")
Back to Journals » OncoTargets and Therapy » Volume 14
A Concise Review: The Role of Stem Cells in Cancer Progression and Therapy
Authors Hayat H, Hayat H, Dwan BF , Gudi M, Bishop JO, Wang P
Received 9 January 2021
Accepted for publication 23 March 2021
Published 20 April 2021 Volume 2021:14 Pages 2761—2772
DOI https://doi.org/10.2147/OTT.S260391
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Federico Perche
Hasaan Hayat,1,2 Hanaan Hayat,1– 3 Bennett Francis Dwan,1,4 Mithil Gudi,1,4 Jack Owen Bishop,1,4 Ping Wang1,5
1Precision Health Program, Department of Radiology, College of Human Medicine, Michigan State University, East Lansing, MI, USA; 2Lyman Briggs College, Michigan State University, East Lansing, MI, USA; 3Institute for Quantitative Health Science and Engineering, Michigan State University, East Lansing, MI, USA; 4College of Natural Science, Michigan State University, East Lansing, MI, USA; 5Department of Radiology, College of Human Medicine, Michigan State University, East Lansing, MI, USA
Correspondence: Ping Wang
Precision Health Program, Department of Radiology, Michigan State University, 766 Service Road, Rm. 2020, East Lansing, MI, 48823, USA
Tel +517-353-3817
Email [email protected]
Abstract: The properties of cancer stem cells (CSCs) have recently gained attention as an avenue of intervention for cancer therapy. In this review, we highlight some of the key roles of CSCs in altering the cellular microenvironment in favor of cancer progression. We also report on various studies in this field which focus on transformative properties of CSCs and their influence on surrounding cells or targets through the release of cellular cargo in the form of extracellular vesicles. The findings from these studies encourage the development of novel interventional therapies that can target and prevent cancer through efficient, more effective methods. These methods include targeting immunosuppressive proteins and biomarkers, promoting immunization against tumors, exosome-mediated CSC conversion, and a focus on the quiescent properties of CSCs and their role in cancer progression. The resulting therapeutic benefit and transformative potential of these novel approaches to stem cell-based cancer therapy provide a new direction in cancer treatment, which can focus on nanoscale, molecular properties of the cellular microenvironment and establish a more precision medicine-oriented paradigm of treatment.
Keywords: stem cell therapy, cancer, exosomes, biomarkers, molecular imaging
Introduction
Conventionally, cancer therapy has relied on various pharmacological and radiation-based interventions, often through means of chemotherapy and radiotherapy.1–3 Current challenges in the clinical success of cancer therapy result from limitations in the interventional mechanisms themselves. Often, this is due to patient incompatibility with treatment, a unique disease phenotype or rapid drug resistance.4 This results in low rates of patient remission and greater rates of mortality.4 A new and developing area of research has opened up the realm of cancer therapy through a deeper focus on a novel interventional paradigm for cancer: stem cell therapy. Although stem cell therapy has remained an ongoing area of research with many new developments in cell-based therapies (CBT) for different diseases including autoimmune disorders and regenerative medicine, the molecular relationship between cancer stem cells (CSCs) and cancer pathogenesis has now grown into a budding realm of interest.5 This is due to various studies that have highlighted the critical role of CSCs in promoting a tumorigenic environment.6 A greater focus on researching the role of stem cells including CSCs in cancer progression and development will permit the creation of novel therapies and technologies that can target cancers at earlier stages of pathogenesis. This can also allow for long-term resolutions to many cancers because the highly transformative properties of stem cells can be repurposed for targeting cancer cells through genetic or phenotypic alteration, a mechanism which is in contrast to short-term remedies like chemotherapy.7 The current reliance on radiation therapy for cancer and the use of chemo drugs which impact healthy, endogenous cellular functions results in greater, more lethal side effects.8,9 To circumnavigate this issue, a focus on genetic and molecular therapies that do not impact normal, healthy cell function can support the development of longitudinal therapies for cancer with reduced side effects and morbidity rates. This approach is made feasible through studying the interactions amongst components of the tumor microenvironment, particularly with a focus on stem cell interactions and cell-conversion in cancer pathogenesis. Hence, due to its potential to transform the current narrative in the approach to cancer therapy, here we explore the current state of the role of stem cells in cancer progression and therapy. Particularly, we summarize numerous studies that explore potential areas of focus when targeting stem cells for cancer therapy, including targeting markers on CSCs, stem cells for immunization against tumors, using stem cells as carriers of therapeutic cargo, quiescent properties of stem cells, and targeting exosomes to prevent (Epithelial Mesenchymal Transition) EMT and metastasis.
Targeting Cell-Surface and Intracellular Markers of Cancer Stem Cells (CSCs) for Cancer Therapy
Primary cancer cells have been found to derive from stem cells, with a subsection of these cells named “cancer stem cells” (CSC’s). CSCs replicate similar characteristics to regular stem cells, such as the ability to proliferate in their microenvironments.10 CSCs sustain the cancer by promoting proliferation, and therefore must be targeted when attempting to eliminate cancer for successful and long-lasting results.11 As do most healthy cells and hematopoietic stem cells, pancreatic, liver, and lung CSCs overexpress CD-47 on their cell surface, which is an immunosuppressive defense signal that inhibits attacks from macrophages on cancer cells.12 Normally, CD-47 plays a positive role of defense in protecting healthy cells in organs and tissue from harmful attack by macrophages.13,14 Similarly, in CSC’s, the CD-47 protein acts as an immune checkpoint blockade for any targeted attempts to diminish the CSC’s by activated macrophages in the tumor microenvironment.15,16 CD-47 elicits its effects via interaction with signal regulatory protein alpha (SIRPα) to inhibit phagocytosis of normal cells, which suppresses the phagocytic activity of immune cells, particularly macrophage phagocytosis.17 CD-47 is overexpressed in many human malignancies.18 One study performed simultaneous silencing of CD-47 and PD-L1 in order to enhance immunotherapy against circulating tumor cells.19 Inhibiting PD-L1 allowed immune cells to locate tumor cells more adequately, and blockade of CD-47 permitted macrophage-mediated destruction of the tumor cells. In vitro flow cytometry confirmed overexpression of CD-47 and PD-L1 in the tumor cell line.19 Compared to the blank controls or single-antibody group, dual inhibition of these immunosuppressive proteins resulted in a more potent reduction of solid tumors in mice.
In order to target CD-47 and silence its downstream effects, various forms of pharmacological and nanomedicine-based approaches have been established. An antibody named Hu5F9-G4 that targets CD-47, allowing macrophages to destroy the cancer cells, has been developed. Another similar antibody, Rituximab, which has been known to positively amplify destruction signals inhibited by CD-47, is highly active and is well tolerated as first-line single-agent therapy for indolent non-Hodgkin lymphoma (NHL).20 Using the application of both antibodies, Hu5F9-G4 and Rituximab, the results of a clinically evaluated study on the treatment outcome in patients of NHL concluded that at least 50% of the test subjects had eliminated most symptoms of cancer.21 The authors did not report directly on tumor size, but clinical evaluation of response to treatment indicated that more than 60% of patients had complete or partial response to the drug, a metric that relates to change in tumor size. PET-CT of patients indicated partial to complete remission of the lymphoma in male and female adult subjects.21 This method of targeting CD-47 protein on Cancer Stem Cells has been shown to result in no detectable side effects in human beings, and thus may pave new routes for immunotherapy towards many forms of cancer by targeting cancer stem cells.22 This is because in the past, various CSCs in cancers such as pancreatic, lung, and breast cancers have been proven to express CD-47.23–25 Hence, targeting this immune blockade molecule expressed on CSCs may provide a new avenue of cancer treatment.
However, current limitations to therapies targeting CD47 result from its presence on an abundance of normal, healthy cells. Thus, the attack on host cells as a side effect of treatment is a potential roadblock in CD47-based therapy. To circumnavigate this issue, several CSC relevant markers have been elucidated in efforts to target CSCs specifically without damaging native cells. CD44 and CD133 are widely used and accepted as relevant cell-surface markers for CSCs.26–30 CD44 has implications in being a CSC marker for breast, pancreatic, and head and neck cancers.28 The enrichment of CD44 cells after drug treatment indicates higher rates of proliferation and a greater resistance to drug-induced death, helping dictate CD44 as a negative prognostic factor.31 CD133 holds implications in acting as a CSC marker for brain, colon, and prostate cancers.32 CD133 has been shown to upregulate the FLIP (FLICE-like inhibitory protein) which aids in CSC resistance to apoptosis.32 CD133 is also a vastly recognized chemo-resistant CSC surface antigen, thus aiding in the prognosis of treatment. By inhibiting CD133, the cell’s proliferation pathways are also inhibited. A larger presence of CD133 on the tumor may require alternative chemotherapeutic agents, or a different treatment.32
The benefit of identifying CSC-surface molecules lies in their potential to act as targets for new cytotoxic therapies,33 often those which are mediated by neutralizing antibodies, in combination with CSC-directed therapy. Several studies have recently explored this form of combined therapy which prevents the repeated formation of tumors and inhibits recurrent population of CSCs. One such study evaluated the effect of a combined therapy on drug-resistant triple-negative breast cancer (TNBC) cells by targeting the TGF-β of the CSC.34 Particularly, the group demonstrated that the TNBC CSC population maintained a unique ability to upregulate IL-8 in response to TGF- β signaling following chemotherapy with Paclitaxel (a mechanism which contributes to drug resistance). Using a TGF- β type 1 receptor kinase inhibitor, a TGF- β type II receptor neutralizing antibody, and SMAD-4 siRNA as forms of combined therapy with Paclitaxel, the subsequent recruitment of IL-8 following chemotherapy was blocked and the expansion of the chemotherapy-resistant CSC populations was inhibited. This study shows the ability of combined therapy to both inhibit primary mammosphere and further prevent drug resistance in the CSC population of the TNBC population. Besides above-mentioned markers, Aldehyde dehydrogenase 1 (ALDH1) has also been reported as a marker of cancer stem cells could be targeted for molecular therapy.35,36 Table 1 provides a list of common cell-surface and intracellular markers pertaining more specifically to CSCs.
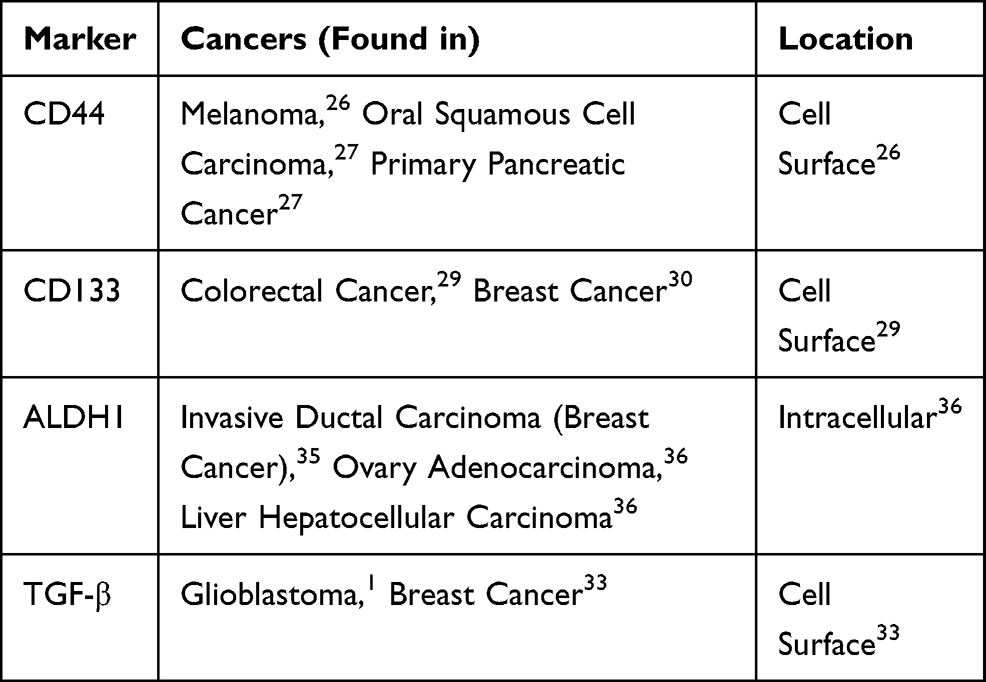 |
Table 1 A List of Common Cell-Surface and Intracellular Markers Pertaining to CSCs |
Stem Cells for Immunization Against Tumors
At the beginning of the 20th century, Frederick Schöne noted that fetal tissue vaccination could suppress transplanted tumor growth in mice.37 However, it took many more years for other groups to further investigate the potential of this discovery. In the 1960s and 70s, research in this area resumed, and investigators reported mice immunized against embryonic material could prevent tumor growth, priming their bodies to recognize and fight cancer cells. However, these results tended to be weak and hard to reproduce.38–40 Furthermore, ethical concerns and technological limitations during this time period made further research in humans impossible. With recent progress involving embryonic cell lines, research into this area has been revisited. These include studies that found very similar RNA transcript profiles and surface antigen expression between embryonic cells and different cancer cell lines, including pancreatic cancer, prostate cancer, breast cancer, myeloid leukemia, and glioblastoma.41,42 Furthermore, ES and cancer cells have both been shown to exhibit similar markers of stemness, particularly when these cancer cells are less differentiated, or more immature.42 In this study by Ben-Porath et al, poorly differentiated breast tumors were shown to display an ESC-like expression signature, more so than further differentiated tumors. As ESCs are known to exhibit stemness, this shows the possibility that more immature tumors (less differentiated) may exhibit higher tumor stemness than mature tumors. On this point of stemness, stemness is usually described as the ability for stem cells to balance between a few different processes: proliferation, quiescence, regeneration, and differentiation.43 Stem cells rely on interactions with and signals within their microenvironment to determine which of these processes to undergo.44 Cancer cells exhibit stemness as well, and can use this stemness to survive stress and treatment, and preserve their lineage.43 Discovering the similarity in gene and surface antigen expression between embryonic stem cells (ESCs) and cancer cells, we now understood why these cells could potentially be used to as an anti-tumor vaccination. Moreover, since the discovery of induced pluripotent stem cells, groups have shown that the transcriptomes and antigens of ESCs and iPSCs are almost identical,45 and that induced pluripotent stem cells (iPSCs) also show potential as an immunization agent. In fact, iPSC is most likely advantageous compared to ESCs for this purpose, as using iPSCs from a specific patient would be more representative of the patient’s own immunogens.46 Furthermore, a theoretically unlimited number of iPSCs can be generated from each patient, given enough time. In 2018, a study used tumor-specific antigens and tumor-associated antigens expressing iPSCs to prime the immune systems of mice, followed by the transplantation of different tumors, including melanoma and breast cancer.47 Significant regression of these tumors was found when compared to the control group. This result was attributed to the upregulation in mature antigen-presenting cells in the lymph nodes, which led to an increase in helper and cytotoxic T-cells. This group then isolated T-cells and tumor-experienced lymphocytes (TELs) from mice with tumor and that had received vaccination and transferred them to mice with tumor that had not received vaccination. Both of these groups experienced tumor regression. However, it is important to note that this iPSC vaccination was only able to slow or prevent the growth of tumors that were transplanted after vaccination; it was not effective in preventing the growth of tumors that have already been established in vivo. This continues to be the shortcoming of using stem cells as a therapeutic treatment; their effect is diminished when tumor transplant and growth takes place before the vaccination. For this reason, they tend to provide better results when used as a prophylactic treatment, as opposed to a therapeutic treatment after disease onset. The safe application of ESC and iPSC–based technologies requires the use of methods of iPSCs production and their directed differentiation which minimize both the possibility of mutations in cell genomes under in vitro culturing and the probability of malignant transformation of the injected cells.48 Some have suggested that vaccinations including CSC lysates would improve outcomes, such as a vaccination that Lin et al formulated.49,50 They combined CSC dendritic cells, which present tumor-associated antigens to T cells, with melanoma and carcinoma tumor models, which showed promise in increasing protective immunity against tumor cell challenge. However, as pointed out in a recent review by Chu et al, the isolation of enough number of CSCs (in this case, CSC-DCs) from tumor tissues is very challenging, which poses difficulties in access and quantity for possible future study in larger animal models, or in the clinic.51 As discussed earlier, iPSCs can be made in large quantities and from each individual patient, but they do not provide adequate tumor suppression when tumor has been established. Combination vaccines with CSCs and tumor cells show promise but provide isolation and collection challenges. A very recent study, published in 2020, provides improvement on cancer vaccines for tumor rejection.52 In this study, this group enriched a whole-cell melanoma vaccine with stem cells (this vaccine also contained a molecular adjuvant, cytokine Hyper-IL6). One vaccine was enriched with melanoma stem-like cells from B16F10 melanospheres, while the other vaccine contained mouse-induced pluripotent stem cells (miPSCs). While both vaccines showed impressive reductions in tumor growth, and in disease-free and overall survival of the immunized mice, the most effective vaccine was the one containing miPSCs. This study provides exciting evidence that vaccines containing iPSCs with tumor cells can be just as effective if not more effective than those containing the harder to obtain tumor stem cells. However, once again, this study was only done with immunization before tumor transplant, so no conclusions about treatment after transplant and tumor establishment can be made. Currently, we can only suggest stem cell vaccine for cancer treatment in addition to other treatments such as surgery, radiation, and chemotherapy, and not as a standalone therapy option, as it has only been seen as effective before tumor formation and progression.47,51,53
Stem Cells as Therapeutic Carriers
Genetic modification enhances the therapeutic potential for stem cells in oncology by facilitating precise secretion of bioactive mediators. Typically derived from bone marrow, endogenous mesenchymal stem cells (MSCs) migrate towards sites of damaged tissue. MSC tropism is propagated by a cascade of signaling mechanisms and chemokines which trigger the recruitment of MSCs towards sites of damaged tissue.54,55 MSCs are able to mobilize effectively as they express numerous chemokine receptors including: CCR1, CCR2, CCR4, CCR7, CCR8, CCR9, CCR10, CXCR1, CXCR2, CXCR3, CXCR4, CXCR5, CXCR6, and CX3CR1.56 Additionally, MSCs possess the ability to produce a diverse array of cell adhesion molecules which facilitate the engraftment to specific target tissue.57 Upon transplantation, MSCs will migrate away from the initial injection site into a tumor microenvironment (TME) before engrafting to various target cells. Therefore, the transduction of MSCs and other multipotent stem cells could potentially facilitate the decisive delivery of a therapeutic payload within a tumor microenvironment. Specifically, virally transduced MSCs and Neural Stem Cells (NSCs) have exhibited the expression of chemotactic cytokines, interleukins, interferons, growth factors and prodrug-converting enzymes.58,59 The latter of which constitutes the technique known as gene-directed enzyme prodrug therapy (GDEPT). This treatment method allows various non-toxic prodrugs to be converted into their active forms via non-endogenous enzymes produced by genetically modified stem cells.60 The aggregate of these characteristics makes GDEPT uniquely qualified to treat gliomas, medulloblastomas, and other brain tumors. Another benefit of this therapy stems from the ability of MSCs to manipulate tight junctions within the blood brain barrier (BBB), temporarily inhibiting its exclusion properties and allowing for the seamless traversal of MSCs into the cortex; MSCs then utilize tumor-tropism mechanisms to infiltrate and destroy tumor cells in the brain.59,61
Another means by which stem cells can serve as therapeutic carriers is by the precise delivery of nanoparticles (NPs) bearing anti-cancer drugs and various other oncolytic mediators. NPs have long been used in the distribution of drugs used to treat cancer. However, the applicability of NPs is limited due to the lack of accurate targeting, their tendency to be internalized by a wide variety of normally function cells, and their rapid excretion from the body.62 One study analyzing the nanodrug deposits provided by MSCs internalized within mice found that NPs exhibited more accurate delivery of therapeutics in a developed orthotopic lung tumor.63 An additional study conducted using rats has demonstrated that MSCs infiltrate tumor tissue uniformly and that this infiltration leads to a more uniform distribution of a therapeutic payload. However, in the same study, they found no evidence to suggest MSCs could engage in long-distance tropism for a series of gliomas.64 Despite this, NPs conjugated to anti-cancer agents can be delivered into a tumor microenvironment reliably using stem cell-mediated tumor tropic delivery. Furthermore, MSCs retain their inherent ability to sense tumors and respond to chemokines following the anchoring of nanoparticles to their surface. In fact, there is no significant difference in tumor tropism between traditional MSCs and those bound to NPs.65 However, in the latter case, the half-life of the nanoparticle is increased exponentially.66,67
MSCs, with their innate tumor tropism characteristics, are ideal agents for this style of theranostic-based therapy. This is because the inherent tumor tropism presents various biomarkers that can be used as targets for nanoparticles which can then be imaged in vivo using clinically relevant imaging modalities such as Magnetic Resonance Imaging (MRI) and Nuclear imaging.53,68–70 Nouri et al demonstrate that theranostic MSCs are a reliable cell-based, non-viral or viral vectors for suicide gene therapy of cancer using enzyme/prodrug systems.71 MSCs were used as a medium to perform the first comparative study that illustrated the impact of subtle differences among various enzyme/prodrug systems such as thymidine kinase/ganciclovir (TK/GCV), yeast cytosine deaminase/5-fluorocytosine (yCD/5-FC) and nitroreductase/CB1954 (NTR/CB1954) on the therapeutic outcome. MSCs were genetically modified to stably express a panel of four suicide genes including TK (TK007 and TKSR39 mutants), yeast cytosine deaminase: uracil phosphoribosyltransferase (yCD:UPRT) and nitroreductase (NTR). Then, they evaluated the anticancer efficacies of the genetically engineered MSCs using SKOV3 cell models in vivo. In addition, all MSCs were engineered to stably express luciferase gene making them suitable for quantitative imaging and dose–response relationship studies in vivo. The study results demonstrated that yCD:UPRT/5-FC was the most effective enzyme/prodrug system among the ones tested with this theranostic imaging platform.71
It is worth noting that theranostics can be applied to nanodrug therapy for cancer. Liu et al reported that a silica-based multifunctional NP system encapsulated a chemotherapeutic agent and magnetic cores and coated with a specific antibody against the lung CSCs was systematically studied in vivo. These NPs were systematically administered and activated for targeted chemotherapy and thermotherapy by using an externally applied alternating magnetic field (AMF).72 The application of an AMF causes localized induced hyperthermia in the areas in which the nanoparticles accumulate.73 This can kill the tumor cells either directly or indirectly. The cytotoxic thermal effects of the localized hyperthermia from the NPs directly ablate the tumor cells. There is also an indirect mechanism which supports dendritic cell activation and immune cell migration to the tumor microenvironment. This is caused by the hyperthermia which induces the production of Heat Shock Protein (HSP) and a subsequent increase in presentation of these antigens on the cell surface, which induces dendritic cells and triggers a greater host immune response towards the cancer cells.74 The antibody-modified NPs targeted to lung CSCs with extended accumulation in tumors after systemic injection. In in vivo models, this hyperthermia and chemotherapeutic combined therapy significantly suppressed tumor growth and metastasis in lung CSC xenograft-bearing mice, with minimal side effects and adverse effects.72 This work demonstrated the feasibility of developing multifunctional nanomedicine targeting CSCs for effective cancer treatment, which can be monitored by magnetic particle imaging or MRI.75–77 Figure 1 highlights some of the aforementioned mechanisms of using stem cells as carriers of therapeutic payload and alternative therapeutic carriers such as NPs for tumor treatment.
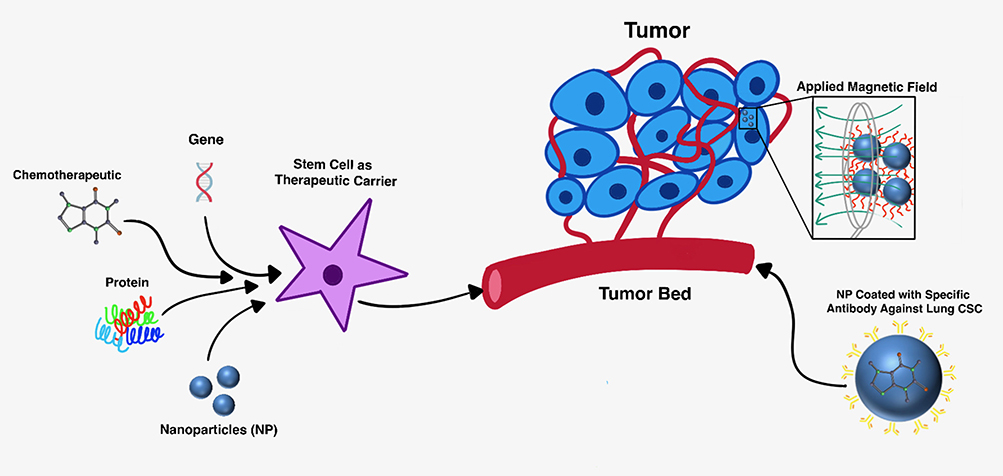 |
Figure 1 Therapeutic carrier models for cancer treatment. (Left) Cancer stem cells (CSCs) loaded with therapeutic cargo can be transplanted or delivered to site of tumor formation (tumor bed). (Right) Nanoparticles (NPs) targeting the CSCs can deliver chemotherapeutic payload and induce hyperthermia with an external applied magnetic field (AMF). |
Quiescent Properties in Treatment
Anti-cancer therapies often utilize a combination of procedures and drugs that target gatherings of tumor cells. CSCs have long been understood now as a group of cells that fuel the growth of tumors and have properties that allow them to persist through the electromagnetic and chemical treatment that are common in contemporary practice.78 CSCs separate themselves among cancer cells due to their ability to maintain a long, slow growing quiescent state. This dormancy allows for the cells to be preserved in spite of conventional cancer treatments that are used to combat their progeny as well as posing long-term tumorigenic potential.78 Previous studies have shown that there maintains a population of chemotherapy-resistant cancer cells that demonstrated unique properties of self-renewal and increased potential for tumor formation. In these studies, CSC-marker expressing cells have survived treatment such as neoadjuvant chemotherapy whereas cells without CSC markers were destroyed.79
Their unique properties have only added on to the need for the development of new therapeutic strategies that exceed the scope of conventional antiproliferative agents and treatments. Much research is focused on CSC’s quiescent properties as a potential target for treatment. Research has shown that this quiescent function is not simply a dormant state, but rather it is actively maintained by the cell by downregulating known regulators of the cell cycle such as cyclin A2 and E2 as well as mitotic regulators such as survivin.80 There are generally three main approaches through which research is attempting to eradicate these quiescent stem cells in tumors. The first involves driving quiescent cells to reenter a normal cell cycle state by stopping cellular mechanisms that drive quiescence and then attack them with chemotherapeutic agents that only function on proliferating cells.81 This theory is primarily supported by the notion that cell quiescence is defined as being in a reversible G0 state that requires maintenance.81 For example, Fbxw7 has been understood to play a crucial role in the maintenance of quiescence, but its ablation along with the introduction of Imatinib treatment, a tyrosine-kinase inhibitor, is shown to interrupt the quiescent state of the studied leukemia-initiating cells (LICs) and contribute to their depletion.82 The second method that has been proposed is one where pharmacological intervention is used to maintain CSCs in their quiescent state throughout the lifetime of their patient so that they will not cause any future tumor growth or metastasis. This strategy attempts to lock these quiescent cells in the G0 state. One such way to accomplish this is to inhibit Src kinase signaling along with inhibiting MEK1/2 as they are both factors in cell cycle progression. This can be achieved through pharmacological means and the prevention of cell cycle progression will induce apoptosis or maintenance in this state without growth.83 The final school of thought looks to eradicate CSCs while they are still in their dormant state.81 However, there are issues with these three potential methods which further highlight why research in these fields has been unsuccessful. Clinical evidence has been inconclusive on whether activating these quiescent cells in order to target them with conventional therapy is even able to be controlled. In addition, these quiescent stem cells are known to be extremely heterogeneous which indicates that activating them could exacerbate the condition by giving them an increased arsenal of mechanisms through which the cancer can develop.84 Difficulties in the latter two methods are due to the need of a more comprehensive understanding of the various pathways and factors associated in CSC quiescence. Normal stem cells are a valuable resource in understanding CSC signaling as they are known to share several pathways such as Notch, Hedgehog, WNT/B-catenin, and NFkB.81,85
Multiple myeloma (MM) is a common cancer of the blood which has an alarming 5-year survival rate of about 54% along with high chances of recurrence and need for further treatment.86 Its resistance to treatments has been attributed to its development of drug resistance. A proposed model of MM malignant stem cell progression suggests that there are two states that cells are in - a quiescent state and a proliferative state. The quiescent state has exhibit properties of increased adhesion and minimal proliferation. Those in the quiescent state also have increased drug-resistant properties. There also exists a small population of cells that are in the proliferative state. These cells have high capacity for growth and mutation as a result of their increased chromosomal instability.87 Impairment of MM cell survival through inducing cell cycle arrest was conducted through the use of anti-sense oligonucleotides (ASOs) to target human interferon regulatory factor 4 (IRF4). IRF4 has been identified as a critical MM cell factor for survival with an important role in disease development and progression. This treatment also reduced mRNA levels and levels of the MYC gene, which has a known impact in stem cell progression and has been studied thoroughly as an oncogene. Increased cleavage rates, apoptosis rates, and decreased colony formation all suggested success in disrupting cell growth. Furthermore, ASO-mediated IRF4 treatment led to cell cycle arrest in G1 and decreased proportion of cells in G2/M phase which signifies significant reduction in cell viability.86 Seeing as though the G1 phase length plays an important factor in maintaining a quiescent state, this ASO treatment could help decrease the risk of quiescent MM cells leading to disease relapse. Further study is necessary to find an effective solution to both quiescent and proliferative states of MM, but studies that combat CSC quiescence show promise for a solution to decrease relapse rates.
Much of the difficulty in identifying, understanding, and treating CSCs originates from their extensive plasticity and asymmetry driven by intra-tumoral heterogeneity.34 Their plasticity allows them to evade therapies by presenting various phenotypes and providing the ability to inhabit different tumor microenvironments. Normal cancer cells within a tumor mass can convert into CSCs in response to chemotherapy and can gain drug resistance as a result of a change in gene expression.88 One such mechanism of drug resistance includes an increase in the expression of ATP-Binding cassette (ABC) transporters that permit greater drug efflux rates. CSCs can also be found in either a quiescent state that is difficult to eradicate or a proliferative state that prioritizes growth and metastasis. This quiescent state, when met with increased drug efflux mechanisms in CSC populations, often contributes to greater relapsed tumors. In hypoxic conditions, cells seeded in the core of the tumor are found to transition to the edge of the tumor where they become more quiescent.89 This is because cells typically found seeded in the tumor core are more proliferative, which is why there is greater cell density in that region as compared to the edge cells which are more invasive, quiescent, and resistant.89 As a result of this core-to-edge migration of tumor cells in response to hypoxia, particularly due to the induction of HIF-1α and HIF-2α, a novel front for cancer intervention and therapy has been unlocked.89 However, the therapeutic potential and efficacy of a combined therapy targeting HIF-1α and HIF-2α in tumor cells, likely to prevent core to edge migration of progressive tumoral cells when presented with hypoxia, needs to be further studied to gain momentum as an effective therapy. The cell division of these quiescent cells can also be either symmetric or asymmetric. Symmetric division either creates a pair of quiescent daughter cells or a pair that is more proliferative and differentiates.90 Asymmetric division results in one quiescent cell and transient amplifying cell that is very proliferative and contributes to most of the tumor.90 It is widely inferred that this asymmetric division is more dangerous for cancer growth and it should be targeted for effective cancer treatment.
CSC Exosomes, New Targets for Cancer Therapy?
Stem cells have been found to play the role of mediators of conversion of healthy cells to cancerous cells.91 This is done through the synthesis and release of exosomes– extracellular vesicles of cellular cargo that are released by a cell for various purposes.92 Exosomes have been previously found to contain various cargo including miRNA, siRNA, transcription factors, and other proteins. Through the use of these extracellular vesicles, cells are able to communicate and exert transformative influence through phenotypic and genotypic alterations of surrounding cells.93 Often used to encourage differentiation, proliferation, or inhibition thereof of cells in the surrounding environment, exosomes role in mediating the transformation of regular, healthy cells into cancerous cells is an area of great research due to its potential to catalyze interventional mechanisms for cancer therapy through targeting exosomes.
Stem cell-derived extracellular vesicles can provide powerful alternatives to cell-based therapies since the former acts as a noninvasive method for in vivo modulation of gene expression, inhibition of cell surface receptors and intracellular signaling molecules, and initiation of cell differentiation or death.92 Instead of transplanting differentiated stem cells or donor cells into the patient, the delivery of extracellular vesicles can allow for transformation of native cells in the subject and transformation of cells that have been transplanted prior. This eliminates the need for surgery and removes the necessity to probe the patient for cell transplantation, which requires some invasive protocol. Stem cell-derived extracellular vesicles can prove to be a valuable tool in cell-based therapy by perhaps altering the nature of the cells that are native to the microenvironment of the subject.
There are various mechanisms through which exosomes released by CSCs exert influence on neighboring cells. One such method is through induction of Epithelial to Mesenchymal Transition (EMT). This is induced through the release of transcription factors such as Snail, Twist, and FOXC2 that are carried by the extracellular vesicle to neighboring cells where these transcription factors exert their effects phenotypically and genotypically.94 In mammary epithelial stem cells, the influence of these factors on neighboring healthy cells is apparent by the CD44 (high)/CD24 (low) antigen phenotype which is typical of CSCs.95 This can contribute to the development of severe tumorigenicity amongst the cells, as noted by the study. Another study observed the role of CD-103+exosomes released by CSCs which promoted EMT in clear cell renal cell carcinoma (CCRCC) cells.96 Particularly, the group observed the importance of one critical factor in promoting EMT in CCRCC: MiR-19b-3p.96 This was done through the repression of PTEN, a protein which is apparently expressed and has been previously studied to play a key role in the promotion of EMT.97 The study was able to confirm the role of MiR-19b-3p in promoting EMT through suppression of PTEN by infecting ACHN and 786-O cells with a miR-19b-3p lentivirus.96 Like the exosomes released by the CSCs, this encouraged the migration and invasion of the CCRCC cells. These findings implicate the role of CD103, since it mediates entry of the miRNA into neighboring cells via use of exosomes, in acting as a potential biomarker or target for cancer therapy. Further studies focusing on CSC-derived exosomes include macrovesicles derived from CD105+ cells which have been found to promote angiogenesis and metastasis with a distinct miRNA profile inside of the vesicles, resulting from presence of CD105.98 This unique composition encouraged greater tumorigenicity amongst the cells, favoring further growth and invasion. Although there are various studies investigating the role of CSC-derived exosomes in promoting tumor development, limited information currently exists on the CSC-specificity of these exosomes and further research must be done in this area to determine entirely which exosomes are specific to only CSCs, or whether it is the content within that can be used as a biomarker. This further emphasizes the importance of researching the role of the CSC-influenced tumor microenvironment and its various components (eg, exosomes) in promoting and accelerating cancer development.
Conclusion and Perspectives
The aforementioned studies permit insight into a previously unexplored interface between stem cells and cancer progression and treatment. Silencing cell surface markers on CSCs can promote immune recognition of tumor sites and inhibit binding of tumor cargo to healthy cells, thus preventing their transformation. Furthermore, iPSCs can be used to vaccinate and immunize individuals against tumor cells in the future due to surface-antigen similarity between iPSCs and cancer cells. In addition to these mechanisms, stem cells have been shown to transfer various genetic and molecular cargo intercellularly through exosomes, another area for drug targeting and disease intervention in cancer. As a result of these various interactions between cancer cells, stem cells and their individual components, there is now a greater need to explore the influence that stem cells have on tumorigenesis. This will enable the innovation and translation of theranostics that are more efficient in nature and do not result in an abundance of unwanted side effects as a result of treatment.
Current organoid models are limited by their inability to mimic mature organ architecture and associated tissue microenvironments. Multilayer bladder ‘assembloids’ were recently created by reconstituting tissue stem cells with stromal components to represent an organized architecture with an epithelium surrounding stroma and an outer muscle layer.99 A urothelial carcinoma assembloids platform has been developed by the same group. These assembloids exhibit characteristics of mature adult bladders and tumors in cell composition and gene expression, and recapitulate in vivo tissue dynamics. This will help advance functional studies in the context of the increasingly recognized importance of tissue stroma and microenvironments.99–101
Disclosure
The authors report no conflicts of interest in this work.
References
1. Dasari S, Tchounwou PB. Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharmacol. 2014;740:364–378. doi:10.1016/j.ejphar.2014.07.025
2. Montero A, Fossella F, Hortobagyi G, Valero V. Docetaxel for treatment of solid tumours: a systematic review of clinical data. Lancet Oncol. 2005;6(4):229–239. doi:10.1016/S1470-2045(05)70094-2
3. Kim N, Choi SH, Chang JS, et al. Use of bevacizumab before or after radiotherapy increases the risk of fistula formation in patients with cervical cancer. Int J Gynecol Cancer. 2020;31:59–65. doi:10.1136/ijgc-2020-002031
4. Lage H. An overview of cancer multidrug resistance: a still unsolved problem. Cell Mol Life Sci. 2008;65(20):3145–3167. doi:10.1007/s00018-008-8111-5
5. Biehl JK, Russell B. Introduction to stem cell therapy. J Cardiovasc Nurs. 2009;24(2):98. doi:10.1097/JCN.0b013e318197a6a5
6. Ayob AZ, Ramasamy TS. Cancer stem cells as key drivers of tumour progression. J Biomed Sci. 2018;25(1):20. doi:10.1186/s12929-018-0426-4
7. Zhang CL, Huang T, Wu BL, He WX, Liu D. Stem cells in cancer therapy: opportunities and challenges. Oncotarget. 2017;8(43):75756–75766. doi:10.18632/oncotarget.20798
8. Garibaldi C, Jereczek-Fossa BA, Marvaso G, et al. Recent advances in radiation oncology. Ecancermedicalscience. 2017;11:785. doi:10.3332/ecancer.2017.785
9. Urruticoechea A, Alemany R, Balart J, Villanueva A, Vinals F, Capella G. Recent advances in cancer therapy: an overview. Curr Pharm Des. 2010;16(1):3–10. doi:10.2174/138161210789941847
10. Brown EJ, Frazier WA. Integrin-associated protein (CD47) and its ligands. Trends Cell Biol. 2001;11(3):130–135. doi:10.1016/S0962-8924(00)01906-1
11. Blazar BR, Lindberg FP, Ingulli E, et al. CD47 (integrin-associated protein) engagement of dendritic cell and macrophage counterreceptors is required to prevent the clearance of donor lymphohematopoietic cells. J Exp Med. 2001;194(4):541–549. doi:10.1084/jem.194.4.541
12. Oldenborg PA, Zheleznyak A, Fang YF, Lagenaur CF, Gresham HD, Lindberg FP. Role of CD47 as a marker of self on red blood cells. Science. 2000;288(5473):2051–2054. doi:10.1126/science.288.5473.2051
13. Jaiswal S, Jamieson CH, Pang WW, et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell. 2009;138(2):271–285. doi:10.1016/j.cell.2009.05.046
14. Olsson M, Bruhns P, Frazier WA, Ravetch JV, Oldenborg PA. Platelet homeostasis is regulated by platelet expression of CD47 under normal conditions and in passive immune thrombocytopenia. Blood. 2005;105(9):3577–3582. doi:10.1182/blood-2004-08-2980
15. Oronsky B, Carter C, Reid T, Brinkhaus F, Knox SJ. Just eat it: a review of CD47 and SIRP-alpha antagonism. Semin Oncol. 2020;47(2–3):117–124. doi:10.1053/j.seminoncol.2020.05.009
16. Uno S, Kinoshita Y, Azuma Y, et al. Antitumor activity of a monoclonal antibody against CD47 in xenograft models of human leukemia. Oncol Rep. 2007;17(5):1189–1194.
17. Majeti R, Chao MP, Alizadeh AA, et al. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell. 2009;138(2):286–299. doi:10.1016/j.cell.2009.05.045
18. Wang CL, Lin MJ, Hsu CY, et al. CD47 promotes cell growth and motility in epithelial ovarian cancer. Biomed Pharmacother. 2019;119:109105. doi:10.1016/j.biopha.2019.109105
19. Lian S, Xie R, Ye Y, et al. Simultaneous blocking of CD47 and PD-L1 increases innate and adaptive cancer immune responses and cytokine release. EBioMedicine. 2019;42:281–295. doi:10.1016/j.ebiom.2019.03.018
20. Hainsworth JD, Litchy S, Burris HA, et al. Rituximab as first-line and maintenance therapy for patients with indolent non-hodgkin’s lymphoma. J Clin Oncol. 2002;20(20):4261–4267. doi:10.1200/JCO.2002.08.674
21. Advani R, Flinn I, Popplewell L, et al. CD47 blockade by Hu5F9-G4 and rituximab in non-Hodgkin’s lymphoma. N Engl J Med. 2018;379(18):1711–1721. doi:10.1056/NEJMoa1807315
22. Eladl E, Tremblay-LeMay R, Rastgoo N, et al. Role of CD47 in hematological malignancies. J Hematol Oncol. 2020;13(1):96. doi:10.1186/s13045-020-00930-1
23. Cioffi M, Trabulo S, Hidalgo M, et al. Inhibition of CD47 effectively targets pancreatic cancer stem cells via dual mechanisms. Clin Cancer Res. 2015;21(10):2325–2337. doi:10.1158/1078-0432.CCR-14-1399
24. Kaur S, Elkahloun AG, Singh SP, et al. A function-blocking CD47 antibody suppresses stem cell and EGF signaling in triple-negative breast cancer. Oncotarget. 2016;7(9):10133–10152. doi:10.18632/oncotarget.7100
25. Liu L, Zhang L, Yang L, et al. Anti-CD47 antibody as a targeted therapeutic agent for human lung cancer and cancer stem cells. Front Immunol. 2017;8:404. doi:10.3389/fimmu.2017.00404
26. Dietrich A, Tanczos E, Vanscheidt W, Schopf E, Simon JC. High CD44 surface expression on primary tumours of malignant melanoma correlates with increased metastatic risk and reduced survival. Eur J Cancer. 1997;33(6):926–930. doi:10.1016/S0959-8049(96)00512-6
27. Thapa R, Wilson GD. The importance of CD44 as a stem cell biomarker and therapeutic target in cancer. Stem Cells Int. 2016;2016:2087204. doi:10.1155/2016/2087204
28. Ranji P, Salmani Kesejini T, Saeedikhoo S, Alizadeh AM. Targeting cancer stem cell-specific markers and/or associated signaling pathways for overcoming cancer drug resistance. Tumour Biol. 2016;37(10):13059–13075. doi:10.1007/s13277-016-5294-5
29. Shmelkov SV, Butler JM, Hooper AT, et al. CD133 expression is not restricted to stem cells, and both CD133+ and CD133- metastatic colon cancer cells initiate tumors. J Clin Invest. 2008;118(6):2111–2120. doi:10.1172/JCI34401
30. Brugnoli F, Grassilli S, Al-Qassab Y, Capitani S, Bertagnolo V. CD133 in breast cancer cells: more than a stem cell marker. J Oncol. 2019;2019:7512632. doi:10.1155/2019/7512632
31. Yoshida K, Tsujimoto H, Matsumura K, et al. CD47 is an adverse prognostic factor and a therapeutic target in gastric cancer. Cancer Med. 2015;4(9):1322–1333. doi:10.1002/cam4.478
32. Barzegar Behrooz A, Syahir A, Ahmad S. CD133: beyond a cancer stem cell biomarker. J Drug Target. 2019;27(3):257–269. doi:10.1080/1061186X.2018.1479756
33. Bellomo C, Caja L, Moustakas A. Transforming growth factor beta as regulator of cancer stemness and metastasis. Br J Cancer. 2016;115(7):761–769. doi:10.1038/bjc.2016.255
34. Bhola NE, Balko JM, Dugger TC, et al. TGF-beta inhibition enhances chemotherapy action against triple-negative breast cancer. J Clin Invest. 2013;123(3):1348–1358. doi:10.1172/JCI65416
35. Rabinovich I, Sebastiao APM, Lima RS, et al. Cancer stem cell markers ALDH1 and CD44+/CD24- phenotype and their prognosis impact in invasive ductal carcinoma. Eur J Histochem. 2018;62(3). doi:10.4081/ejh.2018.2943.
36. Tomita H, Tanaka K, Tanaka T, Hara A. Aldehyde dehydrogenase 1A1 in stem cells and cancer. Oncotarget. 2016;7(10):11018–11032. doi:10.18632/oncotarget.6920
37. Brewer BG, Mitchell RA, Harandi A, Eaton JW. Embryonic vaccines against cancer: an early history. Exp Mol Pathol. 2009;86(3):192–197. doi:10.1016/j.yexmp.2008.12.002
38. Buttle GA, Frayn A. Effect of previous injection of homologous embryonic tissue on the growth of certain transplantable mouse tumours. Nature. 1967;215(5109):1495–1497. doi:10.1038/2151495a0
39. Klavins JV, Mesa-Tejada R, Weiss M. Human carcinoma antigens cross reacting with anti-embryonic antibodies. Nat N Biol. 1971;234(48):153–154. doi:10.1038/newbio234153a0
40. Yaddanapudi K, Mitchell RA, Putty K, et al. Vaccination with embryonic stem cells protects against lung cancer: is a broad-spectrum prophylactic vaccine against cancer possible? PLoS One. 2012;7(7):e42289. doi:10.1371/journal.pone.0042289
41. Ghosh Z, Huang M, Hu S, Wilson KD, Dey D, Wu JC. Dissecting the oncogenic and tumorigenic potential of differentiated human induced pluripotent stem cells and human embryonic stem cells. Cancer Res. 2011;71(14):5030–5039. doi:10.1158/0008-5472.CAN-10-4402
42. Ben-Porath I, Thomson MW, Carey VJ, et al. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet. 2008;40(5):499–507. doi:10.1038/ng.127
43. Aponte PM, Caicedo A. Stemness in cancer: stem cells, cancer stem cells, and their microenvironment. Stem Cells Int. 2017;2017:5619472. doi:10.1155/2017/5619472
44. Mushtaq M, Kovalevska L, Darekar S, et al. Cell stemness is maintained upon concurrent expression of RB and the mitochondrial ribosomal protein S18-2. Proc Natl Acad Sci U S A. 2020;117(27):15673–15683. doi:10.1073/pnas.1922535117
45. Mallon BS, Hamilton RS, Kozhich OA, et al. Comparison of the molecular profiles of human embryonic and induced pluripotent stem cells of isogenic origin. Stem Cell Res. 2014;12(2):376–386. doi:10.1016/j.scr.2013.11.010
46. de Almeida PE, Meyer EH, Kooreman NG, et al. Transplanted terminally differentiated induced pluripotent stem cells are accepted by immune mechanisms similar to self-tolerance. Nat Commun. 2014;5:3903. doi:10.1038/ncomms4903
47. Kooreman NG, Kim Y, de Almeida PE, et al. Autologous iPSC-based vaccines elicit anti-tumor responses in vivo. Cell Stem Cell. 2018;22(4):501–13 e7. doi:10.1016/j.stem.2018.01.016
48. Medvedev SP, Shevchenko AI, Zakian SM. Induced pluripotent stem cells: problems and advantages when applying them in regenerative medicine. Acta Naturae. 2010;2(2):18–28. doi:10.32607/20758251-2010-2-2-18-27
49. Lin M, Chang AE, Wicha M, Li Q, Huang S. Development and application of cancer stem cell-targeted vaccine in cancer immunotherapy. J Vaccines Vaccin. 2017;8(6). doi:10.4172/2157-7560.1000371
50. Ning N, Pan Q, Zheng F, et al. Cancer stem cell vaccination confers significant antitumor immunity. Cancer Res. 2012;72(7):1853–1864. doi:10.1158/0008-5472.CAN-11-1400
51. Chu DT, Nguyen TT, Tien NLB, et al. Recent progress of stem cell therapy in cancer treatment: molecular mechanisms and potential applications. Cells. 2020;9(3):563. doi:10.3390/cells9030563
52. Gabka-Buszek A, Kwiatkowska-Borowczyk E, Jankowski J, Kozlowska AK, Mackiewicz A. Novel genetic melanoma vaccines based on induced pluripotent stem cells or melanosphere-derived stem-like cells display high efficacy in a murine tumor rejection model. Vaccines (Basel). 2020;8(2). doi:10.3390/vaccines8020147
53. Wang P, Aguirre A. New strategies and in vivo monitoring methods for stem cell-based anticancer therapies. Stem Cells Int. 2018;2018:7315218. doi:10.1155/2018/7315218
54. Vanden Berg-Foels WS. In situ tissue regeneration: chemoattractants for endogenous stem cell recruitment. Tissue Eng Part B Rev. 2014;20(1):28–39. doi:10.1089/ten.teb.2013.0100
55. Wang P, Moore A. Molecular imaging of stem cell transplantation for neurodegenerative diseases. Curr Pharm Des. 2012;18(28):4426–4440. doi:10.2174/138161212802481255
56. Hocking AM. The role of chemokines in mesenchymal stem cell homing to wounds. Adv Wound Care (New Rochelle). 2015;4(11):623–630. doi:10.1089/wound.2014.0579
57. Ren G, Roberts AI, Shi Y. Adhesion molecules: key players in Mesenchymal stem cell-mediated immunosuppression. Cell Adh Migr. 2011;5(1):20–22. doi:10.4161/cam.5.1.13491
58. Curfs JH, Meis JF, Hoogkamp-Korstanje JA. A primer on cytokines: sources, receptors, effects, and inducers. Clin Microbiol Rev. 1997;10(4):742–780. doi:10.1128/CMR.10.4.742
59. Nakamizo A, Marini F, Amano T, et al. Human bone marrow-derived mesenchymal stem cells in the treatment of gliomas. Cancer Res. 2005;65(8):3307–3318. doi:10.1158/0008-5472.CAN-04-1874
60. Malekshah OM, Chen X, Nomani A, Sarkar S, Hatefi A. Enzyme/prodrug systems for cancer gene therapy. Curr Pharmacol Rep. 2016;2(6):299–308. doi:10.1007/s40495-016-0073-y
61. Hasan A, Deeb G, Rahal R, et al. Mesenchymal stem cells in the treatment of traumatic brain injury. Front Neurol. 2017;8:28. doi:10.3389/fneur.2017.00028
62. Rosenblum D, Joshi N, Tao W, Karp JM, Peer D. Progress and challenges towards targeted delivery of cancer therapeutics. Nat Commun. 2018;9(1):1410. doi:10.1038/s41467-018-03705-y
63. Layek B, Sadhukha T, Panyam J, Prabha S. Nano-engineered mesenchymal stem cells increase therapeutic efficacy of anticancer drug through true active tumor targeting. Mol Cancer Ther. 2018;17(6):1196–1206. doi:10.1158/1535-7163.MCT-17-0682
64. Bexell D, Gunnarsson S, Svensson A, et al. Rat multipotent mesenchymal stromal cells lack long-distance tropism to 3 different rat glioma models. Neurosurgery. 2012;70(3):731–739. doi:10.1227/NEU.0b013e318232dedd
65. Cheng H, Kastrup CJ, Ramanathan R, et al. Nanoparticulate cellular patches for cell-mediated tumoritropic delivery. ACS Nano. 2010;4(2):625–631. doi:10.1021/nn901319y
66. Chambers E, Mitragotri S. Long circulating nanoparticles via adhesion on red blood cells: mechanism and extended circulation. Exp Biol Med (Maywood). 2007;232(7):958–966.
67. Hoshyar N, Gray S, Han H, Bao G. The effect of nanoparticle size on in vivo pharmacokinetics and cellular interaction. Nanomedicine (Lond). 2016;11(6):673–692. doi:10.2217/nnm.16.5
68. Wang P, Yoo B, Sherman S, et al. Predictive imaging of chemotherapeutic response in a transgenic mouse model of pancreatic cancer. Int J Cancer. 2016;139(3):712–718. doi:10.1002/ijc.30098
69. Zhao H, Richardson R, Talebloo N, Mukherjee P, Wang P, Moore A. uMUC1-targeting magnetic resonance imaging of therapeutic response in an orthotropic mouse model of colon cancer. Mol Imaging Biol. 2019;21(5):852–860. doi:10.1007/s11307-019-01326-5
70. Zhao H, Hayat H, Ma X, Fan D, Wang P, Moore A. Molecular imaging and deep learning analysis of uMUC1 expression in response to chemotherapy in an orthotopic model of ovarian cancer. Sci Rep. 2020;10(1):14942. doi:10.1038/s41598-020-71890-2
71. Nouri FS, Wang X, Hatefi A. Genetically engineered theranostic mesenchymal stem cells for the evaluation of the anticancer efficacy of enzyme/prodrug systems. J Control Release. 2015;200:179–187. doi:10.1016/j.jconrel.2015.01.003
72. Liu D, Hong Y, Li Y, et al. Targeted destruction of cancer stem cells using multifunctional magnetic nanoparticles that enable combined hyperthermia and chemotherapy. Theranostics. 2020;10(3):1181–1196. doi:10.7150/thno.38989
73. Dewhirst MW, Vujaskovic Z, Jones E, Thrall D. Re-setting the biologic rationale for thermal therapy. Int J Hyperthermia. 2005;21(8):779–790. doi:10.1080/02656730500271668
74. Calderwood SK, Theriault JR, Gong J. How is the immune response affected by hyperthermia and heat shock proteins? Int J Hyperthermia. 2005;21(8):713–716. doi:10.1080/02656730500340794
75. Wells J, Twamley S, Sekar A, et al. Lissajous scanning magnetic particle imaging as a multifunctional platform for magnetic hyperthermia therapy. Nanoscale. 2020;12(35):18342–18355. doi:10.1039/D0NR00604A
76. Khizar S, Ahmad NM, Ahmed N, et al. Aminodextran coated CoFe2O4 nanoparticles for combined magnetic resonance imaging and hyperthermia. Nanomaterials (Basel). 2020;10(11):2182. doi:10.3390/nano10112182
77. Brennan G, Bergamino S, Pescio M, Tofail SAM, Silien C. The effects of a varied gold shell thickness on iron oxide nanoparticle cores in magnetic manipulation, T1 and T2 MRI contrasting, and magnetic hyperthermia. Nanomaterials (Basel). 2020;10(12):2424. doi:10.3390/nano10122424
78. Clevers H. The cancer stem cell: premises, promises and challenges. Nat Med. 2011;17(3):313–319. doi:10.1038/nm.2304
79. Borah A, Raveendran S, Rochani A, Maekawa T, Kumar DS. Targeting self-renewal pathways in cancer stem cells: clinical implications for cancer therapy. Oncogenesis. 2015;4:e177. doi:10.1038/oncsis.2015.35
80. Cheung TH, Rando TA. Molecular regulation of stem cell quiescence. Nat Rev Mol Cell Biol. 2013;14(6):329–340.
81. Chen W, Dong J, Haiech J, Kilhoffer MC, Zeniou M. Cancer stem cell quiescence and plasticity as major challenges in cancer therapy. Stem Cells Int. 2016;2016:1740936. doi:10.1155/2016/1740936
82. Takeishi S, Matsumoto A, Onoyama I, Naka K, Hirao A, Nakayama KI. Ablation of Fbxw7 eliminates leukemia-initiating cells by preventing quiescence. Cancer Cell. 2013;23(3):347–361. doi:10.1016/j.ccr.2013.01.026
83. Cho IJ, Lui PP, Obajdin J, et al. Mechanisms, hallmarks, and implications of stem cell quiescence. Stem Cell Rep. 2019;12(6):1190–1200. doi:10.1016/j.stemcr.2019.05.012
84. Sosa MS, Bragado P, Aguirre-Ghiso JA. Mechanisms of disseminated cancer cell dormancy: an awakening field. Nat Rev Cancer. 2014;14(9):611–622. doi:10.1038/nrc3793
85. Schmidt-Kittler O, Ragg T, Daskalakis A, et al. From latent disseminated cells to overt metastasis: genetic analysis of systemic breast cancer progression. Proc Natl Acad Sci U S A. 2003;100(13):7737–7742. doi:10.1073/pnas.1331931100
86. Mondala PK, Vora AA, Zhou T, et al. Selective antisense oligonucleotide inhibition of human IRF4 prevents malignant myeloma regeneration via cell cycle disruption. Cell Stem Cell. 2021. doi:10.1016/j.stem.2020.12.017
87. Yaccoby S. Two states of myeloma stem cells. Clin Lymphoma Myeloma Leuk. 2018;18(1):38–43. doi:10.1016/j.clml.2017.09.020
88. Pattabiraman DR, Weinberg RA. Tackling the cancer stem cells - what challenges do they pose? Nat Rev Drug Discov. 2014;13(7):497–512. doi:10.1038/nrd4253
89. Mortezaee K. Hypoxia induces core-to-edge transition of progressive tumoral cells: a critical review on differential yet corroborative roles for HIF-1alpha and HIF-2alpha. Life Sci. 2020;242:117145. doi:10.1016/j.lfs.2019.117145
90. Najafi M, Mortezaee K, Ahadi R. Cancer stem cell (a)symmetry & plasticity: tumorigenesis and therapy relevance. Life Sci. 2019;231:116520. doi:10.1016/j.lfs.2019.05.076
91. Afify SM, Seno M. Conversion of stem cells to cancer stem cells: undercurrent of cancer initiation. Cancers (Basel). 2019;11(3):345. doi:10.3390/cancers11030345
92. Xu J, Liao K, Zhou W. Exosomes regulate the transformation of cancer cells in cancer stem cell homeostasis. Stem Cells Int. 2018;2018:4837370. doi:10.1155/2018/4837370
93. Li SP, Lin ZX, Jiang XY, Yu XY. Exosomal cargo-loading and synthetic exosome-mimics as potential therapeutic tools. Acta Pharmacol Sin. 2018;39(4):542–551. doi:10.1038/aps.2017.178
94. Mani SA, Guo W, Liao MJ, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133(4):704–715. doi:10.1016/j.cell.2008.03.027
95. Hollier BG, Tinnirello AA, Werden SJ, et al. FOXC2 expression links epithelial-mesenchymal transition and stem cell properties in breast cancer. Cancer Res. 2013;73(6):1981–1992. doi:10.1158/0008-5472.CAN-12-2962
96. Wang L, Yang G, Zhao D, et al. CD103-positive CSC exosome promotes EMT of clear cell renal cell carcinoma: role of remote MiR-19b-3p. Mol Cancer. 2019;18(1):86. doi:10.1186/s12943-019-0997-z
97. Dillon LM, Miller TW. Therapeutic targeting of cancers with loss of PTEN function. Curr Drug Targets. 2014;15(1):65–79. doi:10.2174/1389450114666140106100909
98. Grange C, Tapparo M, Collino F, et al. Microvesicles released from human renal cancer stem cells stimulate angiogenesis and formation of lung premetastatic niche. Cancer Res. 2011;71(15):5346–5356. doi:10.1158/0008-5472.CAN-11-0241
99. Kim E, Choi S, Kang B, et al. Creation of bladder assembloids mimicking tissue regeneration and cancer. Nature. 2020;588:664–669. doi:10.1038/s41586-020-3034-x
100. Lee SH, Hu W, Matulay JT, et al. Tumor evolution and drug response in patient-derived organoid models of bladder cancer. Cell. 2018;173(2):515–28 e17. doi:10.1016/j.cell.2018.03.017
101. Marton RM, Pasca SP. Organoid and assembloid technologies for investigating cellular crosstalk in human brain development and disease. Trends Cell Biol. 2020;30(2):133–143. doi:10.1016/j.tcb.2019.11.004
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.