")
Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 14
A Comprehensive Literature Review of JAK Inhibitors in Treatment of Alopecia Areata
Authors Dillon KL
Received 6 April 2021
Accepted for publication 28 May 2021
Published 25 June 2021 Volume 2021:14 Pages 691—714
DOI https://doi.org/10.2147/CCID.S309215
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Jeffrey Weinberg
Kerry-Ann L Dillon 1, 2
1Department of Microbiology, Immunology and Pharmacology, St. George’s University School of Medicine, True Blue, Grenada; 2Department of Dermatology, School of Health, Sport & Professional Practice, Faculty of Life Sciences and Education, University of South Wales, Pontypridd, South Wales, UK
Correspondence: Kerry-Ann L Dillon
Department of Microbiology, Immunology and Pharmacology, St. George’s University School of Medicine, True Blue, Grenada
Tel +473 5360369
Email [email protected]
Abstract: Alopecia areata (AA) is a chronic, immune-mediated form of nonscarring alopecia that is multifactorial and results in localized patches. It is often described as a self-limiting condition that results in the spontaneous regrowth of hair in most cases. However, this regrowth may take several months or years to occur in some patients, leading to the development of psychoemotional trauma in those that are affected. Although several therapies for AA have been developed and tested, there is no specific treatment that has been approved, leading to the availability of many off-label conventional treatment options, with very limited responses. More recently, with the advancement of pre-clinical and genetic studies, a greater understanding of the pathomechanisms involved in the development of AA has been uncovered. This has resulted in the introduction of targeted therapies that use small molecules to block specific pathways involved in AA pathophysiology. As such, the use of janus kinase (JAK) inhibitors for treatment of AA has emerged. JAK inhibitors block the T-cell mediated inflammatory response thought to be the driving factor behind AA pathogenesis, by inhibiting the janus kinase (JAK) signal transducer and activator of transcription (STAT) signaling pathway, leading to a reversal of hair loss in AA patients. Thus, in an effort to demonstrate the efficacy of JAK inhibitors in the treatment of AA, several studies have been published within recent years. However, the question remains, “Are JAK inhibitors effective and safe in the management of Alopecia Areata?”. This review aims to provide a comprehensive report on the role, efficacy, and outcomes of using JAK inhibitors in the treatment of AA. To competently answer the research question highlighted, the most recent, quality articles published over a 10– 15-year period were sourced using PubMed, NCBI, Research gate, Medline, Cochrane Central Register of Controlled Trials, EMBASE and Google scholar. The literature search was primarily focused on randomized controlled trials (RCTs); however, in the absence of such, only the most recently published case reports, case series, clinical trials and open-label studies published to date were included.
Keywords: janus kinase, JAK-STAT signaling pathway, ruxolitinib, tofacitinib, baricitinib
Introduction/ Background
Alopecia areata (AA), also known as autoimmune alopecia, is a form of nonscarring alopecia and is the most common immune mediated cause of hair loss, worldwide.1 The immune disorder carries a 1–2% lifetime risk, with 10% to 20% of affected persons having a family history of AA.2 AA is independent of ethnicity and affects both males and females of any age. While it has been noted that 80% of cases occur before the age of 40, 50% of these cases begin in childhood and have been frequently associated with other autoimmune disorders such as systemic lupus erythematosus (SLE), atopic dermatitis and autoimmune thyroid disorders, to name a few.3
AA can affect any hair-bearing region and can manifest in various patterns ranging from patchy diffuse alopecia to progression to more severe forms such as alopecia totalis (AT) or alopecia universalis (AU). Although AA has been described as a benign, self-limiting condition with most cases resulting in spontaneous regrowth, this regrowth may take place over a period of several months to years, with approximately 66% of patients showing complete regrowth of hair within 5 years.4 However, relapse is common in AA, and studies have reported that in patients observed over a period of 10–20 years that the overall incidence rate of relapses ranges from 85–100%, with 100% relapse being observed in patients over a 20-year period.4 For patients who progress to the more severe forms of AT and AU, 75% will remain with AT and 34% of adults (44% children) would experience periods of hair growth with AU.4 From this, it is clear that patients may experience many difficulties while dealing with this disease. Affected individuals are at increased risk for emotional and psychological distress, including depression and anxiety. Furthermore, more persistent, or severe cases can significantly disfigure patients, proving to be detrimental to their well-being.
Previously, the etiology of AA was largely unknown. However, recently more research/ studies have focused on gaining a greater understanding of the molecular biology and autoimmune pathways that underlie the pathophysiology of the disease. Various studies have determined that the pathogenesis of AA largely involves hair follicles being attacked by autoreactive CD8 T-cells activated by janus kinase (JAK)-signal transducer and activator of transcription (STAT) signaling.5 Furthermore, evidence from genomic studies using mice models with AA have shown the critical role JAK signaling plays in the pathogenesis of the disease. These studies demonstrated that JAK-STAT signaling, specifically JAK 1/2 and JAK 1/3, led to T-cell mediated inflammatory responses which promoted IFN-γ and IL-15 production in hair follicles and even further amplified inflammatory responses surrounding these hair follicles.6 These findings have resulted in the emergence of targeted therapies involving the use of JAK inhibitors (JAKis), with the hope of providing a more efficient and effective therapeutic strategy for treatment of AA.
To date, there is no known reliable, approved treatment for AA. Several off-label treatments do exist that may result in temporary improvement of the disease, with limited efficacy reported. With the emergence of more studies and small clinical trials, there has been growing evidence that suggests that JAK inhibitors are effective in treating autoimmune disorders, including AA. Several studies on JAK inhibitors such as ruxolitinib, tofacitinib and baricitinib have already been published, attempting to demonstrate the efficacy and outcome of these JAK inhibitor agents. However, because most reports have been reported poorly and most trials being done are on a very small scale, data is limited with regard to efficacy and adverse outcomes. To provide a better understanding and fill any gaps, this review will aim to provide a comprehensive literature report on the evidence that is available on the role, efficacy, and outcomes of JAK inhibitors in the treatment of AA. With special focus on (i) examining the role of JAK inhibitors in treating AA, (ii) summarizing the available evidence on the efficacy of JAK inhibitors (oral as well as topical applications), (iii) determining the adverse outcomes/safety of JAK inhibitors in treating AA, (iv) examining the sustainability of treatment results with JAK inhibitors as well as affordability for long-term treatment, and (v) determining whether JAK inhibitors should be the drug of choice for treatment of AA.
Pathophysiology of Alopecia Areata (AA)
To fully understand the pathogenesis of AA, one must first have an understanding of the human hair growth cycle. The normal human hair growth cycle occurs in distinct phases: anagen (active hair/follicular growth), catagen (regression/follicular involution) and telogen (follicular rest and shedding of hair follicles). In AA, there is damage to hair follicles which occur in the anagen phase, which is then followed by a rapid transformation to catagen and telogen phases, then back to a dystrophic anagen status.2 During the active disease process, hair follicles are unable to progress beyond the early anagen phase. However, due to sparing of the follicular stem cells, no hair follicles are destroyed, ie there is no scarring.2
The pathomechanism surrounding AA is multifactorial and involves very complex interactions between immunologic, genetic as well as environmental factors.
Immunologic Factors
Studies have revealed that AA is an autoimmune disorder that firstly develops through the loss of the immune privileged status of hair follicles.1,7 According to research by Paus et al 2003, in their article entitled “The hair follicle and immune privilege,” it is suggested that the anagen hair follicle is a site of immune privilege. It is through the anagen phase that the proximal hair follicle/hair bulb can generate and maintain a site or region of relative immune privilege. Immune privilege is largely characterized by a number of mechanisms intricately working together to suppress cytotoxic immune attacks on cells and antigens located within specific sites.8 These mechanisms that establish and maintain immune privilege involve (i) the downregulation or absence of classical major histocompatibility complex (MHC) class I expression (this results in sequestering (auto)antigens in tissues, therefore, blocking their presentation to CD8 + T cells), (ii) the production of potent immunosuppressants locally in tissue sites, namely, tissue growth factor beta 1 (TGF β1), interleukin-10 (IL-10) and alpha-melanocyte stimulating hormone (α-MSH), (iii) impairment of the function of antigen presenting cells (APCs), (iv) the expression of non-classical MHC class I molecules (HLA-G, an MHC class Ib molecule in humans) and (v) greater expression of Fas Ligand (FasL) which serves to delete autoreactive T cells expressing Fas.8 The immune privilege of the anagen hair bulb, therefore, serves to maintain a very low level of MHC class Ia antigen expression as well as to locally produce potent immunosuppressive agents (α-MSH, TGF-β1, TGF-β2 and macrophage migration inhibitory factor (MIF) which prevents activation of T cell and NK T cell function).1 In addition to these, anagen hair bulbs have a very low number of APCs (CD 1a + cells and Langerhan cells) which are functionally impaired as they do not express MHC class II antigens.8 The hair bulb itself is devoid of lymphatics and is surrounded by a special extracellular matrix barrier which may prevent immune cell trafficking.8 Recent evidence also suggests that a dysfunction in programmed cell death protein I ligand (PD-LI) may also contribute to the loss of immune privilege as PD-LI functions to establish follicular immune privilege, which protects anagen hair follicles from autoimmune destruction.9
When this immune privilege status collapses or is lost, anagen hair follicles can undergo immune recognition and hence be attacked by autoreactive CD8 + T cells and NK T cells. This leads to a premature transition to catagen and telogen phases in the hair cycle, resulting in alopecia (hair loss). To this day, researchers are challenged with identifying exactly what factors are involved in triggering the collapse or loss of the immune privilege of the hair follicle.7,10 It has been suggested that possible triggers may include bacterial superantigens, infections, skin microtrauma, genetic factors, psychoemotional stressors, pregnancy, mast cell degeneration, hormones, as well as an intrafollicular/perifollicular rise in ectopic interferon-gamma (IFN-γ) secretion.8,10
Continuing with the cytotoxic disease process of AA, it has been noted that activated CD8+ T cells are the primary cytotoxic perpetrators that lead to follicular damage and are helped by CD4+ T cells.7,11 In addition to this, CD8+ T cells, although they are less numerous than CD4+ T cells, have the ability to penetrate to intrafollicular regions, further enabling greater disruption and destruction by these lymphocytes.7 As such, researchers have identified a cytotoxic cluster/subset of differentiation 8-positive (CD8+) NK group 2D-positive (NKG2D+) T cells that contribute to the pathogenesis of AA.6,12 This has been supported by the findings of CD8+NKG2D+T cells surrounding the peribulbar region of the affected hair follicles in humans.6 Additionally, using a mouse model (C3H/HeJ mouse model), researchers Xing et al, 2014, in their study of AA were able to recapitulate the pathologic features of AA and found a marked increased number of CD8+NKG2D+ T cells infiltrating the epithelial layers of hair follicles, further supporting this finding. Concomitant upregulation of MHC class I or ULBP3 (UL-16- binding protein 3), an NKG2D ligand in the hair follicle has also been identified to occur and has been deemed an important factor by genome-wide association studies in the pathogenesis of AA.6 According to Xing et al, 2014 this overexpression of ULBP3 leads to the attack of CD8+NKG2D+ T cells on the hair follicles. Concurrently, a pro-inflammatory response follows, involving a marked increase in IFN-γ response and upregulation of several cytokines (IL-2, IL-7, IL-13, IL-17, IL-15 and IL-21) as well as IFN-γ elements which further promote the activation and survival of IFN-γ producing CD8+NKG2D+ T cells.6,10
The role of CD4+ T cells as helper cells are highlighted in mouse models which demonstrated that when CD8 + T cells in conjunction with CD4+ T cells were transferred to mice, they induced extensive lesions of AA.12,13 Moreover, it was found that when CD8+ T cells were injected alone, they induced localized hair loss, whereas injection of CD4+ T cells alone, induced and promoted systemic AA; highlighting the special roles of both cell types in AA pathogenesis.13 In addition to this, a subpopulation of CD4+ T cells called regulatory T cells (Tregs), in particular CD4+CD25+ natural Treg cells, which are responsible for maintaining tolerance to self-antigens and thus, preventing autoimmunity through the secretion of inhibitory cytokines (IL-10, TGF-β), may have a defect in functioning in AA.7 Although the research on Tregs in AA has not been fully investigated and is therefore limited, it has been observed that in the active autoimmune disease state, a lower number of Tregs is found and produced.7 This has been supported by mouse models where Treg numbers in AA-affected C3H/HeJ mice were markedly reduced.7,13 Defects in Treg activity in humans with AA have also been identified, with low level circulating Tregs found to be incapable of suppressing high levels of serum cytokines (IL-2, IL-13, IL-15, IL-17) and IFN-γ, suggesting a defect in Treg function in patients with AA.7 It is these complex interactions of immunologic events that contribute to, and ultimately results in hair follicle dystrophy, with an acceleration of hair follicles into the catagen phase in AA.
Genetic Factors
It has been widely reported that AA has a strong hereditary component. Observational studies have shown that a high familial incidence of AA exists, ranging between 10% and 42%.14,15 In patients who had their first AA patch before the age of 30, a familial incidence of 37% was reported, and for those who developed a patch after the age 30, an incidence of 7.1% was reported.11 As such, there is some evidence that exists that shows a genetic predisposition to AA. This genetic predisposition has been linked to the major histocompatibility complex (MHC) gene; more specifically, the human leukocyte antigen (HLA) class I and class II genes, with HLA genes accounting for almost half of the genetic predisposition.11,16 HLA genes are mapped on to human chromosome 6p21.3 and encode for cell surface proteins that are important in immune self-recognition and antigen presentation.11 Studies have since identified expressions of HLA heterodimers in the affected scalp tissues of AA patients.11,14 Additionally, genome-wide association studies (GWAS) have discovered many single-nucleotide polymorphisms (SNPs) linked to AA.15–17 HLA class I genes are encoded by various loci (HLA-A, HLA-B and HLA-C) and are specifically recognized by CD8+ T cells, whereas HLA class II genes are bound by CD4+ T cells and are encoded by genes in the HLA-D region (HLA-DP, HLA-DQ and HLA-DR).11 Betz et al, 2015 in their GWAS meta-analysis, found the HLA-DR gene to be a key etiologic driver and having the strongest risk factor for AA, citing the strong association between HLA class II genes and CD4+ and CD8+ effector T cells in the pathogenesis of AA. More specifically, studies performed at a molecular level have identified that DRB1*11:04 allele, when increased in frequency, is linked to early onset AA with a high familial recurrence risk.11
Environmental Factors
In some instances, environmental factors have been cited as triggers that may induce or exacerbate AA, with stress being cited as a major factor. Although much of the evidence relating to stress and AA remains anecdotal, few studies have suggested a relationship between stress and AA. One such study demonstrated that patients affected by AA were found to have an increased frequency of stressful events occurring mostly within their families, when compared to the control groups.18 Another study acknowledged the relationship between stress and AA but found no correlation between the patient’s stress level or psychological state and the severity of AA.11 Most studies, however, remain inconclusive on the matter.14,19 More recently, it has been reported that in patients who may have developed chronic stress due to AA, there may be an alteration of the hypothalamic-pituitary-adrenal axis, resulting in high levels of neuromodulators being produced.11 This has been supported by findings in a C3H/HeJ mouse model study, where activities of the central and peripheral hypothalamic-pituitary-adrenal axis were found to be much higher, when compared to normal mice.15 This increased activity resulted in elevated hormones such as corticosterone, estradiol and adrenocorticotropic hormone, and was correlated to increased pro-inflammatory cytokines in the skin of mice, suggesting that there exists a potential role and relationship between psychological and physiological stressors that cause AA.15 There are other potential environmental factors that may have a role to play in AA, such as infections, diet (soy products, vit D levels), vaccinations as well as change in hormone levels, but these have not been fully investigated, and hence, their impact in the disease pathogenesis remains largely unknown.15
Psychological Burden of AA
For anyone suddenly losing hair in a bizarre pattern, hair loss can be psychologically painful, as hair has a very important role to play in one’s appearance and hence, self-image. Persons affected by AA may have a striking appearance that may result in acquiring intrusive stares from others. This not only limits the social freedom, but overall, affects the quality of life (QoL) of afflicted patients.20 Many patients afflicted by AA experience a tremendous amount of psychological distress. As such, psychological disturbances such as depression, anxiety, sleep disorders and even suicide, when compared to the general population, appear to be more prevalent in AA patients, proving to be very concerning.21 Ghanizadeh and Ayoobzadehshirazi 2014, in their study, reviewed the current knowledge surrounding the link between AA and psychological disorders and found the concordance rate of having at least one psychological disorder in AA patients to be very high (78%). Additionally, the study estimated that 50% of AA patients were affected by major depressive disorder (MDD), and there was a prevalence of 39% of generalized anxiety disorder (GAD) also reported among patients.22 Similar findings were reported in another study where the percentage lifetime prevalence of psychiatric comorbidities in AA patients were found to be 66%- 74%, with a 39%- 62% prevalence of GAD and a 38%- 39% lifetime prevalence of MDD.23 In addition to this, the study found that the age of onset of AA influenced the rate at which psychological comorbidities developed, with patients less than 20 years having an increased risk of, and hence earlier development of depression and anxiety, when compared to AA patients who developed patches after the age of 40 years.23
Given the severe negative psychosocial impact that AA inflicts on its patients, QoL studies have emerged as a means of gaining a better and more complete understanding of the impact of the disease on afflicted individuals. It is estimated that over 50% of AA patients experience diminished health-related QoL.21,23 Factors identified that contributed to low QoL scores were age less than 50 years, female sex, widespread involvement of hair loss (hair loss >25%), familial stress, job-related stress and psychological distress.21
The burden of AA not only impacts patients but has an economic impact as well, on government agencies as well as third-party proprietors. As such, the burden of the disease must be well understood so as to appropriately prioritize health-care resources. In 2010, the World Health Organization (WHO) measured the global disability adjusted life years (DALYs) lost to AA, through the Global Burden of Disease Study which involved 291 diseases across 187 countries in patients of all ages, during the period of 1990–2010.21 WHO measured the global DALYs lost to AA to be 1,332,800.23 This compared to the DALYs for other diseases such as diabetes mellitus and psoriasis were 46,857,100 and 1,050,600, respectively.23 Moreover, AA ranked 137/176 in disability burden which was higher than that of melanoma (138/176), psoriasis (144/176) and nonmelanoma skin cancer (150/176).21 Since 1990, the DALYs for AA have been on a steady linear increase, accounting for a significant global disease burden.23 As such, one can agree that treatment of this disease should be made a top priority.
Current Treatment Regimen for AA
To date, no specific treatment for AA exists. In the United States of America, the Food and Drug Administration (FDA) is yet to approve a specific therapy for AA.9,11 Similarly, in Europe, no universally accepted guidelines exist for AA, other than the guidelines proposed by the British Association of Dermatology (BAD) and the Italian Society of Dermatology.11 This is largely due to the fact that there are very few high quality randomized controlled trials (RCTs) that exist for AA management; although recently, this aspect has begun to change due to the Severity of Alopecia Tool (SALT) which was added to provide more guidelines for clinical research involving AA.15 In addition to this, determining the efficacy of these treatment modalities become difficult, as many patients spontaneously remit, even though the timeframe for this regrowth may take a few months or years.9 As the course of AA can be rather unpredictable, relapses alternating with spontaneous remissions and episodes of regrowth of hair, may not be easily attributed to the efficacy of a specific treatment, but rather to the natural disease course, or even to a placebo effect. As such, these prove as paramount difficulties in assessing what treatments are really effective according to evidence-based medicine.11
Current conventional treatment options for AA mostly include non-specific broad immunosuppressants that are administered either locally or systemically, with the most common approach involving the use of intralesional and topical steroids (first-line therapies).7 These therapies are not without side effects as intralesional and topical steroids may cause telangiectasias, skin atrophy and striae, whereas systemic steroids may lead to more adverse effects such as weight gain, acne, adrenal suppression, glucose intolerance and insomnia, among others.10 As such, they are only approved for short-term use. Other immunosuppressive agents used in treating AA include methotrexate, cyclosporine, and azathioprine. These agents lack adequate RCT evidence to support their use and therefore tend to require very close patient follow-up because of the potentially severe immunosuppression that may result.10 Contact immunotherapy involving the use of squaric acid dibutyl ester (SADBE) and diphenylcyclopropenone (DPCP) has also not been very effective in producing consistent results among patients afflicted with AA.10 The non-specific nature of these treatment modalities along with their variable treatment efficacy for AA should demand greater efforts to achieve more targeted therapies that can better target the pathways involved in the disease process while minimizing side effects. Common therapeutic options used in the treatment of AA are highlighted in Table 1.9,24–26
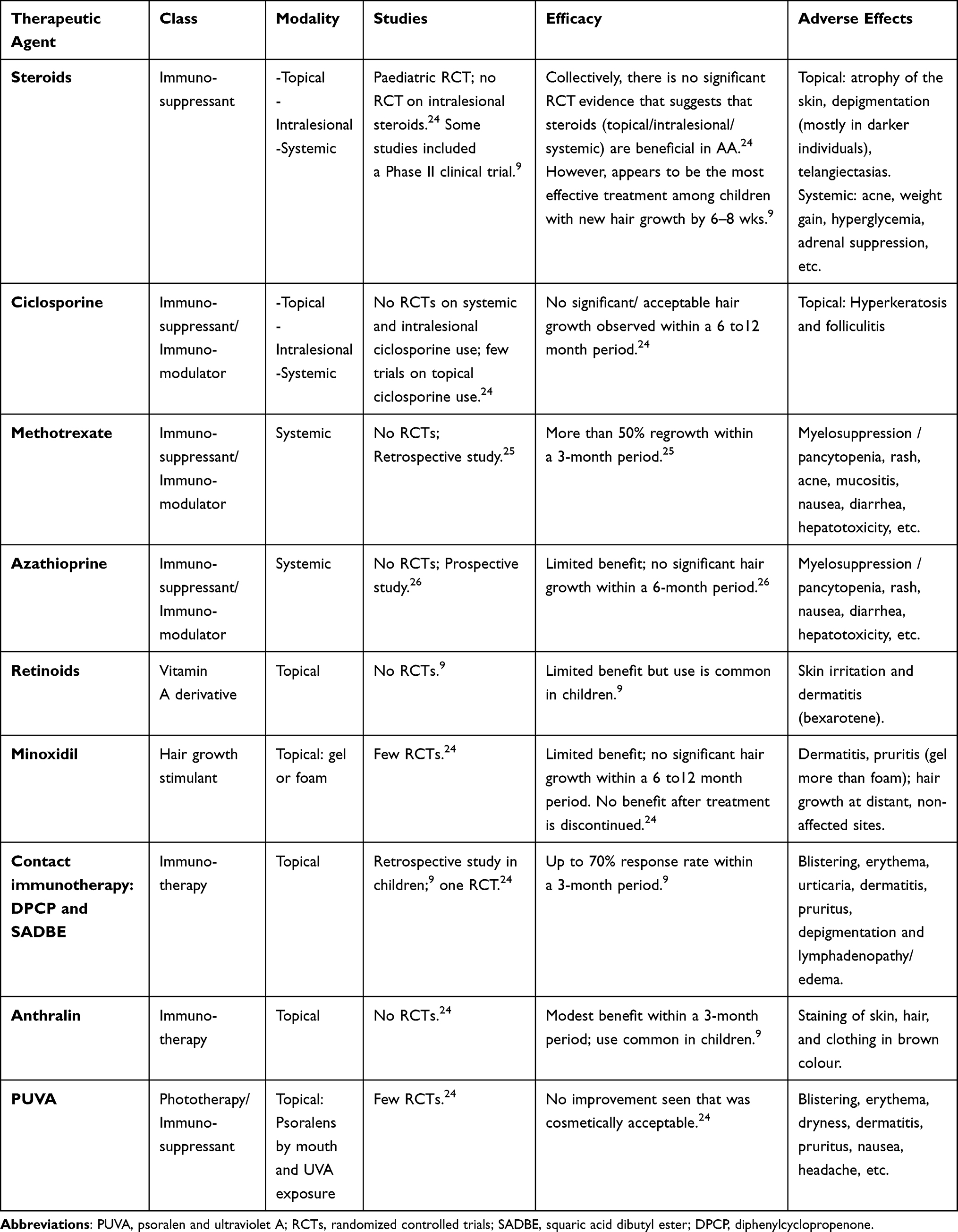 |
Table 1 Therapeutic Options Used in Managing Alopecia Areata |
Choosing therapeutic agents for AA are mostly based on empiric criteria which accounts for the patient’s age, the disease progress (acute or chronic) and the percentage hair loss (< or >50%).11 In the paediatric population, treatment is influenced by the patient’s age. Children that are younger than 10 years are treated with topical steroids, which may include second-line options such as topical minoxidil, topical retinoids, topical anthralin, and in some cases systemic therapy may even be considered.9 For children over the age of 10 years, the same therapeutic options exist with expanded options that include intralesional steroids, excimer laser therapies and contact immunotherapy such as SADBE.9
The Role of the JAK-STAT Signaling Pathway and AA
The JAK-STAT pathway is crucial in maintaining both innate and adaptive immunity. As such, its function was first discovered as a signaling pathway for IFN signaling.6 It is known as an intracellular signaling pathway that mediates the effect of many different pro-inflammatory signaling molecules that include, but are not limited to, interleukins (ILs), interferons (IFNs), colony-stimulating factors, growth factors and hormone-like cytokines.27,28 The JAK-STAT pathway consists of three main components: the receptor, janus kinase (JAK) and the signal transducer and activator of transcription (STAT).6 JAKs are large intracellular cytoplasmic molecules that belong to the family of tyrosine kinases and consist of four members: JAK 1, JAK 2, JAK 3 and tyrosine kinase 2 (TYK 2).27 JAK 1 is commonly known for playing a role in mediating signals for a wide range of inflammatory disorders, while JAK 2 is known for mediating signals for a range of inflammatory cytokines that involve hematopoiesis.27 JAK 3 activity, on the other hand, is restricted to lymphoid lineage and is known to be the cause of severe combined immunodeficiency syndrome due to a loss of function mutation in the JAK 3 molecule, whereas sudden gain of function mutations in JAKs act as oncogenes in a variety of hematologic malignancies and lymphoproliferative disorders such as cutaneous T cell lymphoma.5,27 STATs on the other hand consists of a family of transcription factors that act as downstream regulatory signals of JAKs and consist of seven members: STAT 1, STAT 2, STAT 3, STAT 4, STAT 5A, STAT 5B and STAT 6.29
When ligands become bound to and engage with extracellular receptor components, JAK proteins become activated by phosphorylating its own tyrosine residue (autophosphorylation) in order to activate its kinase function. This in turn, phosphorylates the adjacent STAT component (transphosphorylation), which then dimerizes and activates STATs that will then translocate into the cell nucleus to promote direct regulation of gene expression through DNA transcription (Figure 1).6,27,28 Receptor subunits tend to be associated with a specific JAK; however, there are some that may be associated with more than one JAK molecule. As such, various combinations of JAKs may form a dimer on different cytokine receptors, with one of the seven STAT members, leading to downstream signaling.27
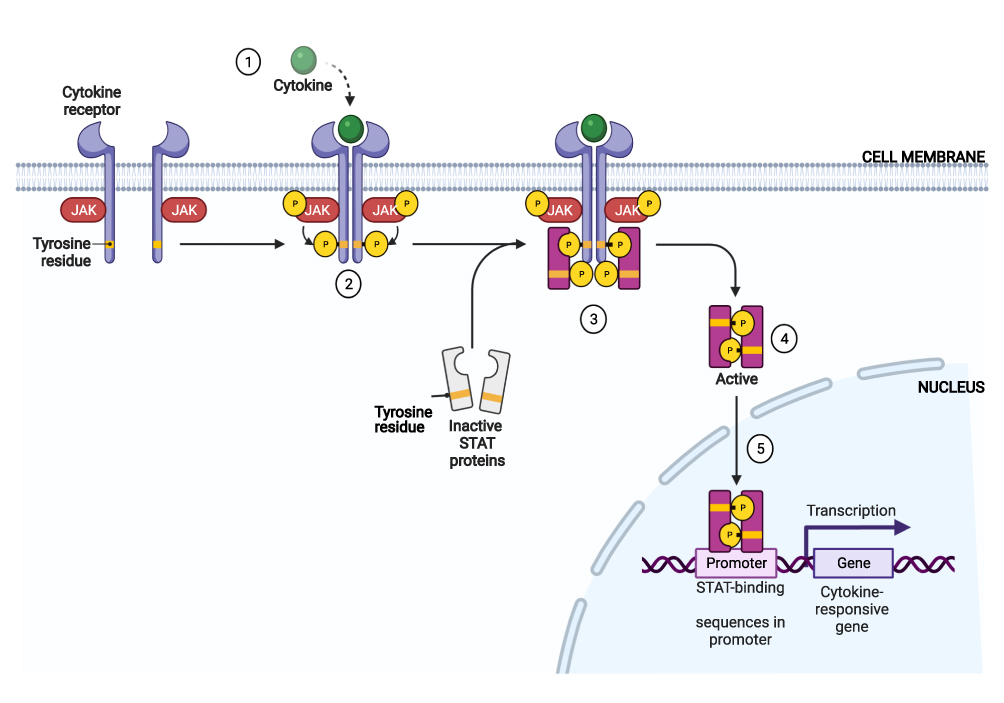 |
Figure 1 The Janus kinase signal transducer and activator of transcription (JAK-STAT) signaling pathway. Abbreviations: JAK, janus kinase; STAT, signal transducer and activator of transcription. Notes: (1) Cytokine binding induces receptor dimerization. (2) Phosphorylation of tyrosine residues by JAK. (3) STATs recruited to phosphorylated cytokine receptor followed by JAK-mediated phosphorylation. (4) Dimerization of STATs. (5) Translocation to nucleus with activation of targeted gene transcription. Created with BioRender.com. |
A vast number of cytokines, mainly γc cytokines, have been discovered that signal through and activate the JAK-STAT pathway, including IL-15 and IFN γ.6 Primarily, IL-15 signals through JAK 1/3, while IFN γ mainly signals through JAK 1/2.27 This is further supported by C3H/HeJ mouse models used in the studying of JAK inhibitors, where it was found that CD8 + effector T cells produced IFN γ which signalled through JAK 1/2 intracellular pathways to produce IL-15 and IL-15r which further mediated activation of the pathway by binding to CD8 + T cell receptors; this in turn led to an increase in the production of IFN γ by CD8 + T cells through JAK 1/3 signaling pathways.7 With this in mind, it can be clearly determined that the JAK-STAT pathway plays a crucial role in mediating and maintaining the cytotoxic CD8+NKG2D+ T cell reaction that occurs in the pathogenesis of AA.6 Moreover, these observations seen in both human and C3H/HeJ mouse models have provided the rationale for the use of JAK inhibitors in treating AA.12,27,30
Mechanism of Action and Role of JAKis
JAK inhibitors (JAKis) belong to a class of immunomodulatory drugs that, over the years have been extensively studied in the treatment of many inflammatory mediated conditions such as psoriasis, rheumatoid arthritis, atopic dermatitis, systemic lupus erythematosus and myelodysplastic disorders.6 More recently, JAKis have received growing attention from researchers and dermatologists in the treatment of AA. JAKis are often classified into first generation and newer JAKis (second generation). First-generation JAKis are less selective and therefore demonstrate activity against three or all four of the members of the JAK enzyme family (pan-JAK inhibitors), whereas newer JAKis display more selectivity for specific JAKs.28 Although there have been many JAKis developed over the years, only three drugs (tofacitinib, ruxolitinib and baricitinib) have been reported in trials and research literature for the treatment of AA and at present, only two (tofacitinib and ruxolitinib) have been US FDA approved for clinical use for other autoimmune conditions (eg psoriasis, rheumatoid arthritis, myelodysplastic disorders).27
The mechanism of action of JAKis in the treatment of AA has been highlighted in several studies. In general, JAKis act as antagonists to the members of the JAK enzyme family and thus, inhibits JAK phosphorylation thereby blocking the downstream regulatory signaling pathway that involves STAT activation (Figure 2).31 In AA, this mechanism helps stimulate and restore hair growth through several means which involve: (i) Terminating the T-cell mediated immune response on the hair follicle by blocking the downstream signaling of inflammatory mediators IFN γ and γc cytokines (IL-2, IL-4, IL-7, IL-9, IL-15, IL-21 and IL-23 (via TYK2)); (ii) Inhibiting the production of inflammatory T helper cells (Th17 cells, Th1 and Th2 differentiation cells); and (iii) Restoring the anagen phase of the hair follicle by promoting activation and/or stimulation of hair follicle stem cells.6,12,31
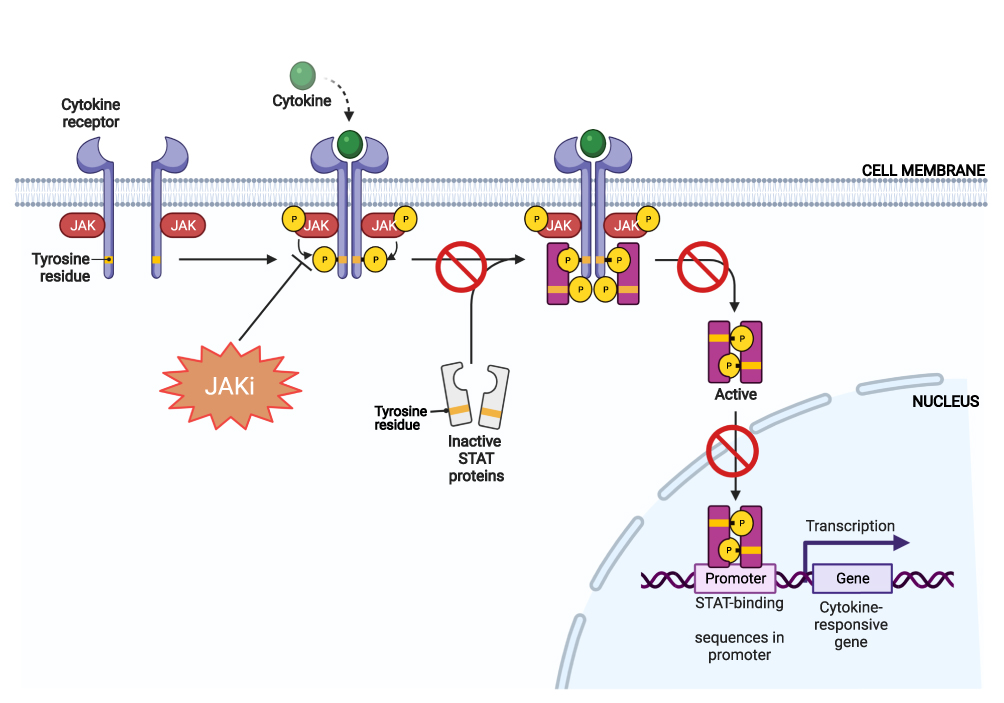 |
Figure 2 Role of JAKis in the JAK-STAT pathway. Abbreviations: JAKis, JAK inhibitors; JAK, janus kinase; STAT, signal transducer and activator of transcription. Notes: JAKis inhibit the JAK-STAT pathway by blocking JAK phosphorylation and hence STAT activation, leading to a downregulation or termination of inflammatory mediators (eg IFN γ, cytokines, chemokines etc). Created with BioRender.com. |
Evidence on the Efficacy and Safety of JAKis
Tofacitinib
Tofacitinib is the first of the first-generation JAK inhibitors. It exhibits greater selectivity for inhibiting JAK 1/3 dependent STAT over JAK 2, with very little to no effects on the TYK 2 pathway.6 Currently, it is US FDA approved for treatment of rheumatoid arthritis, but has been used as an off-label therapy for many inflammatory mediated dermatological conditions that include but are not limited to, vitiligo, psoriasis, dermatomyositis, etc.9 Tofacitinib inhibits the effects of many γc cytokines including IL-2, IL-4, IL-7, IL-15, IL-21 and IFN γ, by blocking the STAT phosphorylation induced by these inflammatory mediators, thereby disrupting the downstream signaling pathway of JAK 1/3 dependent γc receptors, as reported in both human and mice models.6,12
Craiglow and King 2014, were the first to report on the efficacy of tofacitinib in a case report of a young male with AU and psoriasis who was treated with oral tofacitinib (15mg daily) for 8 months and achieved complete regrowth of hair on all affected areas after completion of therapy. Since then, there have been several publications of clinical research studies on the efficacy of tofacitinib (administered orally and topically) in treatment of AA and its variants (AU & AT), in both adult and pediatric patients (Table 2).32–58 Much of these studies highlighted in Table 2 are case reports/series, with a few open-label pilot studies and retrospective studies. These cases consist of patients diagnosed with AA and its variants that had either failed treatment with or were refractory to common conventional therapy such as corticosteroids.
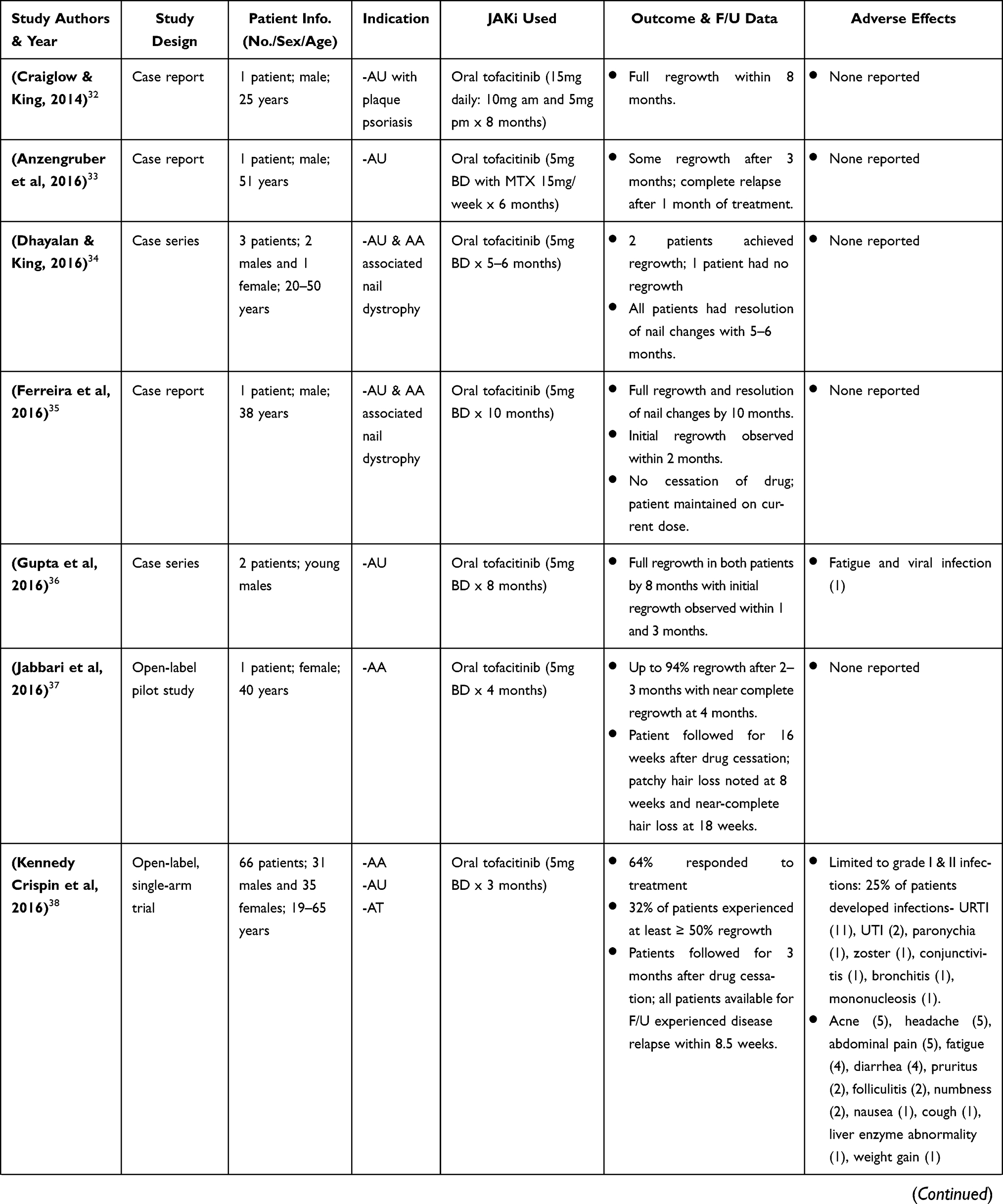 | 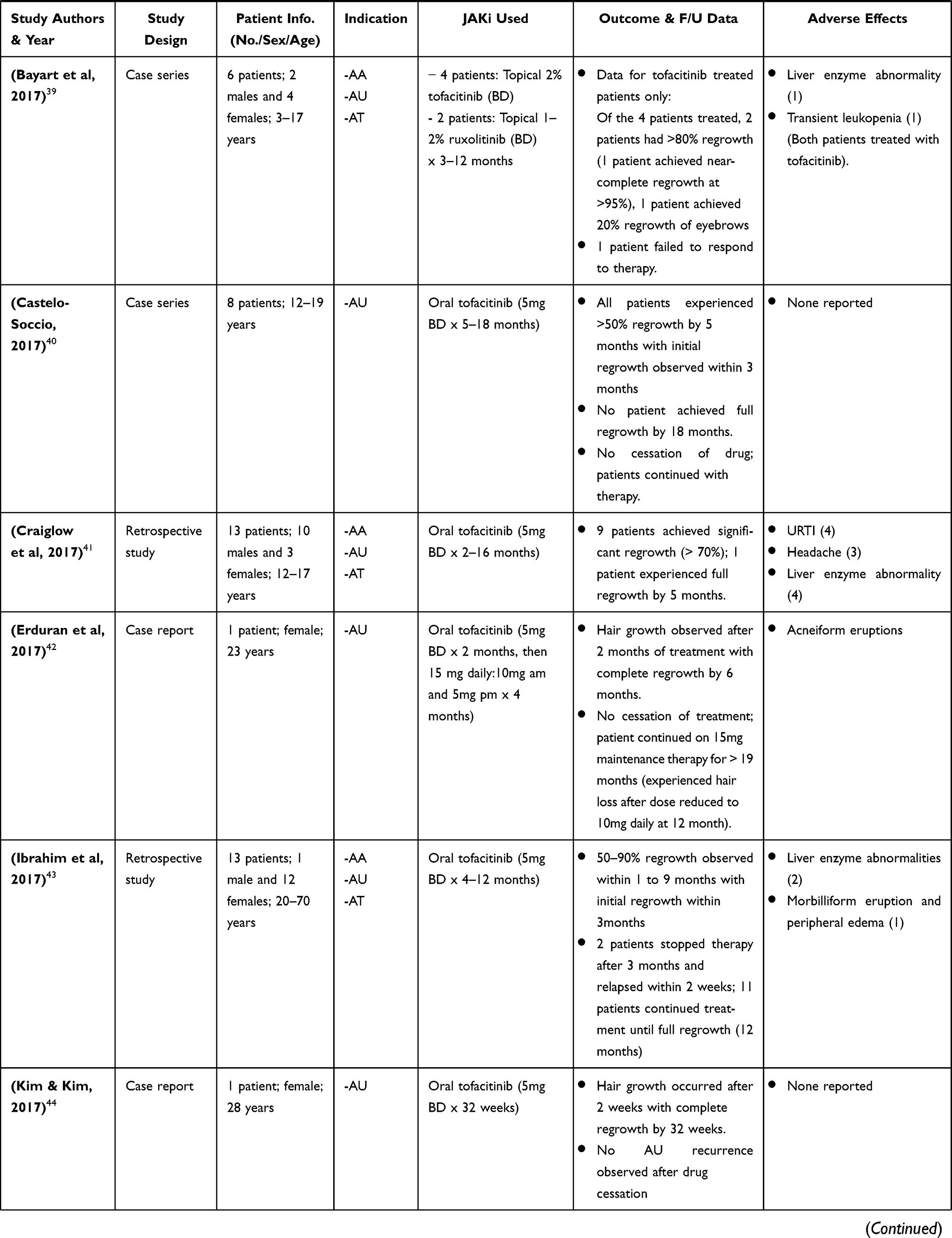 | 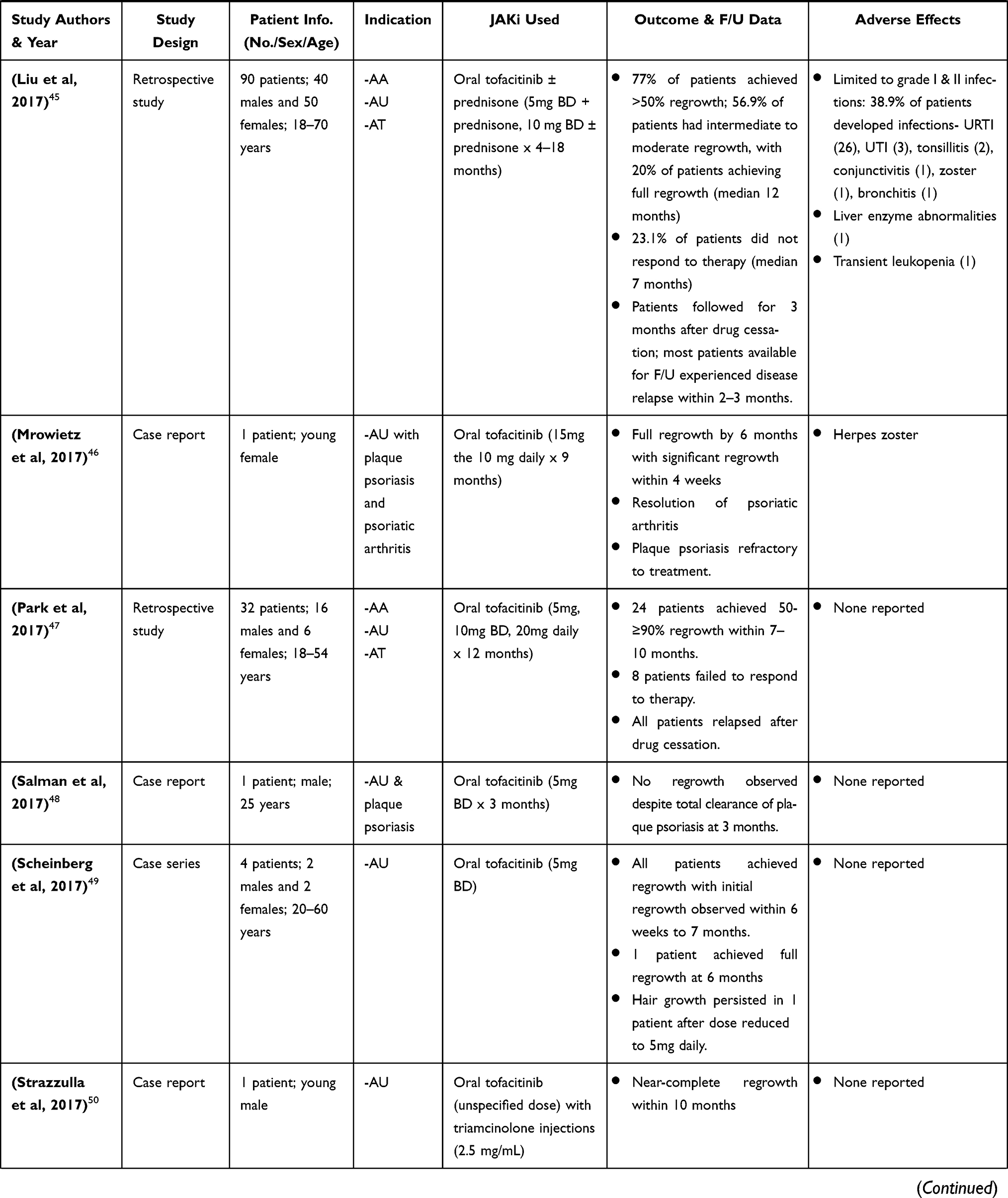 | 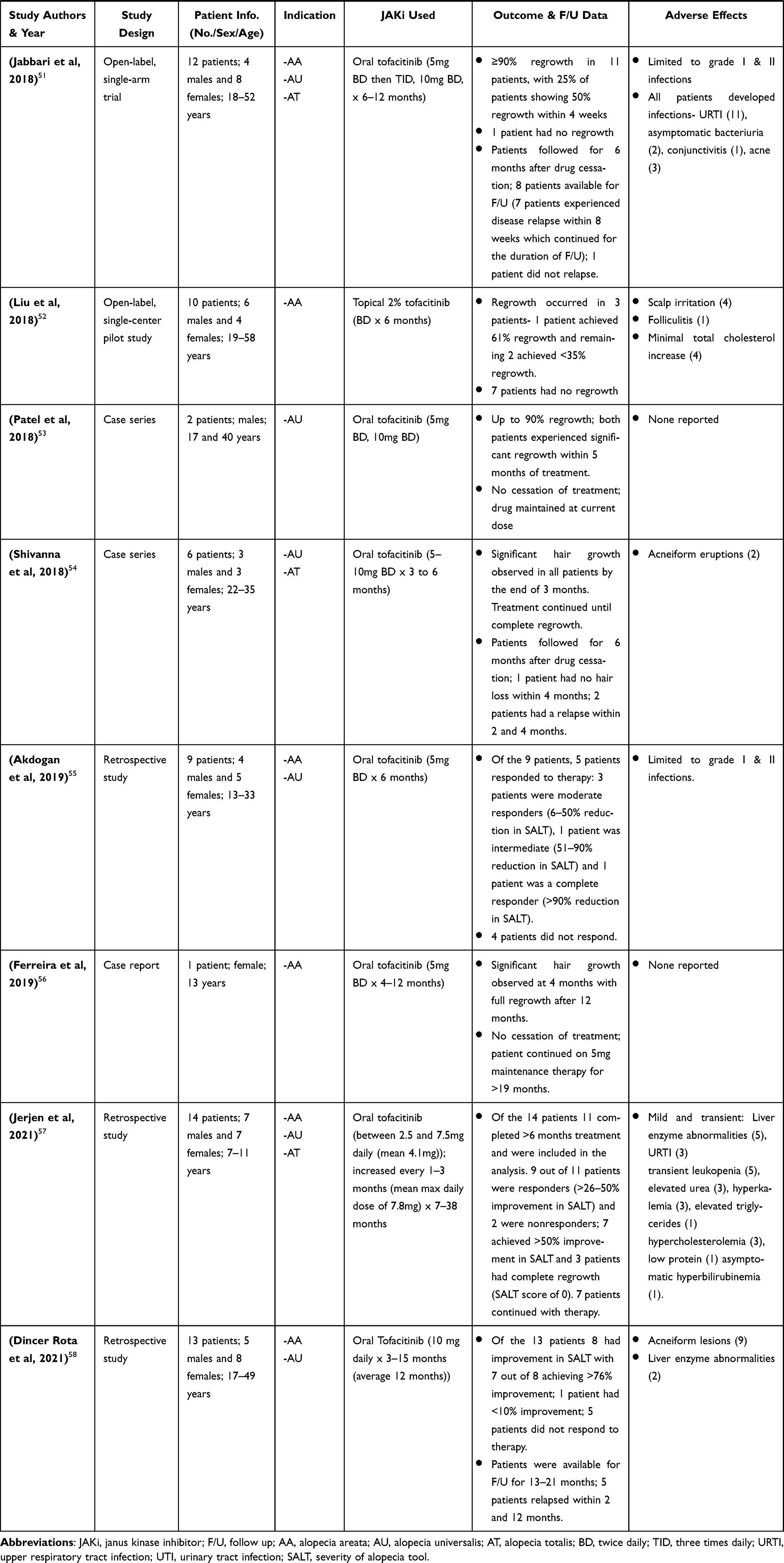 |
Table 2 Clinical Research Studies of Tofacitinib in Alopecia Areata Treatment |
In a most recent case report by Ferreira et al 2019, a 13-year-old female with AA was administered oral tofacitinib 5mg twice daily for 4 to 12 months. The patient had previously been treated with topical and systemic steroids (betamethasone and prednisone) and was refractory to treatment for >5 years.56 Following this, she was started on methotrexate and cyclosporine (duration 9 and 4 months, respectively) but showed no response/improvement.56 Intralesional acetonide triamcinolone was later administered and showed good response, however, later resulted in painful, severe alopecia. After >5 years of failed treatment with common conventional therapy, tofacitinib was administered and the patient was followed up every 4 weeks.56 Significant hair growth was observed at 4 months with complete regrowth after 12 months therapy.56
The largest retrospective study consisting of 90 patients as conducted by Liu et al 2017, demonstrated >50% regrowth in 77% of patients (aged 18–70 years) diagnosed with AA, AU or AT. Patients were administered oral tofacitinib 5mg or 10mg twice daily with or without prednisone and were evaluated using the Severity of Alopecia Tool (SALT) scoring system; first at baseline then at various treatment intervals for a duration of 4–18 months.45 Patients who responded to therapy were identified as intermediate responders, moderate responders and complete responders, whereas those who did not respond were identified as nonresponders. Of the 90 patients, 65 were identified as potential responders based on factors that included, localized disease with AA and the duration of disease with AU or AT being ≤10 years.45 As such, 13 patients (20%) were complete responders and had full regrowth with a >90% reduction in SALT, whereas 37 patients (56.9%) were intermediate to moderate responders (25 patients intermediate and 12 patients moderate) with a 51%-90% reduction in SALT for intermediate responders and a 6%-50% reduction in SALT for moderate responders.45 Additionally, 15 patients (23.1%) were identified as nonresponders with a ≤5% reduction in SALT.45 Other retrospective studies measuring oral tofacitinib administered at ≥10mg daily demonstrated similar results with a significant reduction in SALT and at least 50%-90% (median 70%) regrowth in its patients.41,43,47
Jabbari et al 2018, in their recent open-label single-arm trial consisting of 12 patients (18–52 years) diagnosed with moderate to severe AA or its variants, demonstrated that administering oral tofacitinib ≥10mg daily for 6–12 months resulted in ≥90% regrowth. Patients were first given tofacitinib 5mg twice daily for 1 month and if ≥50% regrowth from baseline was not achieved during this time, the dosage was gradually increased to 15mg then 20 mg daily.51 Of the 12 patients, 1 patient did not complete treatment and had no regrowth; 8 patients had ≥50% regrowth and 3 had ≤50% regrowth from baseline.51
Very limited studies have addressed the efficacy of topical tofacitinib.39,52 In an open-label single-arm pilot study, Liu et al 2018 assessed the efficacy and safety of topical tofacitinib in 10 patients (19–58 years) with AA. Patients were treated with 2% tofacitinib twice daily for 6 months and were assessed periodically for regrowth using SALT.52 Regrowth occurred in 3 patients (significant regrowth in 1 patient at 61% and partial regrowth in 2 patients at 35%) while 7 patients had no regrowth.52
Generally, tofacitinib (oral and topical) appears to be well tolerated, with most studies reporting no adverse effects in patients (Table 2).32–35,37,40,44,47–50,53,56 For studies that have reported adverse effects of tofacitinib, these have been transient and were limited to grade I & II infections (URTI, UTI, zoster, folliculitis, conjunctivitis, etc), with rarer occurrences of liver enzyme abnormalities, thrombocytopenia, neutropenia, hypercholesterolemia and acneiform eruptions.38,39,41,43,45,51,52,57,58 For some patients, discontinuation of tofacitinib resulted in a reversal of adverse effects, whereas for others these resolved spontaneously. No serious adverse effects such as malignancy or hospitalization were reported.
Ruxolitinib
Ruxolitinib is another first-generation JAK inhibitor that is US FDA approved for treatment in myelofibrosis and is currently being tested for used in many other dermatologic conditions.9 Ruxolitinib selectively inhibits JAK1/2 and to a lesser extent TYK2, and has also demonstrated anti-inflammatory effects by means of disrupting IL-17 signaling.6 This has been supported in mice models where ruxolitinib administration was able to reduce cytokine induced phosphorylation of STAT, as well as the levels of circulating inflammatory cytokines (IL-6 and TNF α).6
Multiple case reports have been published to examine the use of ruxolitinib both oral and topical in AA (Table 3).12,39,59–72 Additionally, there has been one open-label, single-arm trial by Mackay-Wiggan et al, 2016. In this trial, 20mg ruxolitinib was administered twice daily for 3–6 months to 12 patients with moderate to severe AA. Of the 12 patients, 9 (75%) had ≥50% regrowth at 12 weeks and at the end of 6 months, 7 of these 9 patients had >95% regrowth (all 9 patients had a 92% reduction in SALT).63
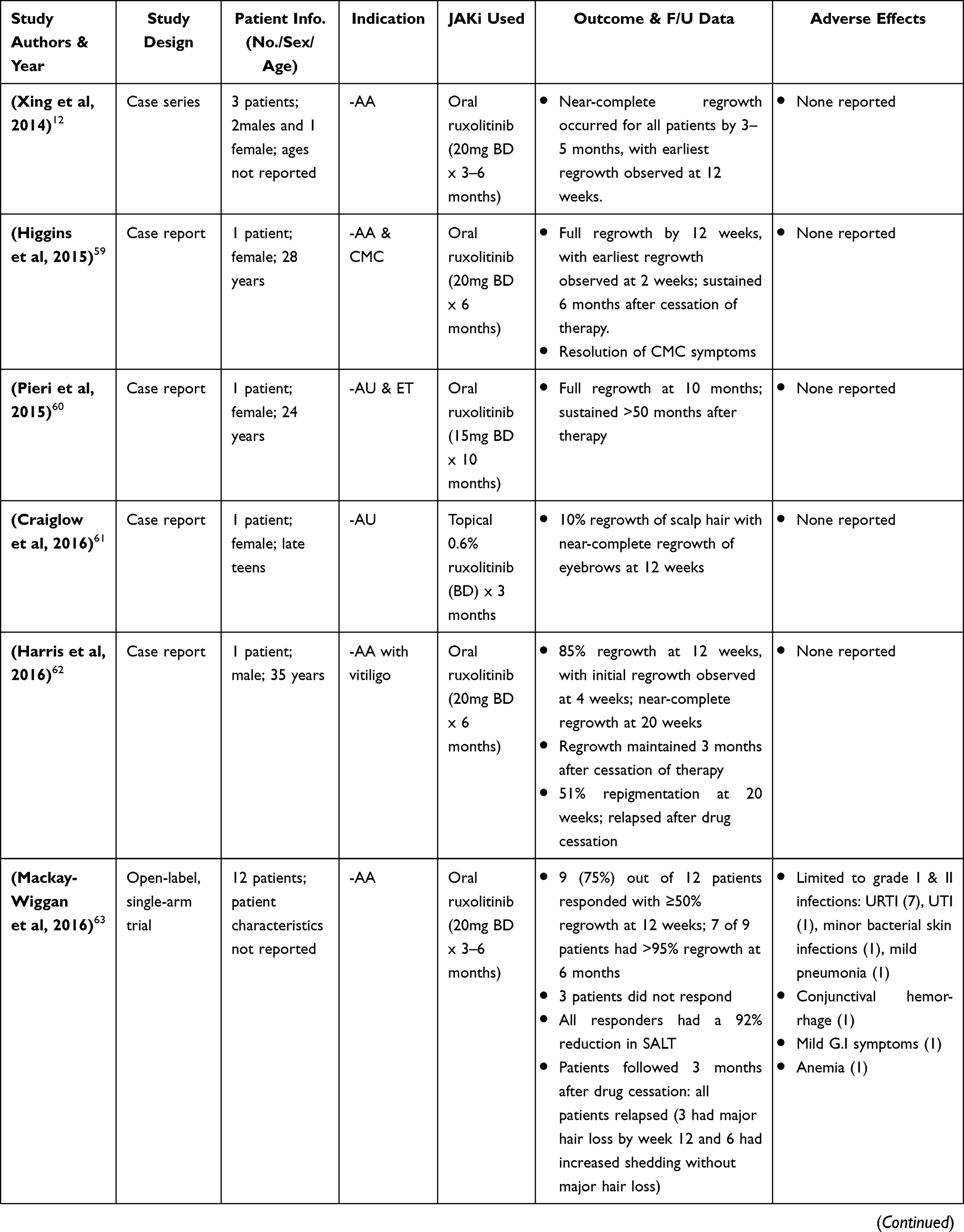 | 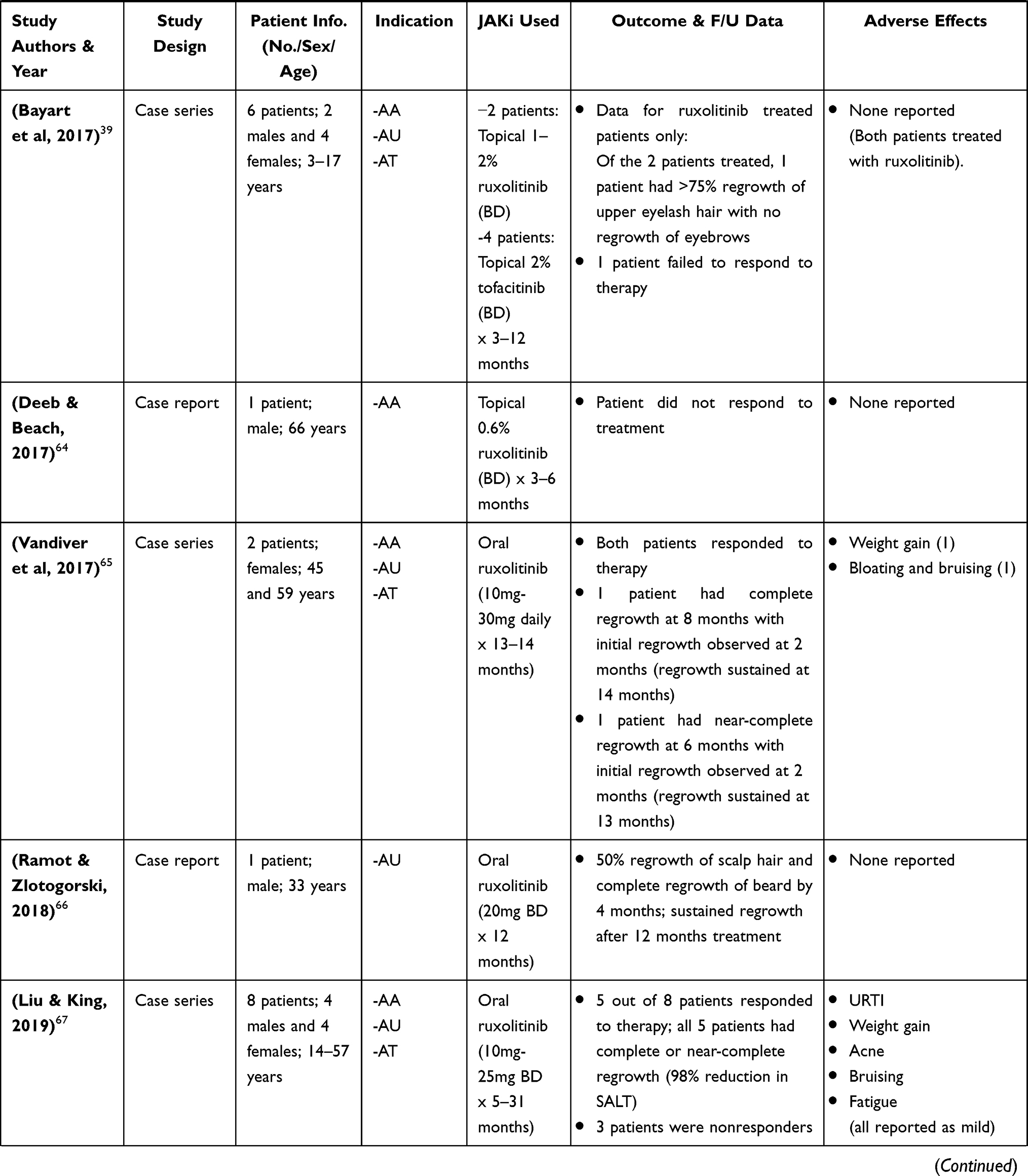 | 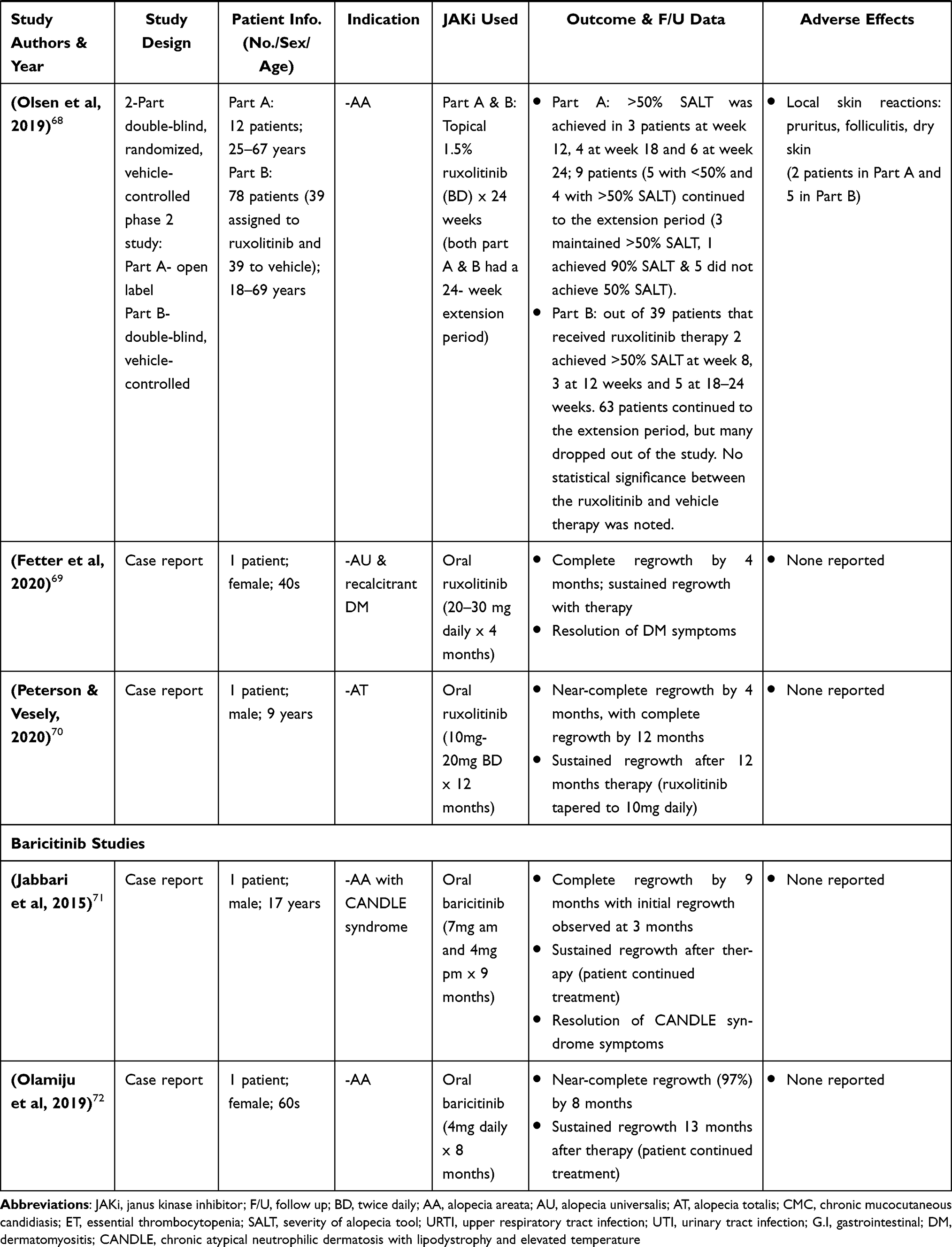 |
Table 3 Clinical Research Studies of Ruxolitinib & Baricitinib in Alopecia Areata Treatment |
Most recently, a case report by Fetter et al 2020, demonstrated the effects of ruxolitinib in a female patient in her 40s with longstanding AU and recalcitrant dermatomyositis. The patient had AA which subsequently progressed to AU for >20 years and was previously treated with a number of immunosuppressive drugs (methotrexate, prednisone, azathioprine, cyclosporine A, etc) for over a period of 10 years without any improvement.69 Ruxolitinib was initiated at 20–30mg daily for 4 months and the patient had complete regrowth at the end of this 4-month period, with a resolution of dermatomyositis symptoms.69 Other case reports have exhibited similar findings of oral ruxolitinib.12,59,60,62,67,70
Topical efficacy of ruxolitinib has not been well studied and has only been demonstrated in a few studies (Table 3).39,61,64,68 Craiglow et al 2016 reported a case of a young female with AU treated with topical 0.6% ruxolitinib, twice daily for 12 weeks. At the end of 12 weeks, the patient had near-complete regrowth of eyebrows with 10% scalp hair regrowth.61 Similar unfavourable efficacy of topical ruxolitinib was reported in Bayart et al 2017 (topical 1–2% ruxolitinib twice daily for 12 months) and Deeb and Beach 2017 (topical 0.6% ruxolitinib twice daily for 6 months). In both case reports, patients failed to respond to treatment.39,64
Similar to tofacitinib, ruxolitinib is well tolerated in AA patients, with only a few studies reporting grade I and II infections and other mild symptoms (Table 3).63,65,67,68 No reports of hospitalization or malignancy related to ruxolitinib have been reported in the literature.
Baricitinib
As one of the newer first-generation JAK inhibitors, baricitinib selectively inhibits JAK1/2 and to lesser extent, JAK 3. It is a very potent inhibitor that does so by disrupting JAK functioning and signaling initiated by inflammatory cytokines IL-6 and IL-23.6 Baricitinib is currently being evaluated in Phase II and Phase III trials for many inflammatory dermatoses including atopic dermatitis, rheumatoid arthritis, psoriasis, and myelofibrosis.9 In the treatment of AA, it is not well studied. As such, only 2 case reports have been published that examine the efficacy of baricitinib (oral administration only) in AA (Table 3).71,72
The first case report, as published by Jabbari et al 2015, reported a case of a young male with AA and chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) syndrome, who was administered baricitinib 11mg daily (7mg in the morning and 4mg in the evening) for a duration of 9 months. The patient had complete regrowth by 9 months, with a resolution of CANDLE syndrome symptoms.71 Similar findings were observed in the most recent case report by Olamiju et al 2019, which reported a case of an elderly female with AA that was administered baricitinib 4mg daily for 8 months at the end of the 8-month period, the patient had near-complete regrowth at 97%.72
In both case reports no adverse effects were reported following baricitinib use. This is supported by a study conducted by Shi et al 2011, that demonstrated that oral administration of baricitinib in healthy volunteers either produced no adverse effects or mild adverse effects (reduced reticulocyte count and neutropenia) in a minority of subjects.73 No malignancy reports related to baricitinib use has been identified. In fact, based on a study that assessed carcinogenicity in mice models, baricitinib was found to be non-carcinogenic.74
In summary, JAK inhibitors appear to be effective in the treatment of AA and its variants. Moreover, oral JAK inhibitors demonstrated a higher efficacy in treatment of AA than topical JAK inhibitors. Phan and Sebaratnam, 2019, in their systematic review and meta-analysis of JAK inhibitors for treatment of AA have supported this finding, as they demonstrated that regardless of the specific agent of oral JAK inhibitor used in treatment, it was associated with a 4 times higher odds of achieving a response, when compared to patients who were administered topical JAK inhibitors.75 Additionally, treatment with oral JAK inhibitors was also associated with a 7 times higher odds of achieving a complete response (50%-100% regrowth) than a partial response (5%-50% regrowth) when compared to topical therapy; with no difference between the specific agents used (ie) tofacitinib, ruxolitinib and baricitinib.75 The study also found that factors such as sex, age, previous failure of systemic therapies, as well as the duration of AA did not appear to influence a patient’s response to therapy.75 As such, the method of JAK inhibitor administration was seen as the major factor that determined response to treatment, with oral JAK inhibitors having the best outcomes.75 Overall, treatment of AA with JAK inhibitors appears to be safe and well tolerated with a low complication rate. The most common adverse effects were limited to grade I and II infections, which mostly presented as upper respiratory tract infections and urinary tract infections. Few cases identified mild, transient laboratory changes in liver transaminases, cholesterol levels and blood cell count, but the prevalence of such was low and completely reversible.38,45,51,52,75 No significant difference in complication profiles were established between topical and oral JAK inhibitors, and no new malignancies or hospitalizations associated with JAK inhibitor use for AA were reported. This, however, may be due to the limited safety data available for JAK inhibitor use in AA.
Currently, there are ongoing human clinical trials, including double-blind placebo-controlled phase II and III trials that are evaluating the efficacy and safety of JAK inhibitors (first generation and second generation/newer JAKis), highlighting the growing importance and interest in these drugs for the treatment of AA and its variants (Table 4).
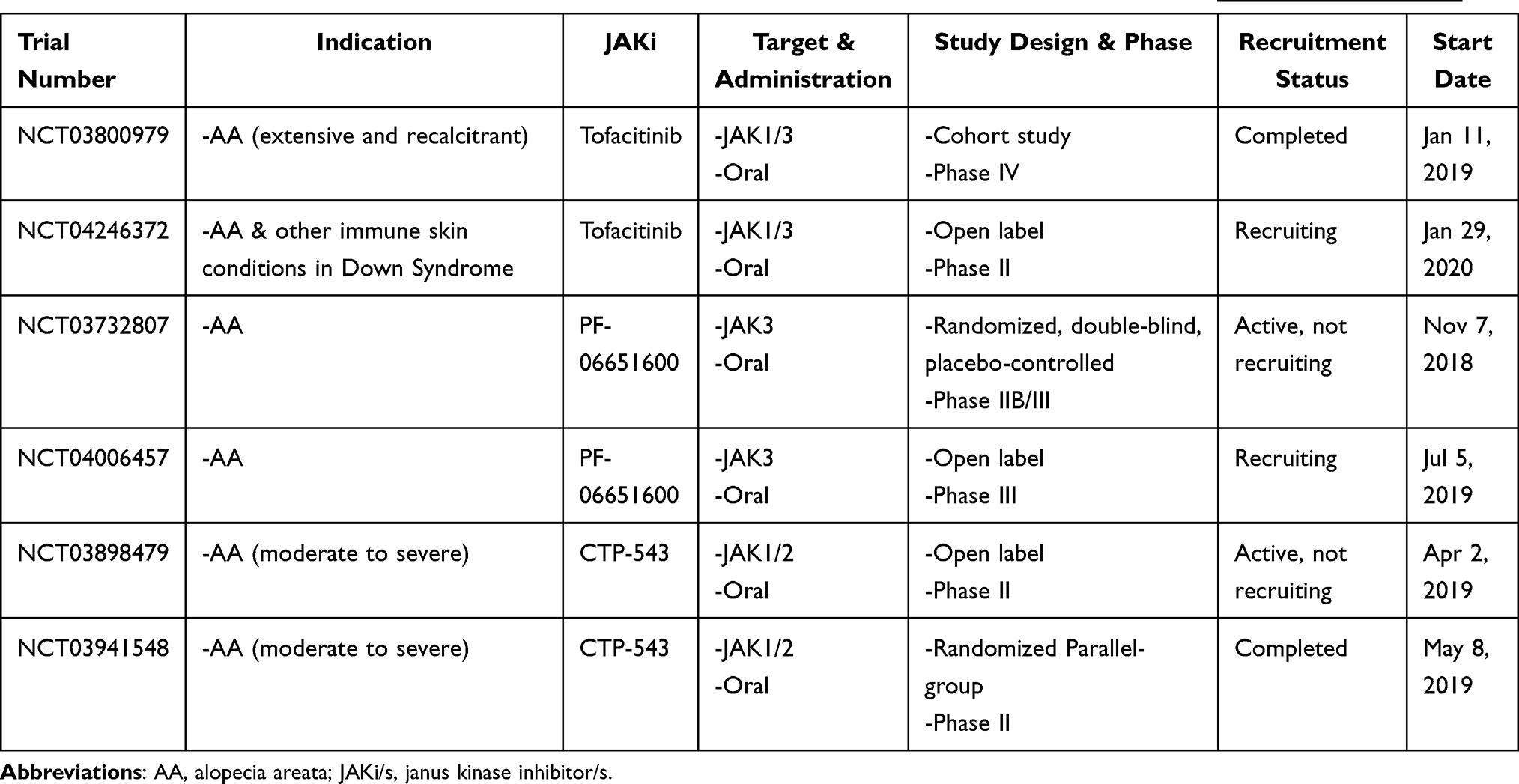 |
Table 4 Current Clinical Trials Listed for AA Treatment- First Generation & Newer JAKis, According to http://clinicaltrials.gov |
Sustainability of Treatment Results with JAKis and Affordability for Long-Term Treatment
The sustainability of treatment results with JAK inhibitors used for AA management has been an area of concern for many dermatologists and researchers alike. In clinical practice, it has been observed that AA frequently recurs after cessation of JAK inhibitor therapy. Liu et al 2017, in their retrospective study where 90 patients were treated with oral tofacitinib (as previously mentioned in this review), followed patients for 3 months after drug cessation and found that all patients who previously responded to treatment, experienced disease relapse within 2–3 months. Jabbari et al 2018, followed 11 patients for 6 months after drug cessation, who had achieved ≥90% regrowth on oral tofacitinib in an open label single-arm trial. Of the 11 patients, 8 were available for follow-up and disease relapse occurred in 7 patients within 8 weeks after drug cessation (1 patient did not relapse).51 Similarly, in another open label single-arm trial by Mackay-Wiggan et al 2016, all patients who responded to oral ruxolitinib treatment were followed for 3 months duration after drug cessation and were all found to have relapsed within 12 weeks. Many other studies have reported similar findings of disease relapse in patients after cessation of JAK inhibitor therapy, including the systematic review by Phan and Sebaratnam 2019, which found that not only were all cases of relapse in the systematic review associated with drug cessation, but there was an average of 2.7 months by which this relapse occurred.38,43,47,75,76
Additionally, for some patients, higher oral doses of JAK inhibitors (≥20mg daily) is required to achieve initial as well as complete regrowth and once this regrowth has occurred, it has been suggested that patients should be continued on maintenance doses to maintain hair growth, which may potentially be reduced over time.27 Several case reports have demonstrated this aspect.42,56,70 Erduran et al 2017 reported a case of a young female patient treated with oral tofacitinib (initially 10mg daily, then increased to 15mg daily) who achieved complete regrowth by 6 months. Treatment was initially maintained on 15mg daily up until the 12th month but was then tapered to 10mg daily. This resulted in patchy hair loss and the dose was increased again to 15mg daily. Since then, the patient has been on 15mg daily maintenance therapy for >19 months without relapse.42 Based on these findings, it can be determined that in order for patients to maintain full regrowth after JAK inhibitor use, patients will most likely be required to continue the therapy indefinitely. This raises concerns as the long-term effects of oral JAK inhibitor agents taken indefinitely for AA treatment are not known. NK cells and interferons play a very important role in tumor surveillance and any drugs that block the actions of these cells, such as JAK inhibitors, may increase the risk of malignancies. Thus, transitioning patients to a topical regiment after achieving complete regrowth may be more ideal as well as sustainable and may mitigate any systemic effects and severe potential adverse effects such as cancer. However, presently there are no topical JAK inhibitors that have yielded results that were found to be satisfactory in any clinical studies.39,52,61,64
Another area of concern is the cost or affordability of JAK inhibitor use for long-term treatment of AA; with some studies highlighting that the extremely high cost of JAK inhibitors act as a barrier for their use in AA.9 As such, Gilhar et al 2019, in their analysis of JAK inhibitors and AA, calls for a “sober risk-cost-benefit evaluation,” since the nature of the disease is non-life-threatening and JAK inhibitors are “prohibitively expensive.”77 GlobalData Healthcare, a fully integrated database for the commercial analysis and research of biotechnology, pharmaceutical and medical device products, has conducted primary research on the annual cost of therapy (ACOT) that JAK inhibitors pose to patients. In their report, it was demonstrated that particularly in the USA, the ACOT of JAK inhibitors if approved for AA treatment, will be approximately $50,000 per patient.78 This, when compared to other treatment options such as cyclosporine, which is currently the most expensive immunomodulatory drug for AA with an ACOT of $1400 per patient, is very high.78 Other pharmaceutical companies within the USA have priced the ACOT of JAK inhibitors similarly at around $59,000 per patient.79 With this in mind, long-term treatment with JAK inhibitors may become financially burdensome for patients and, thus, for future reference, physicians should be aware of the possible financial ramifications that patients may incur once JAK inhibitor therapy is recommended. As a means of circumventing such, broader financial coverage for treatment of this psychoemotional disease is warranted and should be adequately explored by various financial institutions and payers such as healthcare insurance companies.
A Comment on JAKis as the Drug of Choice for AA
AA is a complex autoimmune disease with a multi-factorial etiology that has a severe negative impact on a patient’s psychoemotional well-being as well as their quality of life, and accounts for a significant global disease burden. With greater knowledge and a better understanding into the pathomechanism of AA, the emergence of JAK inhibitor use for AA treatment is in fact very promising and serves as an effective and viable strategy in treating this debilitating disease. In this review, oral and topical formulation of JAK inhibitors tofacitinib, ruxolitinib and baricitinib, were studied in the treatment of AA and its variants. A growing body of scientific publications, mostly in the form of case reports, has demonstrated exceptional efficacy of oral formulations of JAK inhibitors, particularly with tofacitinib and ruxolitinib in treating moderate to severe, as well as refractory or recalcitrant cases of AA; but identifies unfavourable efficacy for topical formulations. Moreover, it was demonstrated that these patients would need to maintain JAK inhibitor treatment in order to maintain hair regrowth. In addition to these, reports also identified adverse effects that were not severe, but mostly mild and transient. Further studies are also required for a better understanding of the efficacy and safety of baricitinib, as the data available is not sufficient to draw conclusions regarding such.
In spite of this, the data suggests that JAK inhibitors may represent the drug of choice for AA, as it has revolutionized the therapeutic outcome of this devastating disease. However, present findings are limited by a few constraints, as much of the evidence available is in the form of case reports/series and uncontrolled trials, which are of all low-quality evidence. These findings, therefore, require confirmation from further studies such as large cohort, well-controlled prospective studies with adequate follow-up and evaluation that will yield higher quality evidence in determining the long-standing efficacy, sustainability of treatment results, long-term safety, as well as cost-effectiveness of treating AA with JAK inhibitors. Thus, until data from more scientifically robust studies are available to assess the risk–benefit ratio more adequately, these current concerns/issues will continue to act as barriers to the use of JAK inhibitors for AA treatment.
Finally, one may argue that it may not be very viable to reduce the treatment of AA to one drug class, as not only may the treatment of AA become very complex and sophisticated overtime, but some patients may achieve remission rates at the same efficacy level of JAK inhibitors with the use of common conventional therapeutic options, which may prove to be less of a financial burden to patients. The current evidence, however, is very promising with regard to the efficacy and safety of JAK inhibitors in the treatment of AA and its variants, and therefore, could be a great addition to dermatologists’ current management strategy, particularly in those cases that are not responsive to standard therapeutic options.
Conclusion
Evidence on the use of JAK inhibitors that target the JAK-STAT pathway in the treatment of AA is still growing. Although existing results show a lot of promise and bring excitement to the field of dermatology, further studies that yield higher quality evidence are needed, to ensure that the safety and efficacy of these agents have met the optimal requirements for their use in clinical practice.
Abbreviations
AA, alopecia areata; ACOT, annual cost of therapy; APCs, antigen presenting cells; AT, alopecia totalis; AU, Alopecia Universalis; DALYs, disability adjusted life years; DPCP, diphenylcyclopropenone; GAD, Generalized Anxiety Disorder; HLA, human leukocyte antigen; IFN, interferon; IL/s, interleukin/s; JAK, janus kinase; JAKis, JAK inhibitors; JAK-STAT, janus kinase, signal transducer and activator of transcription; MDD, Major Depressive Disorder; MHC, major histocompatibility complex; MIF, migratory inhibitory factor; NK cells, natural killer cells; PD-LI, programmed cell death protein I ligand; QoL, quality of life; RCT/s, randomized controlled trial/s; SADBE, squaric acid dibutyl ester; SALT, severity of alopecia areata tool; SLE, Systemic Lupus Erythematosus; TGF, tissue growth factor; Tregs, regulatory T cells; TYK 2, tyrosine kinase 2; ULBP3, UL-16, binding protein 3; URTI, upper respiratory tract infection; US FDA, United States Food and Drugs Administration; UTI, urinary tract infection.
Funding
There is no funding to report.
Disclosure
The author has no conflicts of interest to declare for this work.
References
1. Solimani F, Meier K, Ghoreschi K. Emerging topical and systemic jak inhibitors in dermatology. Front Immunol. 2019;10:2847. doi:10.3389/fimmu.2019.02847
2. Johnson R, Roh E, Saavedra A, Wolff K. Fitzpatrick’s Colour Atlas and Synopsis of Clinical Dermatology.
3. Oakley A. Alopecia Areata. DermNet NZ; 2015. Available from: https://dermnetnz.org/topics/alopecia-areata/.
4. Trueb R, Dutra H, Dias M. A comment on jak inhibitors for treatment of alopecia areata. Int J Trichology. 2018;10(5):193–197. doi:10.4103/ijt.ijt_62_18
5. Damsky W, King B. JAK inhibitors in dermatology: the promise of a new drug class. J Am Acad Dermatol. 2017;76(4):736–744. doi:10.1016/j.jaad.2016.12.005
6. Triyangkulsri K, Suchonwanit P. Role of janus kinase inhibitors in the treatment of alopecia areata. Drug Des Develop Ther. 2018;12:2323–2335. doi:10.2147/DDDT.S172638
7. Guo H, Cheng Y, Shapiro J, McElwee K. The role of lymphocytes in the development and treatment of alopecia areata. Expert Rev Clin Immunol. 2015;11(12):1335–1351. doi:10.1586/1744666X.2015.1085306
8. Paus R, Ito N, Takigawa M, Ito T. The hair follicle and immune privilege. J Invest Dermatol Symposium Proceedings. 2003;8(2):188–194. doi:10.1046/j.1087-0024.2003.00807.x
9. Kranseler J, Sidbury R. Alopecia areata: update on management. Indian J Paediatr Dermatol. 2017;18(4):261–266. doi:10.4103/ijpd.IJPD_80_17
10. Anzai A, Wang E, Lee E, Aoki V, Christiano A. Pathomechanisms of immune-mediated alopecia. Int Immunol. 2019;31(7):439–447. doi:10.1093/intimm/dxz039
11. D’Ovidio R. Alopecia areata: news on diagnosis, pathogenesis and treatment. G Ital Dermatol Venereol. 2014;149(1):25–45.
12. Xing L, Dai Z, Jabbari A, et al. Alopecia areata is driven by cytotoxic T lymphocytes and is reversed by jak inhibition. Nat Med. 2014;20(9):1043–1049. doi:10.1038/nm.3645
13. McElwee K, Freyschmidt-Paul P, Hoffman R, et al. Transfer of CD8(+) cells induces localized hair loss whereas CD4(+)/CD25(-) cells promote systemic alopecia areata and CD4(+)/Cd25(+) cells blockade disease onset in the C3H/HeJ mouse model. J Invest Dermatol. 2005;124(5):947–957. doi:10.1111/j.0022-202X.2005.23692.x
14. Trueb R, Dias M. Alopecia areata: a comprehensive review of pathogenesis and management. Clin Rev Allergy Immunol. 2017;54:68–87. doi:10.1007/s12016-017-8620-9
15. Darwin E, Hirt P, Fertig R, Doliner B, Delcanto G, Jimenez J. Alopecia areata: review of epidemiology, clinical features, pathogenesis, and new treatment options. Int J Trichology. 2018;10(2):51–60. doi:10.4103/ijt.ijt_99_17
16. Petukhova L, Christiano A. Functional interpretation of genome-wide association study evidence in alopecia areata. J Invest Dermatol. 2016;136(1):314–317. doi:10.1038/JID.2015.402
17. Betz R, Petukhova L, Ripke S, et al. Genome-wide meta-analysis in alopecia areata resolves HLA associations and reveals two new susceptibility loci. Nat Commun. 2015;6:5966. doi:10.1038/ncomms6966
18. Manolache L, Benea V. Stress in patients with alopecia areata and vitiligo. J Eur Acad Dermatol Venereol. 2007;21(7):921–928. doi:10.1111/j.1468-3083.2006.02106.x
19. Gulec T, Tanriverdi N, Duru C, Saray Y, Akcali C. The role of psychological factors in alopecia areata and the impact of the disease on the quality of life. Int J Dermatol. 2004;43(5):352–356. doi:10.1111/j.1365-4632.2004.02028.x
20. Habif T. Clinical Dermatology. A Guide to Diagnosis and Therapy.
21. Korta D, Christiano A, Berfield W, et al. Alopecia areata is a medical disease. J Am Acad Dermatol. 2018;78(4):832–834. doi:10.1016/j.jaad.2017.09.011
22. Ghanizadeh A, Ayoobzadehshirazi A. A review of psychiatric disorders comorbidities in patients with alopecia areata. Int J Trichology. 2014;6(1):2–4. doi:10.4103/0974-7753.136746
23. Fricke A, Miteva M. Epidemiology and burden of alopecia areata: a systematic review. Clin Cosmet Investig Dermatol. 2015;8:397–403. doi:10.2147/CCID.S53985
24. Delamere FM, Sladden MJ, Dobbins HM, Leonardi-Bee J, Macbeth A. Interventions for alopecia areata. Cochrane Database of Systematic Reviews. 2008;CD004413. doi:10.1002/14651858.CD004413.pub2
25. Hammerschmidt M, Brenner F. Efficacy and safety of methotrexate in alopecia areata. An Bras Dermatol. 2014;89(5):729–734. doi:10.1590/abd1806-4841.20142869
26. Vano-Galvan S, Hermosa-Gelbard A, Sanchez-Neila N, et al. Treatment of Recalcitrant Adult Alopecia Areata Universalis with Oral Azathioprine. J Am Acad Dermatol. 2016;74(5):1007–1008. doi:10.1016/j.jaad.2015.12.055
27. Wang E, Sallee B, Tejeda C, Christiano A. Montagna symposium 2017- janus kinase inhibitors for treatment of alopecia areata. J Invest Dermatol. 2018;138(9):1911–1966. doi:10.1016/j.jid.2018.05.027
28. Fragoulis G, McInnes I, Siebert S. JAK-inhibitors: new players in the field of immune-mediated diseases, beyond rheumatoid arthritis. Rheumatology. 2019;58(1):43–54. doi:10.1093/rheumatology/key276
29. Aittomaki S, Pesu M. Therapeutic targeting of the JAK/STAT pathway. Basic Clin Pharmacol Toxicol. 2013;114(1):18–23. doi:10.1111/bcpt.12164
30. O’Shea J, Plenge R. JAKs and STATs in immunoregulation and immune-mediated disease. Immunity. 2012;36(4):542–550. doi:10.1016/j.immuni.2012.03.014
31. Renert-Yuval Y, Guttman-Yassky E. The changing landscape of alopecia areata: the therapeutic paradigm. Adv Ther. 2017;34:1594–1609. doi:10.1007/s12325-017-0542-7
32. Craiglow B, King B. Killing two birds with one stone: oral tofacitinib reverses alopecia universalis in a patient with plaque psoriasis. J Invest Dermatol. 2014;134(12):2988–2990. doi:10.1038/jid.2014.260
33. Anzengruber F, Maul J, Kamarachev J, Trueb R, French L, Navarini A. Transient efficacy of tofacitinib in alopecia areata universalis. Case Rep Dermatol. 2016;8(1):102–106. doi:10.1159/000445182
34. Dhayalan A, King B. Tofacitinib citrate for the treatment of nail dystrophy associated with alopecia universalis. JAMA. 2016;152(4):492–493.
35. Ferreira S, Scheinberg M, Steiner D, Steiner T, Bedin G, Ferreira R. Remarkable improvement of nail changes in alopecia areata universalis with 10 months of treatment with tofacitinib: a case report. Case Rep Dermatol. 2016;8(3):262–266. doi:10.1159/000450848
36. Gupta A, Carviel J, Abramovits W. Efficacy of tofacitinib in treatment of alopecia universalis in two patients. J Eur Acad Dermatol Venereol. 2016;30(8):1373–1378. doi:10.1111/jdv.13598
37. Jabbari A, Nguyen N, Cerise J, et al. Treatment of alopecia areata with tofacitinib results in regrowth of hair and changes in serum and skin biomarkers. Exp Dermatol. 2016;25(8):642–643. doi:10.1111/exd.13060
38. Kennedy Crispin M, Ko J, Craiglow B, et al. Safety and efficacy of the jak inhibitor tofacitinib citrate in patients with alopecia areata. JCI Insight. 2016;1(15):e89776. doi:10.1172/jci.insight.89776
39. Bayart C, DeNiro K, Brichta L, Craiglow B, Sidbury R. Topical janus kinase inhibitors for the treatment of pediatric alopecia areata. J Am Acad Dermatol. 2017;77(1):167–170. doi:10.1016/j.jaad.2017.03.024
40. Castelo-Soccio L. Experience with oral tofacitinib in 8 adolescent patients with alopecia universalis. J Am Acad Dermatol. 2017;76(4):754–755. doi:10.1016/j.jaad.2016.11.038
41. Craiglow B, Liu L, King B. Tofacitinib for the treatment of alopecia areata and variants in adolescents. J Am Acad Dermatol. 2017;76(1):29–32. doi:10.1016/j.jaad.2016.09.006
42. Erduran F, Adisen E, Aksakal A. Excellent response to tofacitinib in a patient with alopecia universalis. Acta Dermatovenerol APA. 2017;26:47–49. doi:10.15570/actaapa.2017.15
43. Ibrahim O, Bayart C, Hogan S, Piliang M, Bergfeld W. Treatment of alopecia areata with tofacitinib. JAMA. 2017;153(6):600–602.
44. Kim B, Kim H. Successful hair regrowth in a Korean patient with alopecia universalis following tofacitinib treatment. Singapore Med J. 2017;58(5):279–280. doi:10.11622/smedj.2017039
45. Liu L, Craiglow B, King B, Dai F. Tofacitinib for the treatment of severe alopecia areata and variants: a study of 90 patients. J Am Acad Dermatol. 2017;76(1):22–28. doi:10.1016/j.jaad.2016.09.007
46. Mrowietz U, Gerdes S, Glaser R, Schroder O. Successful treatment of refractory alopecia areata universalis and psoriatic arthritis, but not of plaque psoriasis with tofacitinib in a young woman. Acta Derm Venereol. 2017;97(2):283–284. doi:10.2340/00015555-2491
47. Park H, Kim M, Lee J, Huh C, Kwon O, Cho S. Oral tofacitinib monotherapy in Korean patients with refractory moderate-severe alopecia areata: a Case Series. J Am Acad Dermatol. 2017;77(5):978–980. doi:10.1016/j.jaad.2017.06.027
48. Salman A, Sarac G, Ergun T. Alopecia universalis unresponsive to treatment with tofacitinib: report of a case with brief review of the literature. Dermatol Online J. 2017;23(7):15.
49. Scheinberg M, de Lucena Couto Océa RA, Cruz BA, Ferreira SB. Brazilian experience of the treatment of alopecia universalis with the novel antirheumatic therapy tofacitinib: a case series. Rheumatol Ther. 2017;4(2):503–508. doi:10.1007/s40744-017-0069-z
50. Strazzulla LC, Avila L, Sicco K, Shapiro J. Image Gallery: treatment of refractory alopecia universalis with oral tofacitinib citrate and adjunct intralesional triamcinolone injections. Br J Dermatol. 2017;176(6):e125. doi:10.1111/bjd.15483
51. Jabbari A, Sansarcq F, Cerise J, et al. An open-label pilot study to evaluate the efficacy of tofacitinib in moderate to severe patch type alopecia areata, totalis and universalis. J Invest Dermatol. 2018;138(7):1539–1545. doi:10.1016/j.jid.2018.01.032
52. Liu L, Craiglow B, King B. Tofacitinib 2% ointment, a topical janus kinase inhibitor for the treatment of alopecia areata: a pilot study of 10 patients. J Am Acad Dermatol. 2018;78(2):403–404. doi:10.1016/j.jaad.2017.10.043
53. Patel N, Oussedik E, Grammenos A, Pichardo-Geisinger R. A case report highlighting the effective treatment of alopecia universalis with tofacitinib in an adolescent and adult patient. J Cutan Med Surg. 2018;22(4):439–442. doi:10.1177/1203475418760512
54. Shivanna C, Shenoy C, Priya R. Tofacitinib (selective janus kinase inhibitor 1 and 3): a promising therapy for the treatment of alopecia areata: a case report of six patients. Int J Trichology. 2018;10(3):103–107. doi:10.4103/ijt.ijt_21_18
55. Akdogan N, Ersoy-Evans S, Dogan S, Atakan N. Experience with oral tofacitinib in two adolescents and seven adults with alopecia areata. Dermatol Ther. 2019;32(6):e13118. doi:10.1111/dth.13118
56. Ferreira S, Ferreira R, Scheinberg M. An excellent response to tofacitinib in a Brazilian adolescent patient with alopecia areata: a case report and a review of the literature. Clin Case Rep. 2019;7(12):2539–2542. doi:10.1002/ccr3.2484
57. Jerjen R, Meah N, Trindade de Carvalho L, Wall D, Eisman S, Sinclair R. Treatment of alopecia areata in pre-adolescent children with oral tofacitinib: a retrospective study. Pediatr Dermatol. 2021;38(1):103–108. doi:10.1111/pde.14422
58. Dincer Rota D, Emeksiz MAC, Erdogan FG, Yildirim D. Experience with oral tofacitinib in severe alopecia areata with different clinical responses. J Cosmet Dermatol. 2021;00:1–8.
59. Higgins E, Al Shehri T, McAleer M, Feighery C, Lilic D, Irvine A. Use of ruxolitinib to successfully treat chronic mucocutaneous candidiasis cause by gain-of-function signal transducer and activator of transcription 1 (STAT1) mutation. J Allergy Clin Immunol. 2015;135(2):551–553. doi:10.1016/j.jaci.2014.12.1867
60. Pieri L, Guglielmelli P, Vannucchi A. Roxulitinib-induced reversal of alopecia universalis in a patient with essential thrombocytopenia. Am J Hematol. 2015;90(1):82–83. doi:10.1002/ajh.23871
61. Craiglow B, Tavares D, King B. Topical ruxolitinib for the treatment of alopecia universalis. JAMA. 2016;152(4):490–491.
62. Harris J, Rashighi M, Nguyen N, et al. Rapid skin repigmentation on oral ruxolitinib in a patient with coexistent vitiligo and alopecia areata. J Am Acad Dermatol. 2016;74(2):370–371. doi:10.1016/j.jaad.2015.09.073
63. Mackay-Wiggan J, Jabbari A, Nguyen N, et al. Oral ruxolitinib induces hair-growth in patients with moderate-severe alopecia areata. JCI Insight. 2016;1(15):e89790. doi:10.1172/jci.insight.89790
64. Deeb M, Beach R. A case of topical ruxolitinib treatment failure in alopecia areata. J Cutan Med Surg. 2017;21(6):562–563. doi:10.1177/1203475417716363
65. Vandiver A, Girardi N, Alhariri J, Garza L. Two cases of alopecia areata treated with ruxolitinib: a discussion of ideal dosing and laboratory monitoring. Int J Dermatol. 2017;56(8):833–835. doi:10.1111/ijd.13598
66. Ramot Y, Zlotogorski A. Complete regrowth of beard with roxulitinib in an alopecia universalis patient. Skin Appendage Disord. 2018;4(2):122–124. doi:10.1159/000479722
67. Liu L, King B. Ruxolitinib for the treatment of severe alopecia areata. J Am Acad Dermatol. 2019;80(2):566–568. doi:10.1016/j.jaad.2018.08.040
68. Olsen EA, Kornacki D, Sun K, Hordinsky MK. Ruxolitinib cream for the treatment of patients with alopecia areata: a 2-part, double-blind, randomized, vehicle-controlled Phase 2 study. J Am Acad Dermatol. 2020;82(2):412–419. doi:10.1016/j.jaad.2019.10.016
69. Fetter T, Cuevas Rios G, Niebel D, Bieber T, Wenzel J. Unexpected hair regrowth in a patient with longstanding alopecia universalis during treatment of recalcitrant dermatomyositis with the janus kinase inhibitor ruxolitinib. Acta Derm Venereol. 2020;100(10):adv00144. doi:10.2340/00015555-3481
70. Peterson D, Vesely M. Successful treatment of alopecia totalis with ruxolitinib in a preadolescent patient. J Am Acad Dermatol. 2020;6(4):257–259.
71. Jabbari A, Zhenpeng D, Xing L, et al. Reversal of alopecia areata following treatment with the JAK1/2 inhibitor baricitinib. EBioMedicine. 2015;2(4):351–355. doi:10.1016/j.ebiom.2015.02.015
72. Olamiju B, Friedmann A, King B. Treatment of severe alopecia areata with baricitinib. J Am Acad Dermatol. 2019;5(10):892–894.
73. Shi J, Chen X, McGee R, et al. The pharmacokinetics, pharmacodynamics, and safety of orally dosed INCB018424 phosphate in healthy volunteers. J Clin Pharmacol. 2011;51(12):1644–1654. doi:10.1177/0091270010389469
74. Carfagna M, Cannady E, Ryan T, et al. Carcinogenicity assessment of baricitinib in Tg.rasH2 mice and Sprague-Dawley (Crl: CD)rats. Regul Toxicol Pharmacol. 2018;92:458–471. doi:10.1016/j.yrtph.2017.11.020
75. Phan K, Sebaratnam DF. JAK inhibitors for alopecia areata: a systematic review and meta-analysis. J Eur Acad Dermatol Venereol. 2019;33(5):850–856. doi:10.1111/jdv.15489
76. Guo L, Feng S, Sun B, Jiang X, Liu Y. Benefit and risk profile of tofacitinib for the treatment of alopecia areata: a systemic review and meta-analysis. J Eur Acad Dermatol Venereol. 2020;34(1):192–201. doi:10.1111/jdv.15937
77. Gilhar A, Keren A, Paus R. JAK inhibitors and alopecia areata. The Lancet. 2019;393(10169):318–319. doi:10.1016/S0140-6736(18)32987-8
78. GlobalData Healthcare. Can JAK inhibitors penetrate the alopecia areata market effectively. GlobalData Healthcare, Pharmaceutical Technology; 2019. Available from: https://www.pharmaceutical-technology.com/comment/alopecia-areata-treatment-2019/#:~:text=Primary%20research%20conducted%20by%20GlobalData,inhibitor%20is%20just%20under%20%241%2C400.
79. Xconomy. With FDA’s Eye on JAK Drugs, Concert Pharma Moves One Ahead for Hair Loss; 2019. Available from: https://xconomy.com/boston/2019/09/03/with-fdas-eye-on-jak-drugs-concert-pharma-moves-one-ahead-for-hair-loss/.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.