, 0.25, 0.5, 1.5, 1.8, 2.0). The cellular uptakes of TF3.4K-SSL (coumarin-6) with different TF densities were investigated on BMVECs by FCM. The optimal density of TF was the maximal density that prompted the best cellular uptake of TF3.4K-SSL. It has been reported that TF-Rs are expressed abundantly on the C6 cell line.24 To investigate whether the optimization of TF density in TF3.4K-SSL was related to cell species, the cellular uptake of TF3.4K-SSL by glioma C6 cells was also measured with FCM.
After the TF modification density was fixed, TF3.4K-CPP2K-SSL with 0, 1%, 2%, 4%, 6%, and 8% of CPP densities were each prepared by regulating the molar ratio of SPC/cholesterol/DSPE-PEG3.4K-MAL/DSPE-PEG2K/TF-SH/DSPE-PEG2K-CPP =100:50:7.2:(8-m):1.8:m (m=0, 1, 2, 4, 6, and 8). Cellular uptake of TF3.4K-CPP2K-SSL with different densities of CPP was investigated on free TF presaturated glioma C6 cells by FCM as described previously.
Characterization of SSL, CPP-SSL, TF-SSL, and TF-CPP-SSL
SSL, CPP2K-SSL, TF3.4K-SSL, and TF3.4K-CPP2K-SSL with 1.8% TF and 4% CPP, either blank or drug-encapsulated, were diluted with distilled water, respectively. After that, they were filtered through a 0.22 μm filter membrane, size distribution (in nm), polydispersity index (PDI), and zeta potential were determined at 25°C using a Nano ZS ZEN3600 Zetasizer (Malvern Instruments, Ltd., Worcester, UK). Assays were run 15 times with an equilibrium time of 60 s. The morphological examinations of SSL and TF-CPP-SSL were performed on Multimode atomic force microscopy (AFM; Bruker Dimension FastScan, Germany). Encapsulation efficiency (ee%) was determined by comparing the FI of eluted liposome solutions after removing free drugs through a Sephadex G-50 column, with the primary liposome suspensions in equal volumes before application to the columns.25
Drug release profiles of SSL, CPP-SSL, TF-SSL, and TF-CPP-SSL-loaded DOX
The stability of liposomes was evaluated with the release profile of DOX from liposomes against PBS (pH 7.4) or PBS containing 10% horse serum (HS). For this, 1 mL of SSL (DOX), CPP-SSL (DOX), TF-SSL (DOX), and TF-CPP-SSL (DOX) was sealed into dialysis bags (molecular weight cutoff [MWCO], 8,000–14,000 Da), respectively, and incubated in 30 mL of release medium at 37°C in a thermal bath, stirring continuously for 36 h. A 1 mL sample was extracted at predetermined time points, and 1 mL of fresh medium was supplemented to maintain volumes. The released DOX concentration (csample, μg/mL) was determined with a spectrofluorometer (Cary Eclipse, Palo Alto, CA, USA) at excitation and emission wavelengths of 470 and 580 nm, respectively. The cumulative release proportion (CRP), indicating the proportion of total DOX being released from liposomes, was calculated and plotted at the given time.26
Permeability of liposomes crossing the BBB
In vitro BBB models were established according to our previous work.27 BMVECs were seeded at a density of 2×104 cells/cm2 in a six-well Transwell (Corning, Steuben County, NY, USA), 0.4 μm pore size, 24 mm diameter, and precoated with 2% gelatin. The transendothelial electrical resistance (TEER) during culture was monitored by an epithelial volt-ohm meter (Millicell ERS®; Millipore, Bedford, MA, USA). Once the TEER value exceeded 200 Ωcm2 and the distance of liquid between donor chamber and acceptor chamber was retained in a 4-h leakage test, the in vitro BBB model was considered to be established successfully and could be used for the permeability assay.
For comparing the effects of different ligands on the permeability of liposomes across the BBB, each liposomal formulation-loaded coumarin-6 at the concentration of 0.2 mg/mL−1 SPC was added to the donor chamber. Samples extracted from the acceptor chamber at 0.5, 1.0, 2.0, 4.0, and 6.0 h were analyzed by enzyme linked immunosorbent assay (ELISA) (Infinite 200; Tecan, Hombrechtikon, Switzerland) at λex =466 nm, λem =504 nm, and volume was replaced by 200 μL of fresh DMEM medium without phenolsulfonphthalein. Cells were kept under culture conditions (37°C, 5% CO2, and saturated humidified atmosphere) during the transportation experiment. The permeability efficiency (Pe%) was calculated according to the following equations, respectively:27
|
|
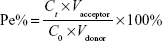
where Ct and C0 are the concentrations of the drug in the acceptor chamber at a given time and the original concentration of the drug in the donor chamber, respectively. They were calculated from the standard equation between FI and the concentration of liposome-loaded coumarin-6. Vacceptor and Vdonor are 2.0 and 1.0 mL, respectively, so that the liquid inside and outside of the Transwell was at the same level to avoid hydrostatic pressure.
Integrity evaluation of liposomes crossing the BBB
FRET techniques were used to further confirm the integrity of liposomal lipid bilayers after crossing the BBB.28 FRET donor dye (DiO) and acceptor dye (DiI) were individually or simultaneously encapsulated into liposomes to produce TF-CPP-SSL (DiO), TF-CPP-SSL (DiI), or TF-CPP-SSL (DiO-DiI). The permeability of SSL and TF-CPP-SSL on in vitro BBB models was measured as described earlier. Samples extracted from the acceptor chamber were analyzed by fluorescence spectrophotometer (Cary Eclipse) at λex =549 nm and λem =565 nm for DiI, λex =488 nm and λem =501 nm for DiO, and λex =488 nm and λem =565 nm for the fluorescent molecule pair DiO-DiI. Furthermore, the fluorescence spectrum of the sample from the acceptor chamber at 6 h was recorded. The integrity of liposomes after crossing the BBB was evaluated by the penetrating efficacy of TF-CPP-SSL encapsulated with different fluorescent probes and comparison of the fluorescence spectrum of samples from the acceptor chamber with the original liposomal formulation.
To investigate whether the ligands were still retained on the liposomes after being transported across the BBB and the structure of liposomes was maintained before entering the glioma, permeability and cellular uptake experiments were conducted on the coculture model of BBB and glioma C6 cells. SSL, TF-SSL, and TF-CPP-SSL loaded with coumarin-6 at the mass ratio of SPC to coumarin-6 =200:3 were each prepared. The tested liposomes with a concentration of 0.2 mg/mL SPC were added to the donor chamber of the in vitro BBB model for the 6-h penetration assay in a humidified 5% CO2 incubator. Samples were extracted from the acceptor chamber and diluted with DMEM until the concentration of coumarin-6 was 80 ng/mL. Then, C6 cellular uptake of samples from the acceptor chamber was measured by FCM as described previously. C6 cellular uptake of the original liposomal formulation in which the concentration of coumarin-6 was diluted with DMEM to 80 ng/mL directly was performed as a control.
Cellular internalization and lysosome escape of TF-CPP-SSL observed by CLSM
Glioma C6 cells at a density of 1×105 cells per dish were cultured onto glass-based dishes for cellular uptake studies of TF-CPP-SSL. The cells were rinsed with PBS twice after a 12-h culture and incubated with sterile SSL (DOX), TF-SSL (DOX), and TF-CPP-SSL (DOX) at a concentration of 10 μg/mL DOX for 2 h. Cellular uptake was terminated with ice-cold PBS, and lysosomal staining was performed with Lysotracker Green (500 nmol/L) for 20 min. After the cells were rinsed three times with PBS, the colocalization of DOX-loaded liposomes with lysosomes was observed under a Leica TCS SP8 confocal laser scanning microscope (CLSM, Leica, Heidelberg, Germany). The emission wavelength of Lysotracker Green and DOX was 520 nm and 584 nm, respectively, while their excitation wavelength was 488 nm.
For further investigating the transport mechanisms of the TF-CPP-SSL, cellular internalization and lysosomal escape of TF-CPP-SSL were observed by real-time confocal microscopy. The procedure for cell culture was same as mentioned earlier. During the observation of cellular internalization of TF-CPP-SSL, lysosomes were transfected with 0.1 mL CellLight® Lysosomes-RFP for 12 h, and then rinsed with PBS three times, with each rinse lasting 5 min. The prepared liposome solution with a final concentration of 200 ng/mL coumarin-6 was added to the cell cultures and the response measured using CLSM at λex =555 nm, λem =584 nm of RFP and λex =466 nm, λem =504 nm of coumarin-6. Images were collected every 5 min for 2 h. The parameters such as gain and laser intensity were never changed once an observing field was selected to promise that the comparing of FI intensity in each picture at interval time is valid.
For observation of the lysosomal escape of TF-CPP-SSL, glioma C6 cells at a density of 1×105 cells per dish were cultured for 12 h and then washed with PBS three times. The cells were transfected with CellLight® Lysosomes-GFP for 12 h and then rinsed with PBS three times. Then cells incubated with DOX-loaded liposome suspensions for 2 h were rinsed with PBS and observed by confocal laser scanning microscopy under cold PBS. Photographs were collected every 5 min using CLSM at λex =485 nm and λem =520 nm of GFP and λex =493 nm and λem =584 nm of DOX.
In vivo images of TF-CPP-SSL in the in situ model of glioma
BALB/c nude mice were anesthetized with 10% chloral hydrate (0.4 mL/100 g body weight). They were placed on a stereotaxic apparatus (RWD Life Science, Inc., Shenzhen, China). A midline sagittal incision was made to expose the cranium. Glioma C6 cells (5×105 cells/7.5 μL) were injected into the right brain of each mouse at the coordinate of 0.7 mm anterior to the right coronal suture, 1.5 mm lateral to the bregma, at 2.5 mm depth from the dura (3.0 mm depth followed by 0.5 mm withdrawal).29
For visual observation of the biodistribution of TF-CPP-SSL in vivo, fluorescent probe DiR-labeled liposomes were prepared; SSL and TF-SSL served as the control groups. These animals were intravenously injected with SSL (DiR), TF-SSL (DiR), or TF-CPP-SSL (DiR) at a dose of 1 mg lipid/20 g body weight after the glioma tumor was implanted into the brain of nude mice for 7 days. Then, at 1, 2, 4, 8, 12, 24, and 48 h postinjection, whole body optical imaging was conducted using a Kodak In vivo Imaging System FX PRO (Carestream Health, Inc., Rochester, NY, USA) with an excitation band-pass filter at 748 nm and an emission wavelength of 780 nm. The exposure time was 20 s per image.
Statistical analysis
Statistical data were processed using Microsoft Excel 2010 software and presented as mean ± standard deviation (SD) of at least three independent experiments. The differences and correlations between the two groups were analyzed using Student’s t-test, in which data were considered statistically significant if P<0.05(*) and very significant if P<0.01(**).
Results
Optimization of molecular weight of PEG linked to ligands in TF-CPP-SSL
Modifications of liposomal surfaces with PEG derivatives have served as a milestone in the development of liposomes.20 PEG acts not only as a coating to protect the liposomes from quick clearance by the reticuloendothelial system but also a linker for coupling targeting moieties to the liposomes. The cellular uptake results in BMVECs, measured with flow cytometry, showed that the cellular uptake ratio of TF3.4K-SSL (coumarin-6) was 1.38-fold greater than that of TF2K-SSL (coumarin-6) (Figure 1A), indicating that relatively larger molecular weight of PEG increased the targeting ability of TF and thus facilitated the cellular uptake process.
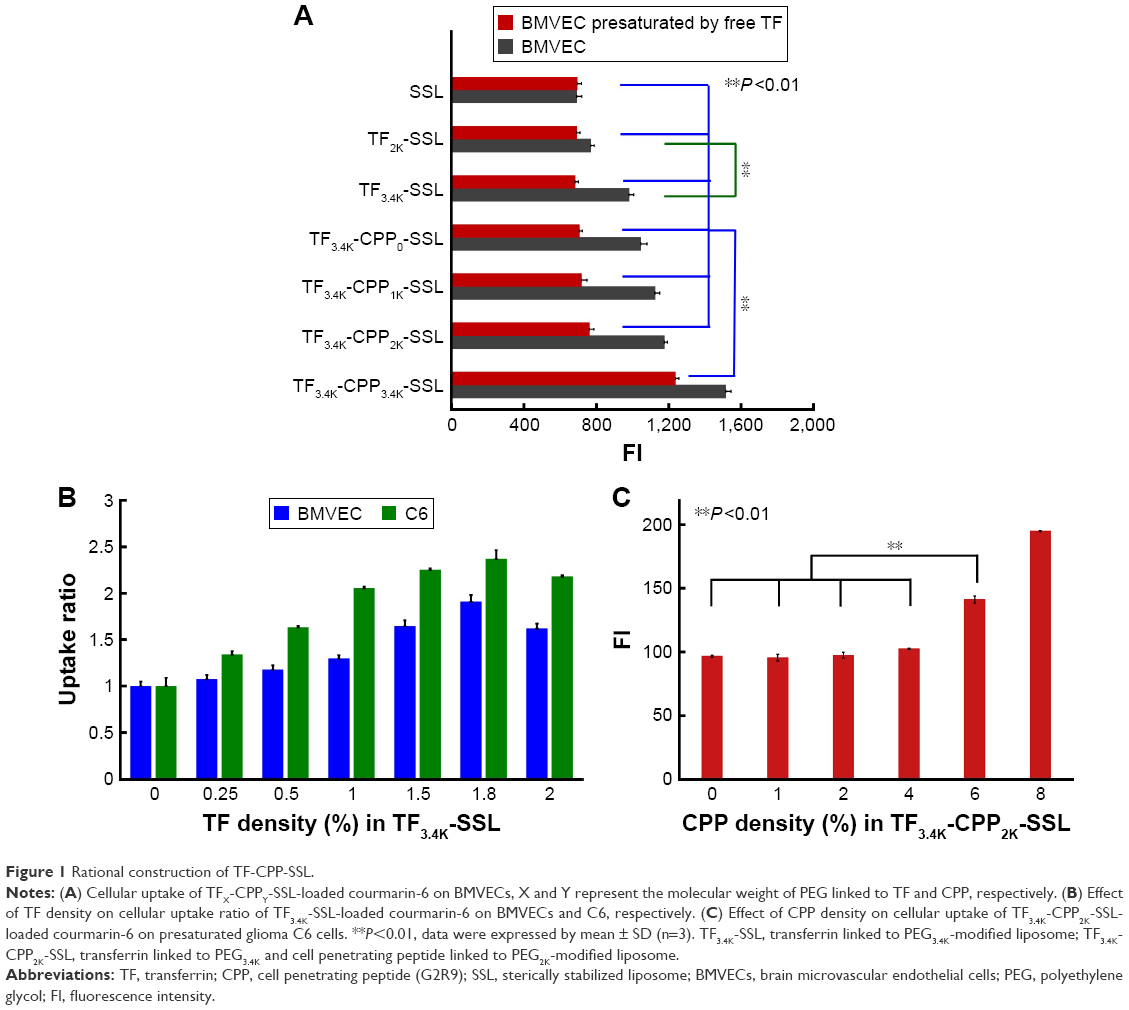 | Figure 1 Rational construction of TF-CPP-SSL. |
In order to define the best molecular weight of PEG for linking to CPP in TF-CPP-SSL, the TF’s PEG molecular weight was initially fixed to 3.4K. With increasing PEG molecular weight (from 0 to 3.4K) for the CPP linker, the cellular uptake of TF3.4K-CPPY-SSL (coumarin-6) increased, showing that the longer the PEG linked to CPP, the stronger the synergetic targeting and penetrating effect. However, on BMVECs preincubated with 1.0 mg/mL free TF to saturate TF-Rs, the cellular uptake results of TF3.4K-CPPY-SSL (coumarin-6) (Y =0, 1K, 2K) indicated that as long as Y was smaller than 2K, the cellular uptake of TF3.4K-CPPY-SSL (coumarin-6) was not affected by the molecular weight of the PEG and was equivalent to that of SSL (Figure 1A). When Y increased to 3.4K, BMVECs presaturated TF-R exerted the highest cellular uptake efficiency of TF3.4K-CPP3.4K-SSL (coumarin-6) (Figure 1A), indicating that TF3.4K-CPP3.4K-SSL had a stronger penetrating effect on cells that expressed fewer TF-R, lacking selectivity to targeting cells. Therefore, by balancing the penetrating ability of CPP and the shielding efficacy of PEG, the optimized dual-mediated liposome is seen to be TF3.4K-CPP2K-SSL.
Optimization of modification density of TF and CPP in TF-CPP-SSL
The effect of modification density of TF in TF3.4K-SSL (coumarin-6) on the cellular uptake of BMVECs and C6 is shown in Figure 1B. The results indicated that FI in the BMVECs captured TF3.4K-SSL (coumarin-6) improved with increasing density of TF as it approached 1.8%, followed by a decrease after FI of 2%; the same observations were recorded in relation to glioma C6 cells. We confirmed that the optimal molar ratio of TF was 1.8% of SPC in TF3.4K-SSL for the highest cellular uptake, and it did not change with the cell species.
Under the situation of the TF-R on glioma C6 cells presaturated with free TF, CPP in TF3.4K-CPP2.0K-SSL did not contribute any penetrating ability to C6 until its density was in excess of 4% (Figure 1C). This meant that the hydrated layer of PEG3.4K could shield CPP completely, leading to TF3.4K-CPP2K-SSL never being captured by normal cells at 4% of CPP density. At the same time, the cellular uptake of TF3.4K-CPP2K-SSL (coumarin-6) on C6 with abundant TF-R was positively correlated with the density of CPP. According to the criterion of the highest targeting efficacy for the BBB and glioma, the optimum CPP density in TF3.4K-CPP2K-SSL was determined to be 4%.
Based on systematic investigations of structure–activity effects on the cellular level, we constructed dual-mediated liposome TF-CPP-SSL rationally by conjugating TF and CPP moieties to the liposomes via PEG3.4K and PEG2.0K, respectively, with optimum density of TF and CPP of 1.8% and 4%, respectively. In the following studies, SSL, 4% CPP2K-SSL, and 1.8% TF3.4K-SSL were prepared as controls. Furthermore, the density of PEG in each liposome was 11.2% to control for the effect of PEG on the liposomes.
Characterization of SSL, CPP-SSL, TF-SSL, and TF-CPP-SSL
The average size of each blank liposome was ~100 nm, with PDI <0.2. Compared with SSL, the size of liposomes increased slightly after the addition of TF and CPP (Figure 2A) and the liposomal surface roughened (Figure 2B). The zeta potential of the liposomes changed remarkably after modification with CPP (P<0.01; Figure 2A), indicating the effect of positive charges of CPP. The incorporation of TF linked with PEG3.4K relieved the positive charge effect of CPP in TF-CPP-SSL as a result of the shielding function of PEG3.4K. The size and zeta potential did not change significantly after the liposome-encapsulated drugs such as coumarin-6, DOX, and DiR. The ee% of liposomes exceeded 90% because of the hydrophobic features of the fluorescent probes or active loading of DOX driven by the pH gradient.
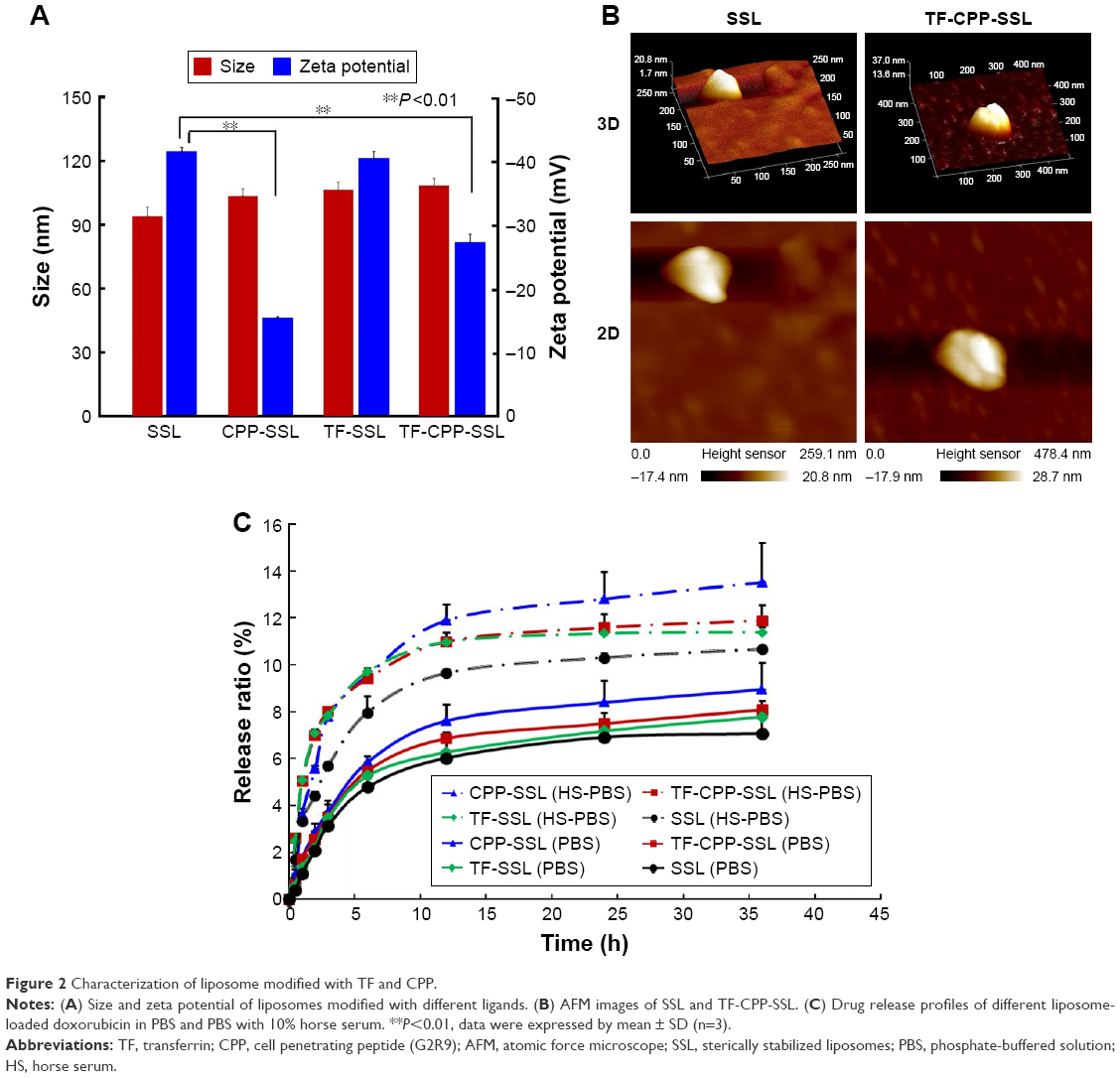 | Figure 2 Characterization of liposome modified with TF and CPP. |
Drug release profiles of DOX-loaded SSL, CPP-SSL, TF-SSL, or TF-CPP-SSL
The release profiles of DOX in vitro are shown in Figure 2C. The release rates of all liposomes were <3% within the initial 0.5 h and <15% over 48 h, indicating that DOX encapsulated in the liposomes was relatively stable for further evaluation. The release trend of all the liposomes increased when PBS was replaced with PBS with HS. The sequence of the DOX release rate for TF-CPP-SSL, TF-SSL, CPP-SSL, and SSL in the two types of medium was as follows: CPP-SSL > TF-CPP-SSL > TF-SSL > SSL. Our previous work demonstrated that the interaction between CPP and DOX enhanced DOX efflux from the liposomal medium,30 depending on the cation-pi interaction and presence of hydrogen bonds when the guanidinium group of CPP was parallel to the planar aromatic chromophore of DOX. The incorporation of a TF moiety might reduce the interaction to make the liposomes more stable.
Permeability of liposomes crossing the BBB
The TEER of each well administered with SSL (coumarin-6), TF-SSL (coumarin-6), CPP-SSL (coumarin-6), and TF-CPP-SSL (coumarin-6) was monitored to keep them above 200 Ωcm2 throughout the permeability detection (Figure 3A), indicating that the BBB (confluent BMVEC monolayers) had not been disrupted by liposomes. The Pe% of each liposome across the BBB is shown in Figure 3B. Decorated with CPP or TF, the permeability coefficient of CPP-SSL and TF-SSL was double and 1.3-fold higher than that of SSL, respectively; the permeability of the liposomes modified with both TF and CPP was enhanced 3.6-fold over SSL, suggesting a synergistic effect by dual mediation of liposomes.
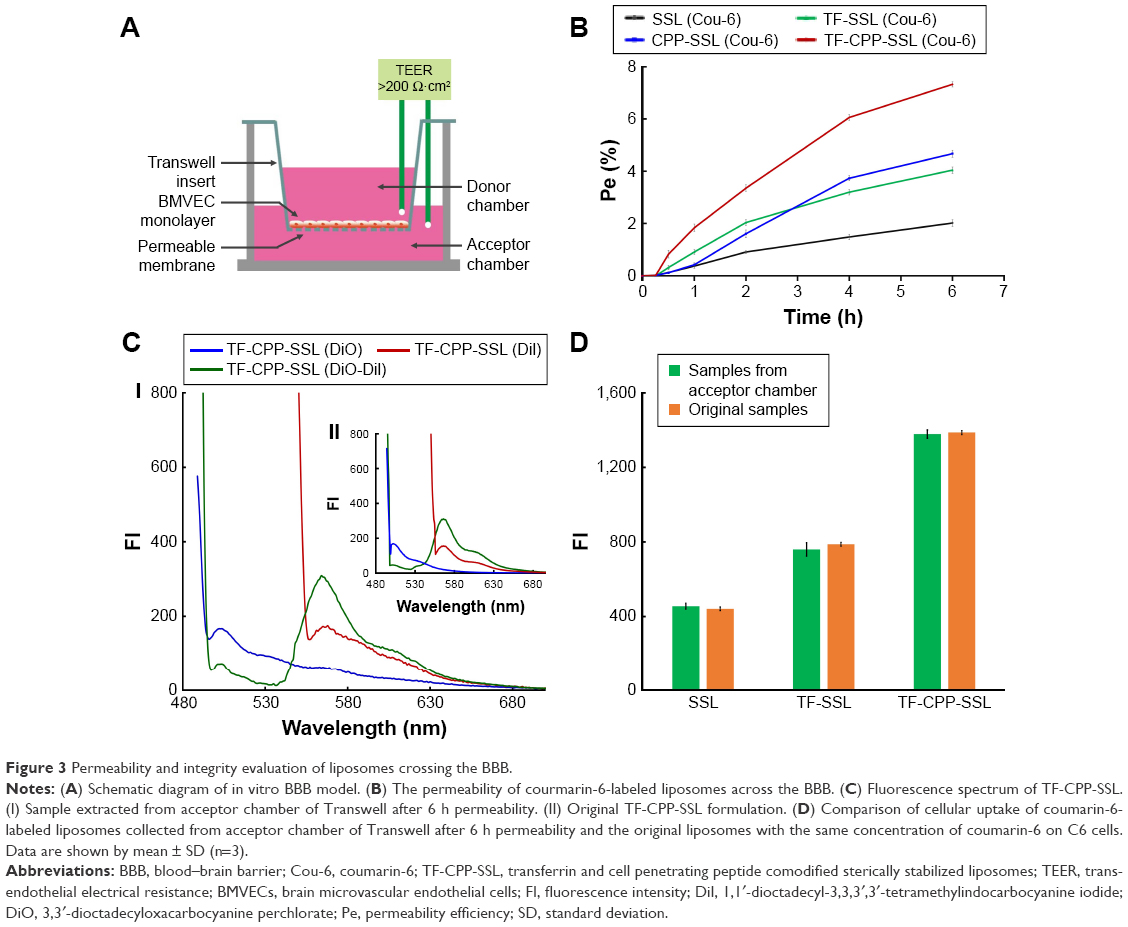 | Figure 3 Permeability and integrity evaluation of liposomes crossing the BBB. |
Integrity evaluation of liposomes crossing the BBB
The transportation of TF-CPP-SSL across the BBB was evaluated with FRET technology. The standard curve showed that the FI of TF-CPP-SSL (DiI-DiO) was higher than that of TF-CPP-SSL (DiO) or TF-CPP-SSL (DiI) at the same concentration of DiI or DiO (Figure S4A), indicating that FRET appeared when DiI and DiO were coencapsulated in liposomes. The permeability coefficient of TF-CPP-SSL (DiO), TF-CPP-SSL (DiI), and TF-CPP-SSL (DiI-DiO), calculated from the corresponding standard equation, was almost the same (P>0.05; Figure S4B), which is consistent with that of TF-CPP-SSL (coumarin-6). This indicates that the permeability of TF-CPP-SSL was independent of drugs or that there was no drug leakage during the liposomal penetration of the BBB. The fluorescence spectrum of samples from the Transwell acceptor chamber illustrated that the FRET effect still happened and was consistent with the original formulation of TF-CPP-SSL loaded with DiO and/or DiI (Figure 3C), further indicating that TF-CPP-SSL crossed the BBB in the form of intact liposomes.
TF-Rs are highly expressed on both BMVECs and brain glioma C6 cells. On the coculture model of BBB and glioma C6 cells, C6 cellular uptake treated with samples from the Transwell acceptor chamber was the same with that of the original liposomal formulations (Figure 3D). We confirmed that liposomes were transported across the BBB, based on the integrity of the form without drug leakage, liposomes breaking up, and even the cleavage of ligands. The advantages of TF-CPP-SSL crossing the BBB offered strong evidence of their capability to enter glioma cells.
Cellular internalization and lysosomal escape of TF-CPP-SSL observed by CLSM
DOX is a typical antitumor drug that destroys the structure of DNA in nuclei and emits fluorescence afterward. After 2-h incubation with a concentration of 10 μg/mL DOX, the CLSM results revealed that SSL (DOX) mainly appeared in the cytoplasm, colocalized with lysosomes to yield yellow light (Figure 4A) and failed to deliver DOX molecules into the cell nuclei. With TF modified onto the liposome, TF-SSL (DOX) facilitated DOX transport into the tumor cells, but most of the DOX was still in the cytoplasm, colocalized with lysosomes to yield orange-yellow fluorescence. Our results also demonstrated that TF-CPP-SSL (DOX) exhibited substantially higher red FI in cell nuclei and lower red intensity in the cytoplasm, while the lysosomes exhibited bright green fluorescence. It was also observed that the cell shape gradually became oval, suggesting that TF-CPP-SSL (DOX) had internalized into the tumor cells and delivered DOX to the cell nuclei after successfully escaping from the lysosomes.
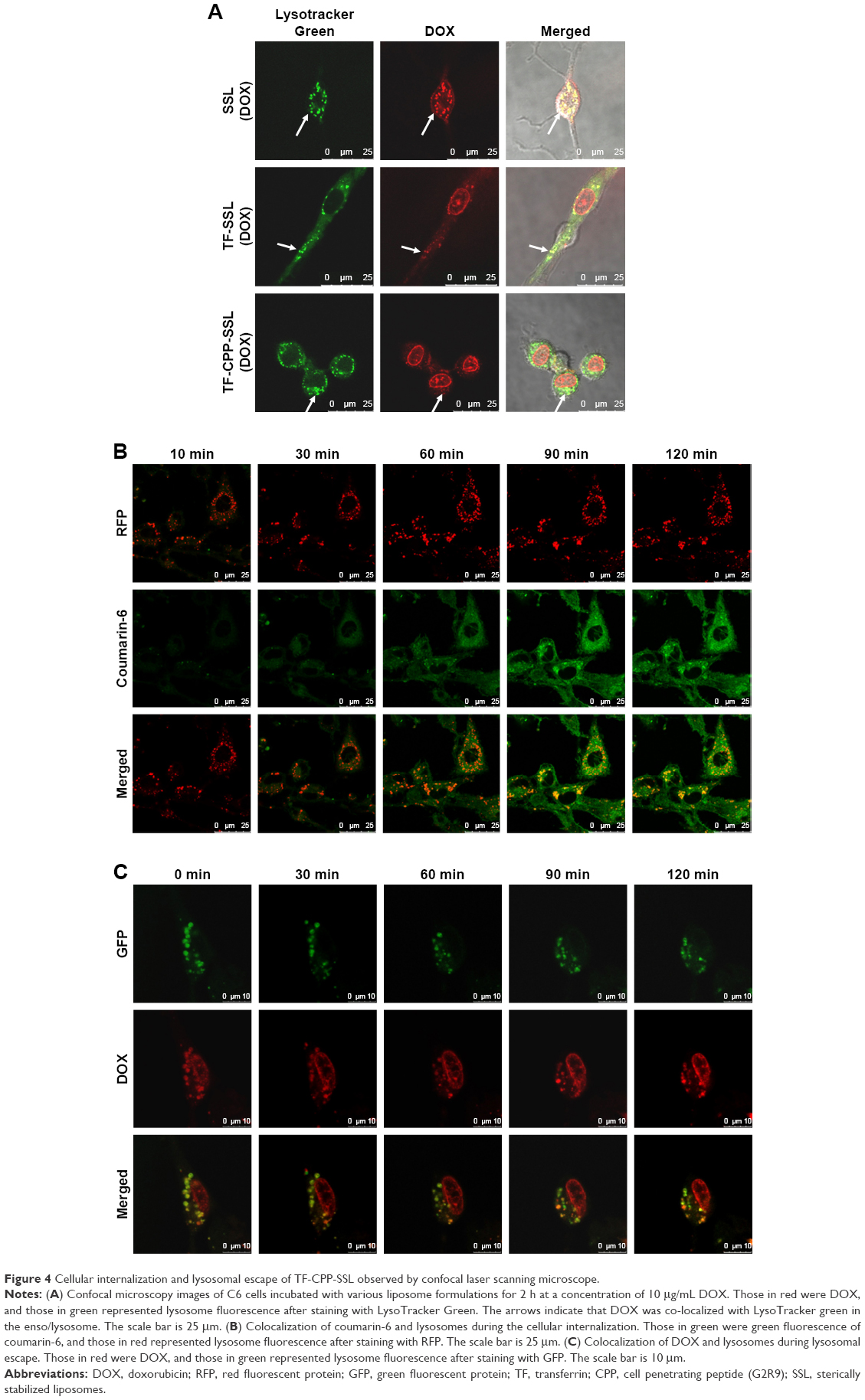 | Figure 4 Cellular internalization and lysosomal escape of TF-CPP-SSL observed by confocal laser scanning microscope. |
For further investigating the cellular internalization of TF-CPP-SSL, coumarin-6 with green fluorescence and lysosome-RFP were used to label TF-CPP-SSL and endosomes/lysosomes, respectively. As shown in Figure 4B, a little amount of TF-CPP-SSL (cou-6) appeared in the cytoplasm, and lysosomes within the initial 10 min and green fluorescent intensity of cou-6 gradually increased after 30 min, suggesting that it is a dynamic process for TF-CPP-SSL (cou-6) transporting into the cytoplasm. A large amount of red fluorescence accumulated in endosomes/lysosomes and the FI did not change for the duration of the experiments, while the liposomes gradually adsorbed on the cell membrane, preparing for the uptake process. As time passed, the green FI was augmented. With increased incubation time, the resulting yellow fluorescence demonstrated that the red fluorescence was overlaid with green fluorescence, which meant the liposome was trapped by the endosomes/lysosomes.
Real-time observations of endosome/lysosome escape in the TF-CPP-SSL experiments are shown in Figure 4C. DOX with red fluorescence and lysosome-GFP were used to label TF-CPP-SSL and lysosomes, respectively. It was evident that TF-CPP-SSL (DOX) color localized well with endosomes/lysosomes to exhibit bright yellow fluorescence at the initial time of escaping. At 30 min, as DOX began to escape from the lysosomes, the color in the lysosomes turned yellow-green, followed by more red fluorescence in the nuclei. Then, from 60 to 90 min, an increasing amount of DOX escaped from the lysosomes, and the color of the lysosomes became green again, while the red fluorescence of the nuclei enhanced, which demonstrated that a great portion of the DOX escaped from the endosomes/lysosomes and entered into the nuclei.
In vivo images of TF-CPP-SSL in nude mice bearing in situ glioma tumor
To evaluate the in vivo distribution of dual-modified liposomes in tissue, DiR-labeled SSL, TF-SSL, and TF-CPP-SSL were prepared for in vivo imaging on the in situ glioma-bearing BALB/c mouse model. As shown in Figure 5, the results indicated that all three liposomes could distribute to liver and spleen. SSL (DiR) and TF-SSL (DiR) had no distribution into the brain until 48 h after administration, whereas the distribution of TF-CPP-SSL (DiR) gradually increased in the brain 8 h after administration, indicating that the dual-modified liposome had strong efficacy in brain targeting.
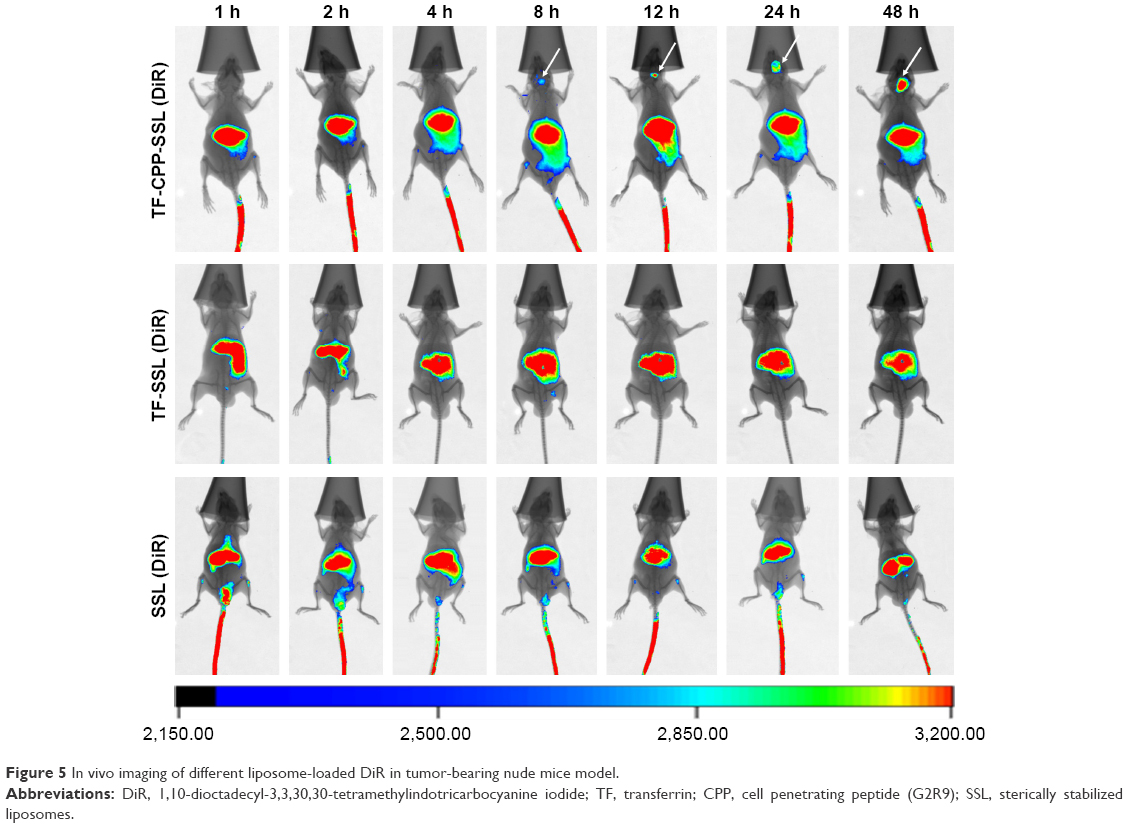 | Figure 5 In vivo imaging of different liposome-loaded DiR in tumor-bearing nude mice model. |
Discussion
The BBB presents a significant impediment, restricting the entry of most chemotherapeutic agents and proteins into the brain parenchyma.31 Based on the fact that both BMVECs and glioma C6 cells overexpress TF-Rs32 and are negatively charged by phospholipid phosphatidylserine,33 we designed a dual-ligand liposomal DDS, named TF-CPP-SSL, and anticipated that it would promote high-efficiency transit across the BBB and aid delivery of antitumor drugs to neoplastic tissues combining receptor mediation with adsorption mediation.
Rational construction of TF-CPP-SSL improves the efficiency of drug delivery to the target cells. Coupling ligands to liposomes by PEG derivatives is a major new strategy for the construction of actively targeted liposomes.12 Recent research shows that the conformation of PEG chains on the liposomal surface relies not only on the PEG molecular weight but also on the density of the PEG chains. Generally, PEGs with a higher coverage density or a larger molecular weight present a brush-like conformation and increased steric hindrance, resulting in more hindrance of the coupling interaction of the modified ligands with the liposomes with the corresponding receptors on the cell surface.34
In this study, we utilized the properties of PEG to construct dual-mediated liposomes, in which the chains and the molecular weight of PEG linked to TF or CPP were optimized to improve targeting efficacy. As shown in Figure 6A, when TF3.4K-CPP2K-SSL circulates in the blood, TF with a longer PEG chain could mask the nonspecificity of CPP. After arriving at the targeting site, longer PEG chains enhance TF binding with the TF-Rs. Simultaneously, flexible PEG linkers shorten during the interaction of TF ligands with their receptors, which brings CPP closer to the cell membrane to enhance cellular uptake through the electrostatic interactions. We call the behavior of TF3.4K-CPP2K-SSL at the targeting cell a “hand in hand” effect. The longer the PEG that is linked with CPP, the stronger the penetrating effect will be. When TF-R expressed on the cell surface was presaturated with free TF, the binding sites for TF-CPP-SSL were blocked. The cellular uptake of TF3.4K-CPPY-SSL was the same as that of SSL when Y was lower than 2K. This demonstrated that CPP moieties were “hidden” by the large volume of long PEG3.4K linker of TF. However, when Y increased to 3.4K, CPP exerted its penetrating efficiency because it could not be sterically shielded, and that led to a significant increase in normal cellular uptake for TF3.4K-CPP3.4K-SSL. Therefore, to achieve maximal efficacy at the target cells while avoiding normal cell capture, PEG3.4K and PEG2K were selected to link TF and CPP with liposomes for the construction of TF-CPP-SSL.
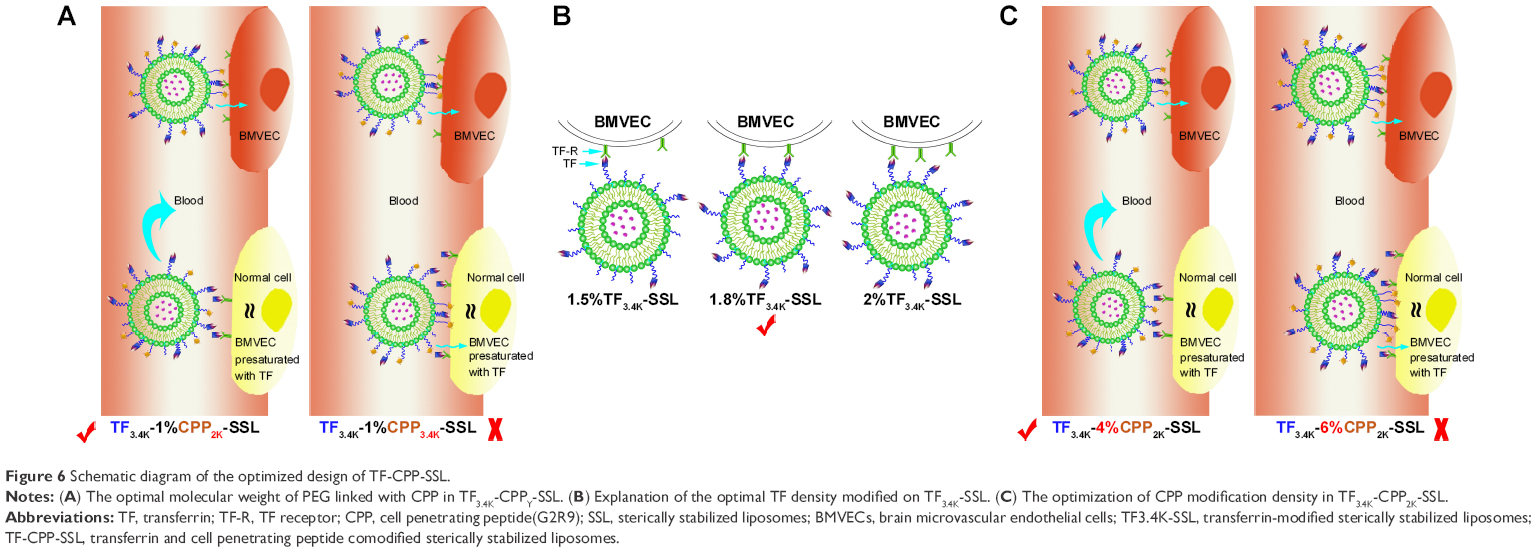 | Figure 6 Schematic diagram of the optimized design of TF-CPP-SSL. |
Although the distribution of receptor proteins is generally stable, it is now well established that extracellular ligands can induce the aggregation, or clustering, of receptors on the cell membrane.35 When the ligand-bearing liposomes closely approach the cell, the membrane proteins on the cell surface aggregate to cause receptor clustering. The optimal TF density modified on TF-SSL was selected as 1.8%, probably because TF bound well with TF-Rs for suitable binding positions, and thus contributed to the highest cellular uptake efficiency (Figure 6B). The same optimization results occurred in glioma C6 cells, further demonstrating that receptor protein clustering also occurred in this cell species.
The arginine-rich CPPs carried a high density of positive charge in physiological or slightly acidic environments, suggesting that it could activate cellular uptake through multisite binding and electrostatic attraction binding.36 The adsorptive interaction was generally direction independent and concentration independent. When TF-CPP-SSL arrived at the targeted cells, CPP with a higher density would enhance more cellular uptake based on the “hand in hand” effect (Figure 6C). When TF-R was presaturated with free TF, the CPP with a lower modified density could be shielded by PEG3.4K-TF (Figure 6C) but exert its penetrating efficiency again when the density exceeded 4% (Figure 6C). Hence, TF3.4K-CPP2K-SSL with 1.8% TF and 4% CPP only targeted cells such as BMVECs and C6 and did not interact with normal cells, resulting in maximum targeting uptake efficiency and long circulation in the blood.
The BBB is the first barrier preventing drugs entering the brain via the blood. Through an intricate experimental design, we proved that TF-CPP-SSL maintains not only the integrity of liposomes but also TF and CPP moieties on the liposomes after crossing the BBB. After these liposomes arrived at a glioma site, the intact dual-modified liposome could further promote glioma cellular uptake through receptor mediation and adsorption mediation. The dual-modified liposomes are initially endocytosed into clathrin-coated vesicles inside the tumor cells, and then, transferrin interacts with TFR and subsequently leaves the surface of the liposome after liposomes are sorted into early endosomes that mature into late endosomes and lysosomes,37–39 where CPP adsorbs more H+ for the lumen’s low pH and then promotes the lipid fusion between cationic CPP-H+ liposomes and anionic components in the lysosomal membrane via electronic attraction. Fusion pore is subsequently formed to release the liposomal cargoes into the cytosol.10 Entrapped constituents, such as DOX, gain opportunities to enter the nucleus and exert pharmacological effects. The transport process of TF-CPP-SSL loaded with DOX in vivo is illustrated in Figure 7.
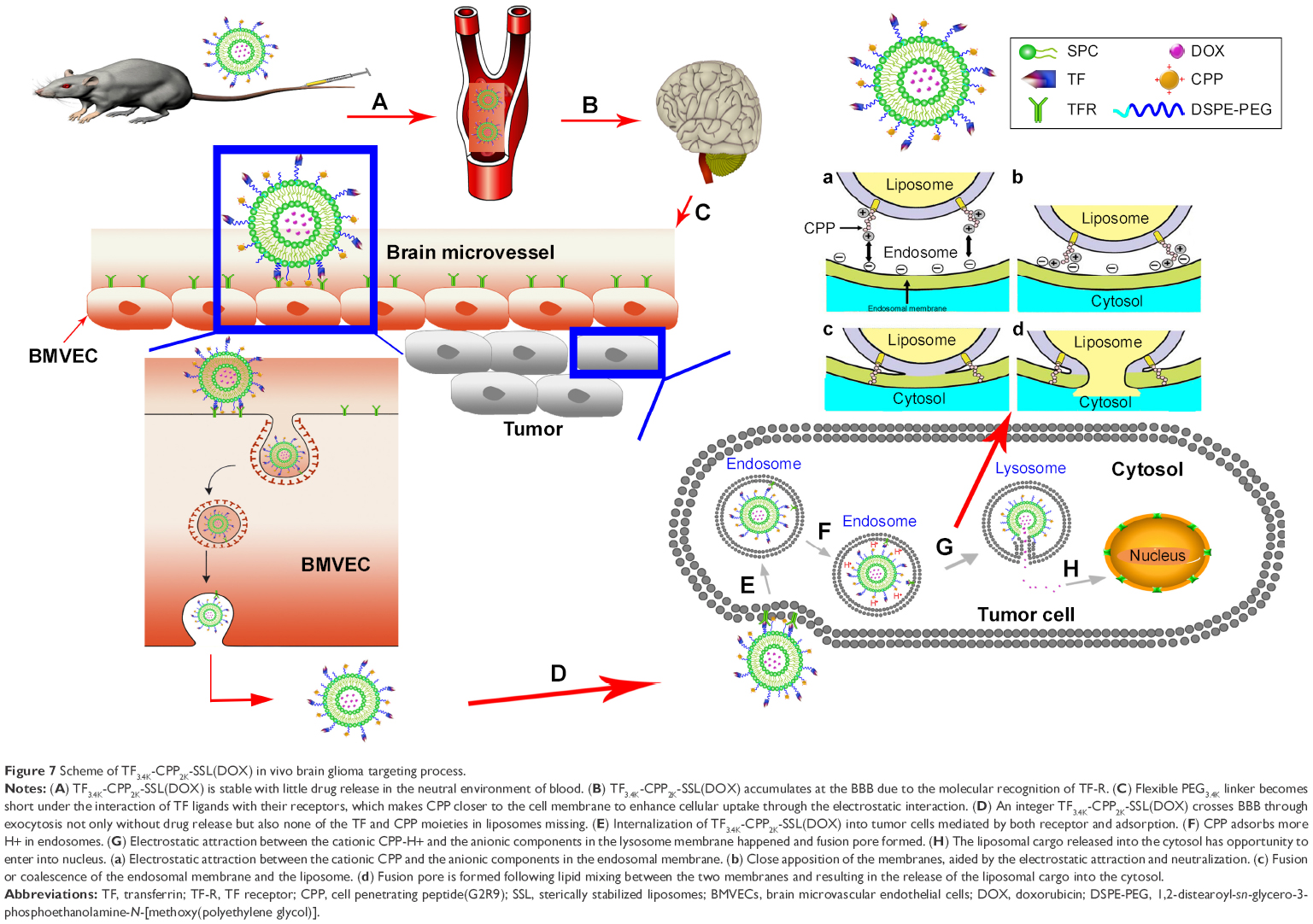 | Figure 7 Scheme of TF3.4K-CPP2K-SSL(DOX) in vivo brain glioma targeting process. |
As described herein, we rationally constructed the dual-mediated liposomes to improve the penetrating ability of vectors across the BBB and enhance effective treatment of glioma cells. The TF-CPP-SSL DDS demonstrated the efficacy of liposomes crossing the BBB, prolonged circulation in vivo, and specific tumor targeting, as well as high cell penetrating and lysosomal escape ability, which accomplished our goal of targeting subcellular compartments and identifying potential antitumor drug carriers for lesions in the CNS.
Conclusion
We constructed dual-mediated liposome TF-CPP-SSL by conjugating TF and CPP moieties to the liposomes via PEG3.4K and PEG2K, respectively, and found the optimum densities of TF and CPP were 1.8% and 4%, respectively. Such TF-CPP-SSL DDSs demonstrated the efficacy of liposome penetration of the BBB, long-term blood circulation in vivo, and specific tumor targeting, as well as high cell penetration and intracellular lysosomal escape capability, which accomplished our goal of targeting subcellular compartments and identifying potential antitumor drug carriers for neoplasms in the CNS.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant No 81202469 and No 91330103) and the National Key Research and Development Program of China (No 2016YFC0103605 and No 2016YFC0103600).
Disclosure
The authors report no conflicts of interest in this work.
References
Colman H, Aldape K. Molecular predictors in glioblastoma: toward personalized therapy. Arch Neurol. 2008;65(7):877–883. | ||
Langner M, Kral TE. Liposome-based drug delivery systems. Pol J Pharmacol. 1999;51(3):211–222. | ||
Smith MW, Gumbleton M. Endocytosis at the blood-brain barrier: from basic understanding to drug delivery strategies. J Drug Target. 2006;14(4):191–214. | ||
Ying X, Wen H, Lu WL, et al. Dual-targeting daunorubicin liposomes improve the therapeutic efficacy of brain glioma in animals. J Control Release. 2010;141(2):183–192. | ||
Du J, Lu WL, Ying X, et al. Dual-targeting topotecan liposomes modified with tamoxifen and wheat germ agglutinin significantly improve drug transport across the blood-brain barrier and survival of brain tumor-bearing animals. Mol Pharm. 2009;6(3):905–917. | ||
Varkouhi AK, Scholte M, Storm G, Haisma HJ. Endosomal escape pathways for delivery of biologicals. J Control Release. 2011;151(3):220–228. | ||
Frankel AD, Pabo CO. Cellular uptake of the tat protein from human immunodeficiency virus. Cell. 1988;55(6):1189–1193. | ||
Fang J, Nakamura H, Maeda H. The EPR effect: unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev. 2011;63(3):136–151. | ||
Koren E, Torchilin VP. Cell-penetrating peptides: breaking through to the other side. Trends Mol Med. 2012;18(7):385–393. | ||
El-Sayed A, Futaki S, Harashima H. Delivery of macromolecules using arginine-rich cell-penetrating peptides: ways to overcome endosomal entrapment. AAPS J. 2009;11(1):13–22. | ||
Dietz GP, Bähr M. Delivery of bioactive molecules into the cell: the Trojan horse approach. Mol Cell Neurosci. 2004;27(2):85–131. | ||
Zalipsky S. Chemistry of polyethylene glycol conjugates with biologically active molecules. Adv Drug Deliv Rev. 1995;16(2–3):157–182. | ||
Almeda D, Wang B, Auguste DT. Minimizing antibody surface density on liposomes while sustaining cytokine-activated EC targeting. Biomaterials. 2015;41:37–44. | ||
Agarwal S, Manchanda P, Vogelbaum MA, Ohlfest JR, Elmquist WF. Function of the blood-brain barrier and restriction of drug delivery to invasive glioma cells: findings in an orthotopic rat xenograft model of glioma. Drug Metab Dispos. 2013;41(1):33–39. | ||
Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nat Rev Cancer. 2006;6(8):583–592. | ||
Silvius JR. Fluorescence energy transfer reveals microdomain formation at physiological temperatures in lipid mixtures modeling the outer leaflet of the plasma membrane. Biophys J. 2003;85(2):1034–1045. | ||
Gao JQ, Lv Q, Li LM, et al. Glioma targeting and blood–brain barrier penetration by dual-targeting doxorubincin liposomes. Biomaterials. 2013;34(22):5628–5639. | ||
Szoka F Jr, Papahadjopoulos D. Procedure for preparation of liposomes with large internal aqueous space and high capture by reverse-phase evaporation. Proc Natl Acad Sci U S A. 1978;75(9):4194–4198. | ||
Sharma V, Anandhakumar S, Sasidharan M. Self-degrading niosomes for encapsulation of hydrophilic and hydrophobic drugs: an efficient carrier for cancer multi-drug delivery. Mater Sci Eng C Mater Biol Appl. 2015;56:393–400. | ||
Barenholz Y. Doxil® – the first FDA-approved nano-drug: lessons learned. J Control Release. 2012;160(2):117–134. | ||
Kolate A, Baradia D, Patil S, Vhora I, Kore G, Misra A. PEG – a versatile conjugating ligand for drugs and drug delivery systems. J Control Release. 2014;192:67–81. | ||
Rabanel JM, Hildgen P, Banquy X. Assessment of PEG on polymeric particles surface, a key step in drug carrier translation. J Control Release. 2014;185:71–87. | ||
Cheng L, Huang FZ, Cheng LF, et al. GE11-modified liposomes for non-small cell lung cancer targeting: preparation, ex vitro and in vivo evaluation. Int J Nanomedicine. 2014;9(1):921–935. | ||
Lv Q, Li LM, Han M, et al. Characteristics of sequential targeting of brain glioma for transferrin-modified cisplatin liposome. Int J Pharm. 2013;444(1–2):1–9. | ||
Dipali SR, Kulkarni SB, Betageri GV. Comparative study of separation of non-encapsulated drug from unilamellar liposomes by various methods. J Pharm Pharmacol. 1996;48(11):1112–1115. | ||
Jin Y, Liang X, An Y, Dai Z. Microwave-triggered smart drug release from liposomes co-encapsulating doxorubicin and salt for local combined hyperthermia and chemotherapy of cancer. Bioconjug Chem. 2016;27(12):2931–2942. | ||
Xie Y, Ye L, Zhang X, et al. Transport of nerve growth factor encapsulated into liposomes across the blood–brain barrier: in vitro and in vivo studies. J Control Release. 2005;105(1–2):106–119. | ||
Zou P, Chen H, Paholak HJ, Sun D. Noninvasive fluorescence resonance energy transfer imaging of in vivo premature drug release from polymeric nanoparticles. Mol Pharm. 2013;10(11):4185–4194. | ||
Zong T, Mei L, Gao H, et al. Synergistic dual-ligand doxorubicin liposomes improve targeting and therapeutic efficacy of brain glioma in animals. Mol Pharm. 2014;11(7):2346–2357. | ||
Liu C, Luo Q, Tu Y, Wang G, Liu Y, Xie Y. Drug-carrier interaction analysis in the cell penetrating peptide-modified liposomes for doxorubicin loading. J Microencapsul. 2015;32(8):745–754. | ||
Zhang CX, Zhao WY, Liu L, et al. A nanostructure of functional targeting epirubicin liposomes dually modified with aminophenyl glucose and cyclic pentapeptide used for brain glioblastoma treatment. Oncotarget. 2015;6(32):32681–32700. | ||
Jefferies WA, Brandon MR, Hunt SV, Williams AF, Gatter KC, Mason DY. Transferrin receptor on endothelium of brain capillaries. Nature. 1984;312(5990):162–163. | ||
Yeung T, Gilbert GE, Shi J, Silvius J, Kapus A, Grinstein S. Membrane phosphatidylserine regulates surface charge and protein localization. Science. 2008;319(5860):210–213. | ||
Koren E, Apte A, Jani A, Torchilin VP. Multifunctional PEGylated 2C5-immunoliposomes containing pH-sensitive bonds and TAT peptide for enhanced tumor cell internalization and cytotoxicity. J Control Release. 2012;160(2):264–273. | ||
Bray D, Levin MD, Morton-Firth CJ. Receptor clustering as a cellular mechanism to control sensitivity. Nature. 1998;393(6680):85–88. | ||
Trabulo S, Cardoso AL, Mano M, De Lima MC. Cell-penetrating peptides – mechanisms of cellular uptake and generation of delivery systems. Pharmaceuticals (Basel). 2010;3(4):961–993. | ||
Lakadamyali M, Rust MJ, Zhuang X. Ligands for clathrin-mediated endocytosis are differentially sorted into distinct populations of early endosomes. Cell. 2006;124(5):997–1009. | ||
Bleil JD, Bretscher MS. Transferrin receptor and its recycling in HeLa cells. EMBO J. 1982;1(3):351–355. | ||
Hopkins CR, Trowbridge IS. Internalization and processing of transferrin and the transferrin receptor in human carcinoma A431 cells. J Cell Biol. 1983;97(2):508–521. |
Supplementary materials
Synthesis and characterization of Thiolated transferrin (SH-TF)
Synthesis of SH-TF
The synthetic route of SH-TF was shown as following (Figure S1).1 Briefly, sulfhydryl groups were introduced to the transferrin (TF) by the reaction of TF and Traut’s reagent (2-iminothiolane hydrochloride, ITH; BioVision, San Francisco, CA, USA) in Tris-HCl buffer (pH 8.0) at the molar ratio of 1:20 for 2 h, and unreacted reactant was removed by ultrafilter tube the cutoff molecular weight of which was 10 kDa under centrifugation of 8,000 rpm for 10 min at 4°C.
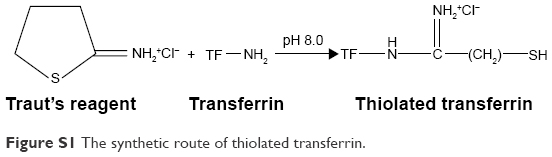 | Figure S1 The synthetic route of thiolated transferrin. |
Quantitative determination of sulfhydryl groups number on SH-TF
5,5′-Dithio-bis-(2-nitrobenzoic acid) (DTNB) has been widely applied on protein research due to its specificity for SH groups (Figure S2), high molar extinction coefficient (14,150 M−1cm−1 at 412 nm) and short reaction time (10 min).
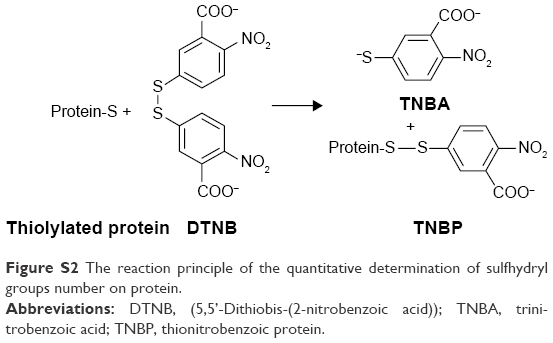 | Figure S2 The reaction principle of the quantitative determination of sulfhydryl groups number on protein. |
Seven samples with different concentrations (0.40, 0.20, 0.10, 0.05, 0.025, 0.0125, 0.00625 mmol/L−1) of Traut’s reagent were dissolved in Tris-HCl buffer (pH 8.0) to prepare standard solution samples. A total of 5 mL of DTNB (1.0 mmol/L−1) was then incubated with those solution samples with different concentrations at 25°C for 10 min. The absorbance of each sample was measured at 412 nm while DTNB solution was applied as blank control. The standard curve was A =1.7508 c (mmol/L−1) −0.0241 (R2=1).
Meanwhile, the concentration of transferrin was measured by BCA kit. After incubating 200 μL Bradford regent (Beijing CellChip Biotechnology Co., Ltd, Beijing, China)with 20 μL of standard transferrin solution (5.13, 2.56, 1.28, 0.64, 0.32, 0.16, 0.08 nmol/mL) for 30 min at room temperature, the samples were measured at 580 nm by spectrophotometer (Thermo Fisher, Waltham, MA, USA) with three replications, with Bradford reagent served as blank control. The standard curve was A =0.0522 c (nmolL−1) −0.0068 (R2=0.9995).
After 20 μL of synthesized SH-TF was reacted with DTNB and BCA kit, respectively, their absorbance was measured at 412 and 580 nm. As the total amount of sulfhydryl groups and the concentration of TF can be addressed according to standard curve, the result indicated that there are eight sulfhydryl groups coupled to TF in SH-TF.
Synthesis and identification of 1,2-distearoyl-sn-glycerol-3-phosphoethanolamine-N-[poly(ethylene glycol)-Y] (DSPE-PEG(Y))-cell penetrating peptide (CPP)
DSPE-PEG(Y)-CPP (Y =1K, 2K, 3K) were synthesized and purified according to the established procedure with some modifications.2 Briefly, the purchased DSPE-PEG(Y)-hydroxy succinamide (NHS; Y =1K, 2K, 3K) were reacted with twofold molar excess of CPP (amino acid sequence: G2R9) in the solvent of DMF, respectively, containing threefold molar excess of triethylamine in darkness at room temperature with DMAP as a catalyzer. After 48 h stirring, the reaction was traced by Thin-Layer Chromatography until DSPE-PEGx-NHS was fully consumed. As the organic solvent and excessive unreacted reactant or catalyzer was separated from the product using a cellulose ester membrane filter (MWCO, 3,500 Da), the products were identified by Matrix-Assisted Laser Desorption/ Ionization Time of Flight Mass Spectrometry (Figure S3) after being freeze-dried.
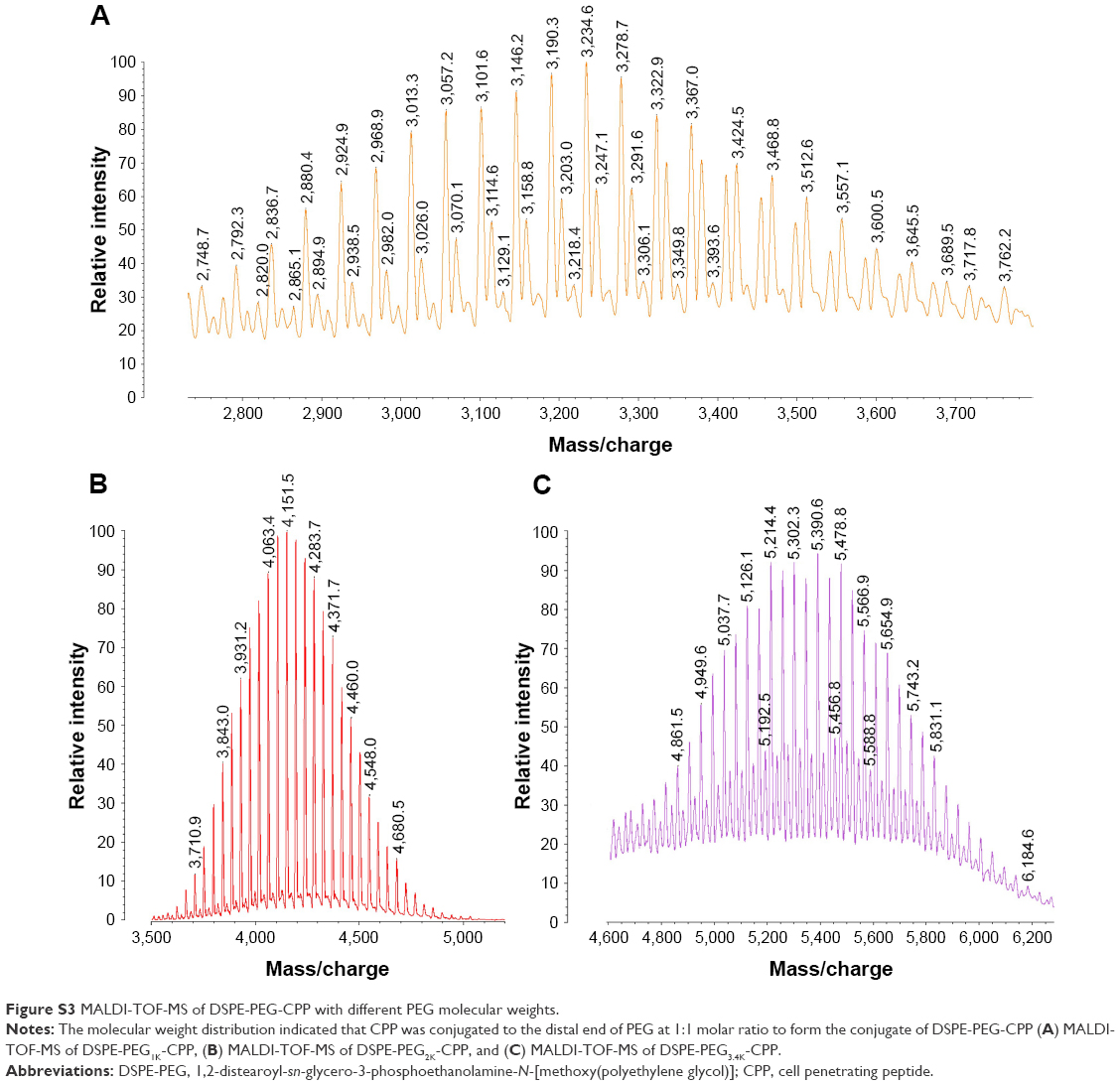 | Figure S3 MALDI-TOF-MS of DSPE-PEG-CPP with different PEG molecular weights. |
Characterization of different liposome-loaded drugs
The size and zeta potential did not change significantly after the liposome-encapsulated drugs such as coumarin-6, DOX, coumarin-6, and 1,10-dioctadecyl-3,3,30,30-tetramethylindotricarbocyanine iodide (DiR). The encapsulated efficiency % (ee%) of liposomes exceeded 90% because of the hydrophobic features of the fluorescent probes or active loading of DOX driven by the pH gradient (Table S1).
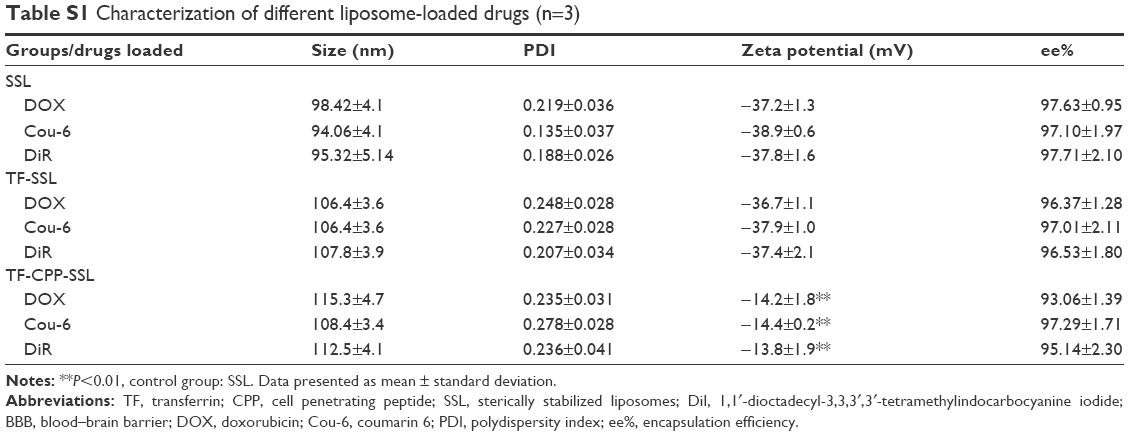 | Table S1 Characterization of different liposome-loaded drugs (n=3) |
Permeability of liposome-loaded 3,3′-dioctadecyloxacarbocyanine perchlorate (DiO), 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine iodide (DiI), or DiO-DiI crossing the blood–brain barrier (BBB)
Fluorescence resonance energy transfer (FRET) techniques were used to further confirm the integrity of liposomal lipid bilayers after crossing the BBB. FRET donor dye (DiO) and acceptor dye (DiI) were individually or simultaneously encapsulated into liposomes to produce TF-CPP-sterically stabilized liposome (SSL; DiO), TF-CPP-SSL (DiI), or TF-CPP-SSL (DiO-DiI). The permeability TF-CPP-SSL on in vitro BBB models was measured as before.3 Each liposomal formulation mentioned earlier at the concentration of 0.2 mgmL−1 soybean phosphatidylcholine (SPC) was added to the donor chamber. Samples extracted from the acceptor chamber at 0.5, 1.0, 2.0, 4.0, and 6.0 h were analyzed by ELISA (Tecan Infinite 200, Switzerland) at λex =466 nm, λem =504 nm, and volume was replaced by 200 μL of fresh DMEM medium without phenolsulfonphthalein. Cells were kept under culture conditions (37°C, 5% CO2, and saturated humidified atmosphere) during the transportation experiment. The permeability efficiency (Pe%) was calculated according to the following equation, respectively:
|
|
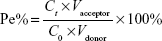
where Ct and C0 are the concentrations of the drug in the acceptor chamber at a given time and the original concentration of the drug in the donor chamber, respectively. They were calculated from the standard equation between fluorescence intensity and the concentration of liposome-loaded DiO/DiI/DiO-DiI (Figure S4A). Vacceptor and Vdonor are 2.0 and 1.0 mL, respectively, so that the liquid inside and outside of the Transwell was at the same level to avoid hydrostatic pressure.
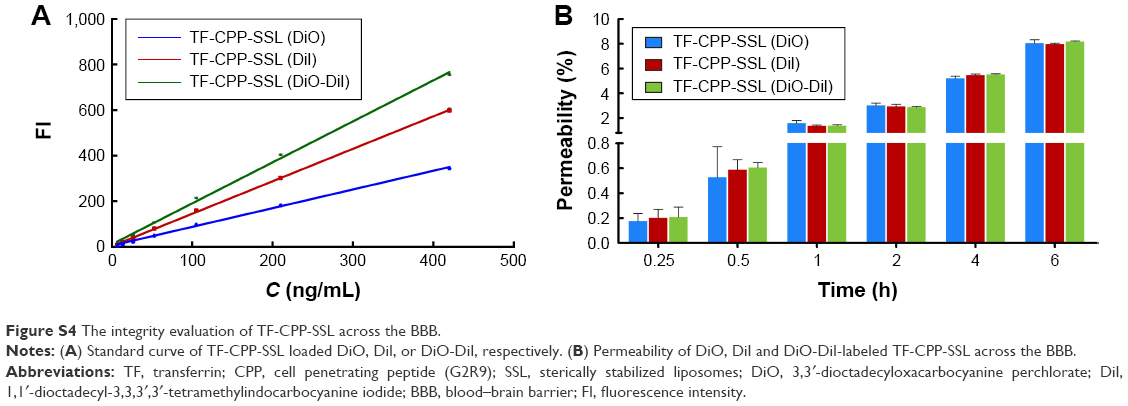 | Figure S4 The integrity evaluation of TF-CPP-SSL across the BBB. |
The permeability (Pe%) of TF-CPP-SSL (DiO), TF-CPP-SSL (DiI), and TF-CPP-SSL (DiI-DiO), calculated from the corresponding standard equation, was almost the same (P>0.05; Figure S4B), which indicates that the permeability of TF-CPP-SSL was independent of drugs.
References
Xu S, Liu Y, Tai HC, Zhu J, Ding H, Lee RJ. Synthesis of transferrin (Tf) conjugated liposomes via Staudinger ligation. Int J Pharm. 2011;404(1–2):205–210. | ||
Ding Y, Sun D, Wang GL, et al. An efficient PEGylated liposomal nanocarrier containing cell-penetrating peptide and pH-sensitive hydrazone bond for enhancing tumor-targeted drug delivery. Int J Nanomedicine. 2015;10:6199–6214. | ||
Xie Y, Ye L, Zhang X, et al. Transport of nerve growth factor encapsulated into liposomes across the blood–brain barrier: In vitro and in vivo studies. J Control Release. 2005;105(1–2):106–119. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.