")
Back to Journals » International Journal of Nanomedicine » Volume 13
A complex micellar system co-delivering curcumin with doxorubicin against cardiotoxicity and tumor growth
Authors Zhang D, Xu Q, Wang N, Yang Y, Liu J, Yu G , Yang X, Xu H , Wang H
Received 3 April 2018
Accepted for publication 25 May 2018
Published 10 August 2018 Volume 2018:13 Pages 4549—4561
DOI https://doi.org/10.2147/IJN.S170067
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Di Zhang,1 Qian Xu,1 Ning Wang,1 Yanting Yang,1 Jiaqi Liu,1 Guohua Yu,2 Xin Yang,3 Hui Xu,1 Hongbo Wang1
1School of Pharmacy, Key Laboratory of Molecular Pharmacology and Drug Evaluation, (Yantai University), Ministry of Education, Collaborative Innovation Center of Advanced Drug Delivery System and Biotech Drugs in Universities of Shandong, Yantai University, Yantai, People’s Republic of China; 2Department of Pathology, Yantai Yuhuangding Hospital, Yantai, People’s Republic of China; 3School of Chemistry and Chemical Engineering, Yantai University, Yantai, People’s Republic of China
Background: Dose-dependent irreversible cardiac toxicity of doxorubicin (DOX) becomes a major obstacle for the clinical use. Nowadays much attention is being paid to combination therapy with DOX and antioxidant agents, which would improve the clinical efficacy by protecting from cardiotoxicity along with the maintained performance as an antitumor drug. With the assistance of nanoscience and polymer engineering, herein a complex polymeric micellar system was developed for co-loading DOX and a premium natural antioxidant curcumin (CUR), and we investigated whether this new formulation for DOX delivery could achieve such a goal.
Methods: The dually loaded micelles co-encapsulating DOX and CUR (CPMDC) were prepared through thin-film rehydration by using the amphiphilic diblock copolymer monomethoxy poly(ethylene glycol) (mPEG)–poly(ε-caprolactone) (PCL)–N-t-butoxycarbonyl-phenylalanine (BP) synthesized by end-group modification of mPEG–PCL with BP. Quantitative analysis was conducted by HPLC methods for drugs in micelles or biosamples. Molecular dynamics simulation was performed using HyperChem software to illustrate interactions among copolymer and active pharmaceutical ingredients. The safety and antitumor efficacy were evaluated by in vitro viability of H9C2 cells, and tumor growth inhibition in tumor-bearing mice respectively. The protection effects against DOX-induced cardiotoxicity were investigated according to several physiological, histopathological and biochemical markers concerning systemic and cardiac toxicity.
Results: CPMDC were obtained with favorable physicochemical properties meeting the clinical demand, including uniform particle size, fairly high encapsulation efficiency and drug loadings, as well as good drug release profiles and colloidal stability. The result from molecular dynamics simulation indicated a great impact of the interactions among copolymer and small molecules on the ratiometrical co-encapsulation of both drugs. MTT assay of in vitro H9C2 cells viability demonstrated good safety of the CPMDC formulation, which also showed definite signs of decrease in xenograft tumor growth. The studies on pharmacokinetics and tissue distribution further revealed that DOX delivered by CPMDC could result in prolonged systemic circulation and increased DOX accumulation in tumor but decreased level of the toxic metabolite doxorubicinol in heart tissue compared to free DOX alone or the cocktail combination.
Conclusion: The findings from present study substantiated that such a complex micellar system codelivering DOX with CUR does produce the effect of killing two birds with one stone via distinctive nanocarrier-modified drug-drug interactions.
Keywords: doxorubicin, curcumin, co-delivery, polymeric micelles, combination therapy, drug-drug interactions
Introduction
Doxorubicin (DOX) is one of the first anthracycline antibiotics derived from the fungus Streptomyces peucetius with broad spectrum anticancer activity. It has remained a most frequently prescribed component in several currently used chemotherapy drug regimens to treat various hematological and solid malignancies, including leukemias, lymphomas, soft-tissue sarcomas, breast carcinoma and osteosarcoma.1 In spite of the effectiveness in blocking cancer progression, DOX administration may cause the development of toxicity related to its specific chemical structure. Indeed, the clinical use of DOX as a chemotherapeutic agent is usually associated with multiple adverse effects, particularly the risk of cardiac complications, which include congestive heart failure, dilated cardiomyopathy, and early death, and may affect ~11% of the patients under treatment.2 Furthermore, DOX-induced toxic effect on the heart has been found to be a correlation of degree of DOX administration in such a way that high-dose infusion or higher cumulative doses increase the presentation of cardiac toxic effects, thus resulting in limited cumulative tolerable dose and lowered therapeutic efficacy. At a cumulative dose of 550 mg/m2 of DOX, even 26% of patients develop congestive heart failure, a condition that is lethal in ~50% of cases.3,4
Although the exact mechanism of DOX-induced cardiotoxicity still remains unclear, there is already extensive evidence that DOX-mediated production of reactive oxygen species (ROS) may play a pivotal role in the development of severe cardiac complications.1,5 DOX-induced cardiomyopathy occurs chiefly due to the oxidation–reduction cycle, the rupture of the cell membrane, the progressive loss of myofibrils, and mitochondrial vacuolization, which denote lipid peroxidation.6 It has been demonstrated that DOX could stimulate ROS generation through two pathways, namely, the enzymatic pathway leading to the synthesis of a semiquinone radical following a reduction of DOX by the enzyme nicotinamide adenine dinucleotide phosphate (NADPH)–cytochrome P450 reductase7 and the nonenzymatic pathway that results in increased production of free radicals via DOX molecule being put in contact with iron.8
Some related studies have reported ROS as a therapeutic target for DOX-induced toxicity, particularly the DOX-dependent toxic effects on the cardiac tissue, which is vulnerable to the impairment caused by free radicals for its strong oxidative metabolism and weakened antioxidant defense in comparison with other organs, including the liver and kidneys.1,6 Accordingly, much attention is being paid to find novel antioxidant for combination regimens against the oxidative stress-triggered cardiomyopathy in DOX therapy, especially those plant-derived small molecules with considerable safety.9 Curcumin (CUR) is a kind of natural antioxidant derived from the perennial herb Curcuma longa, and some research has highlighted its potency as an ROS scavenger.10 Available laboratory evidence also confirmed that cardiotoxicity as a result of acute DOX exposure could be reduced by CUR supplementation both in vitro and in vivo.6,11 However, the full potential of treatments utilizing this phytochemical has not been realized in the clinic, primarily due to the poor systemic bioavailability of free drug outside the lower gastrointestinal tract.6,12,13
In recent decades, a couple of papers have proposed that co-delivering DOX and CUR may result in less toxicity and improved drug distribution in tumor tissue.14–16 However, the cocktail combination of both free drugs could not provide better antitumor efficacy or lower systemic toxicity because of the rapid drug elimination in vivo. Currently, several co-delivery systems have been developed for simultaneously delivering both drugs, including liposomes, nanoparticles, and polymeric micelles.
However, self-assembled polymeric micelles have been investigated widely for their potential functions in cancer therapy. Polymeric micelles have a core–shell structure, where hydrophobic drugs can be solubilized and stabilized in the hydrophobic core of micelles, and hydrophilic shell can prolong the circulation time and improve the steric stabilization by reducing opsonization in blood circulation. Furthermore, polymeric micelles could selectively and effectively accumulate in tumor through enhanced permeability and retention (EPR) effect, thereby enhancing the therapeutic effects of chemotherapeutic drugs. Hence, co-delivery micellar systems have attracted more attention.17
By using new technology with the assistance of nanoscience and polymer engineering,18–21 we have recently formulated polymeric micelles for CUR and its derivative that display favorable pharmacokinetic (PK) characteristics, including significant enhancement of the systemic bioavailability. In the present study, a dually loaded complex polymeric micellar system was developed for co-delivering DOX with this potent antioxidant agent using a classic thin-film hydration method. Compared to those reported co-delivery systems of DOX and CUR, the preparation process is more controllable and simple. Moreover, the optimal formulation may provide great advantage in adequate drug loading efficiency and perfect molar ratio of both drugs that would meet the demands of clinical application. We proposed that the complex polymeric micelles could harness the benefit of CUR against dose-dependent cardiotoxicity associated with DOX administration, thus successfully settling with the major limitation of DOX-based chemotherapy. A variety of strategies in vivo and in vitro were performed to confirm whether the combinatorial formulation would produce desired effect of killing two birds with one stone via nanocarrier-modified drug–drug interactions.
Materials and methods
Materials
The amphiphilic diblock copolymer of monomethoxy poly(ethylene glycol) (mPEG) and poly(ε-caprolactone) (PCL) and the end-group modification products of mPEG–PCL by converting its hydroxyl terminal into phenylalanine (Phe) or N-t-butoxycarbonyl-phenylalanine (BP) were synthesized in our laboratory as reported previously.22 CUR was obtained from Beijing Huafeng United Technology Co., Ltd. (Beijing, China). DOX and doxorubicinol (DOXol) were both purchased from Sigma-Aldrich Co. (St Louis, MO, USA), and bovine serum albumin (BSA) was provided by Thermo Fisher Scientific (Waltham, MA, USA). All the kits for biochemical assay were purchased from Nanjing Jiancheng Biochemistry Co. Ltd. (Nanjing, China). Other chemicals were of analytical grade or higher and were used as received.
Cells and animals
The rat cardiomyoblast cell line H9C2 and human lung cancer cell line A549 were supplied by the Cell Culture Center of Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences (Beijing, China). All the cells were cultured in DMEM supplemented with 10% fetal calf serum, penicillin (100 U/mL) and streptomycin (10 μg/mL) (Thermo Fisher Scientific) incubated at 37°C in a humidified air atmosphere containing 5% CO2, and harvested for use in their exponentially growing phase. Male Sprague Dawley (SD) rats (240–260 g) and inbred female C57BL/6J mice (20–25 g) were obtained from the Laboratory Animal Services Center of Luye Pharma Group Ltd. (Yantai, China). The animals were housed in well-ventilated cages under standard room temperature, pressure and humidity conditions and were provided with free access to normal mouse chow and water throughout the studies. All the animal experiments described conformed to the National Institutes of Health (NIH) guidelines (NIH Pub. No 85-23, revised 1996) and were approved by the Animal Care and Use Committee at Yantai University (Yantai, China).
Formula optimization and preparation of complex polymeric micelles
A set of experiments based on L9 (34) orthogonal arrays were performed for the optimal formula of complex polymeric micelles co-encapsulating DOX and CUR (CPMDC). The three factors (A for the amphiphilic diblock copolymer, B for the molar ratio of DOX to CUR in CPMDC and C for the mass ratio of drug to copolymer) and their levels are shown in Table 1. The classic thin-film hydration method was used for CPMDC preparation according to a two-step way23 as illustrated in Figure 1A. Briefly, the copolymer and CUR were dissolved in acetone at first. After the solvent was slowly evaporated under water bath at 50°C–55°C to form a thin-layer film, phosphate-buffered solution (PBS; 10×, pH 7.4) and DOX aqueous solution were added successively, and the mixture was stirred for 20 min to obtain CPMDC.
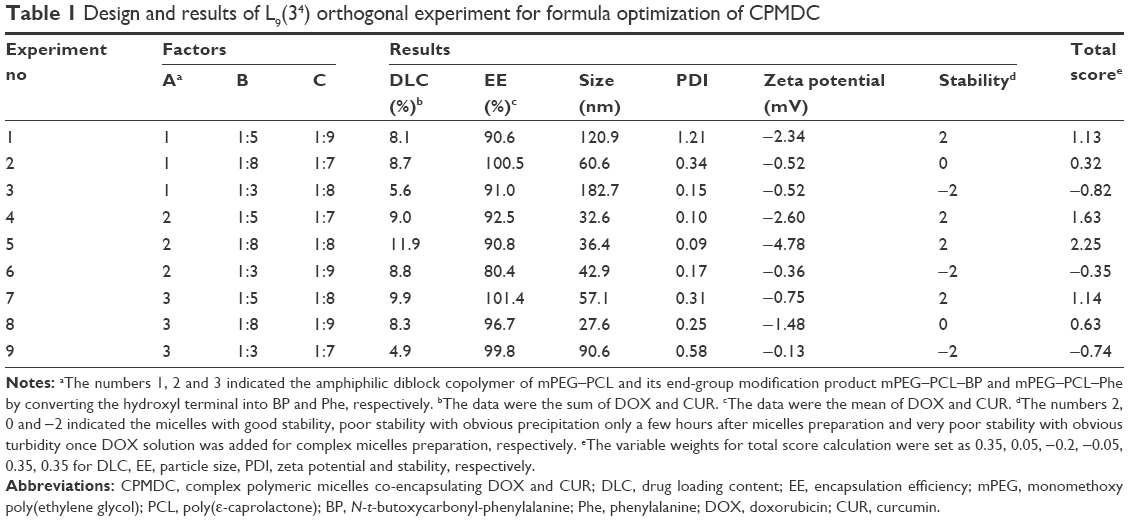 | Table 1 Design and results of L9(34) orthogonal experiment for formula optimization of CPMDC |
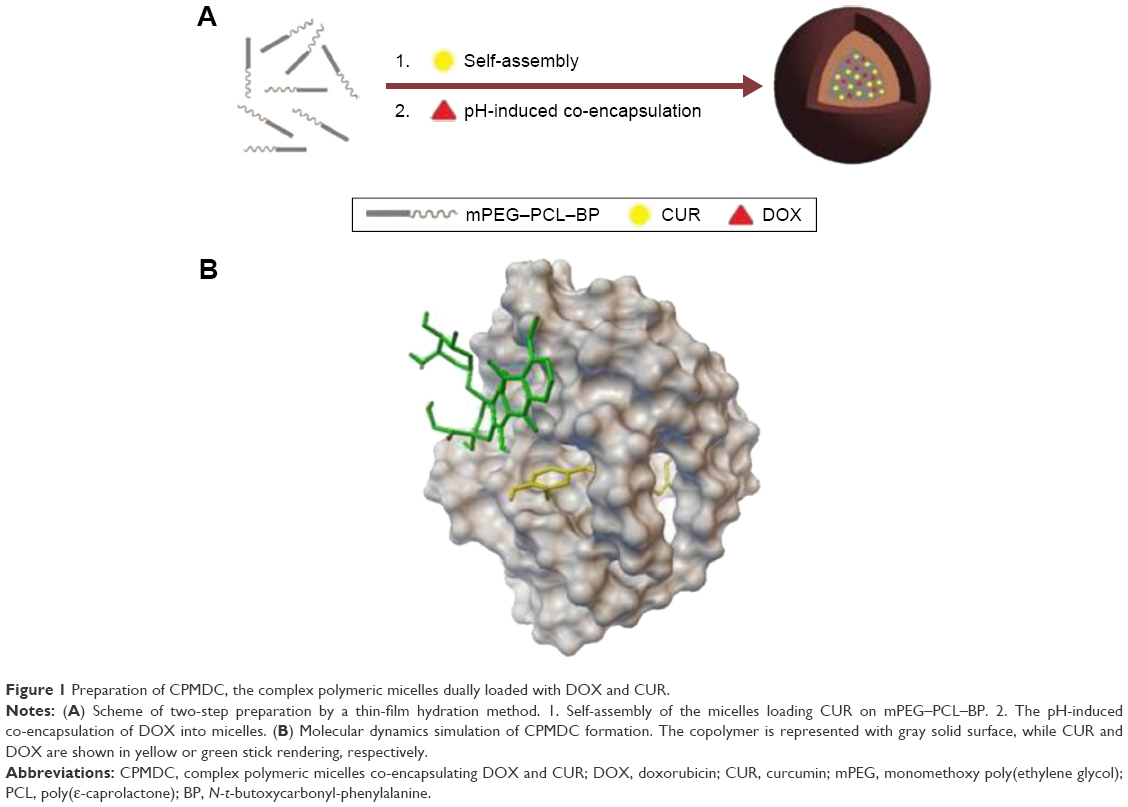 | Figure 1 Preparation of CPMDC, the complex polymeric micelles dually loaded with DOX and CUR. |
Characterization of CPMDC
Micellar particle size, size distribution and zeta potential were measured by dynamic light scattering (DLS) using a Beckman-Coulter zeta potential/submicron size analyzer. The micellar drug loading content (DLC) and particle size (EE) were assayed by high-performance liquid chromatography (HPLC), and the colloidal stability of micelles was evaluated by measuring the changes in particle size and zeta potential after incubation with PBS (pH 7.4) containing 1% or 10% BSA at 37°C for 24 h. Assessment of in vitro drug release from CPMDC was performed using a dialysis bag (molecular weight cut-off [MWCO], 14 kDa) in PBS (pH 3.5, 0.5% polysorbate 80) as reported previously.24
In vitro cell viability assay
The in vitro viability of cells was evaluated using an MTT assay as reported previously with a slight modification.25 After a 24 h preculture (10,000 cells/well) at 37°C with 5% CO2, the H9C2 cells were treated with various formulations, namely, free DOX alone, CUR micelles, the cocktail or CPMDC. Each was dissolved in normal saline, and the final concentration was 1 μM for DOX and 5 μM for CUR. The vehicle was normal saline and used as a normal control (NC), while the blank micelle without drug loading was used as a blank control (BC). Upon treatment, the plates were incubated for an additional 24 h. Then, the cells were subjected to incubation for 2 h with addition of MTT. After the medium was removed and DMSO was added, optical density measurement was performed at 570 nm using a microplate reader (Wellscan MK3; Labsystems Dragon, Finland). All data were presented as mean of three replicates.
Xenograft model in mice
The xenograft tumors of murine melanoma B16 were established using C57BL/6 mice following our previous publication.26 Briefly, the tumors were isolated from donor mice and implanted by subcutaneous injection. The day after implantation, the animals with successfully engrafted xenografts were randomized into five groups (n=9) and i.p. administered once every 2 days with various formulations, namely, free DOX solution, CUR micelles, the cocktail of free DOX and CUR micelles, CPMDC formulation, or the vehicle. For each group, normal saline was used as a vehicle with an injection volume of 10 mL/kg, and the dosage was 2.5 mg/kg for DOX and 8.0 mg/kg for CUR equivalent. At the end of experiment, the mice were sacrificed and tumor tissues were harvested, weighed and stored at −80°C until further analysis.
Rat model of DOX-induced acute cardiotoxicity
The DOX-induced acute cardiotoxicity in SD rats was performed following our previous publication.27 Briefly, the rats were randomly divided into five groups (n=6), namely, NC, DOX, CUR, cocktail and CPMDC, then weighed and i.p. administered a single dose of various formulations that were the same as mentioned earlier. The vehicle was normal saline with an injection volume of 10 mL/kg for each group, and the dosage was 20 mg/kg for DOX and 64 mg/kg for CUR equivalent. Within 48 h after dosing, blood samples were periodically collected via orbital venous plexus and immediately centrifuged to obtain plasma stored at −80°C for further chemical and biochemical estimations. At 48 h after dosing, all the animals were euthanized and cardiac tissues were immediately dissected out and weighed. Then, the similar sections of left ventricles were fixed with 4% formaldehyde and embedded in paraffin, and the remaining sections were stored at −80°C for further analysis.
Chemical, biochemical and histological analyses
Concentrations of CUR, DOX and its major metabolite DOXol in micelles or biosamples were determined by HPLC methods as previously reported.28,29 Using commercial kits, colorimetric assays were performed using a microplate reader (VersaMax; Molecular Devices LLC, Sunnyvale, CA, USA) for the known markers of cardiotoxicity, such as lactate dehydrogenase (LDH), creatine kinase (CK) and cardiac troponin T (cTnT) in plasma and malondialdehyde (MDA), superoxide dismutase (SOD) and glutathione peroxidase (GSH-Px) in cardiac tissue.30 For histological examination, the paraffin-embedded sections of the left half heart of rat were sliced (5 μm) and then HE staining for light microscope observations was performed. All specimens were analyzed, and the representative images were captured by two pathologists with blind investigation.
Statistics
One-way analysis of variance (ANOVA) and two-tailed Student’s t-test were performed for significance tests. A p-value of <0.05 was considered as statistically significant.
Results
Optimal formulation of CPMDC and characterization
The design and results of orthogonal experiment for formula optimization of CPMDC are shown in Table 1. According to the principle that a higher total score stands for a better formulation, direct analysis of the total scores provided an optimal formulation of CPMDC as A2B1C3, indicating that DOX and CUR at a molar ratio of 1:5 could be simultaneously encapsulated using amphiphilic diblock copolymer mPEG–PCL–BP as a micellar carrier at a fairly high mass ratio of 1:8. The range of total score was calculated as 0.97, 1.94 and 0.45 for the factors A, B and C, respectively, suggesting the impact of these factors on construction of CPMDC in the order of B>A>C. ANOVA results further demonstrated that the molar ratio showed significant impact on the total score (p<0.05).
Molecular dynamics simulation was conducted to demonstrate the interactions among drugs and copolymer using HyperChem software (HyperChem Professional 80; Hypercube, Inc., Gainesville, FL, USA) based on the OPLS method and conjugate gradient algorithms.31 mPEG–PCL–BP would change its initial curve-like conformation into ball shape to acquire suitable binding sites for interaction with the small molecules. As shown in Figure 1B, the stable complex micelles formed with CUR bound into the cavity and those formed with DOX on the surface of copolymer.
Under the optimal formulation, CPMDC was prepared for an overall characterization. The results showed a DLC of 8.1%±0.79% for CUR and 2.6%±0.25% for DOX, and the values of EE were >98% for both CUR and DOX. The particle size of freshly prepared CPMDC averaged 35.7±1.1 nm with a mean PDI of 0.09 and zeta potential of −2.28 mV. As shown in Figure 2A, when incubated in the simulated body fluid of PBS containing 1%–10% BSA (pH 6.8), the complex micelles could remain stable for at least 24 h with no significant change in the particle size (p>0.05). Meanwhile, the zeta potential of these micellar particles became more negative as the content of BSA increased (Figure 2B), suggesting the coating effect of BSA that contributes to the colloidal stability of CPMDC in PBS with a pH value higher than the isoelectric point of BSA (pH 4.7).
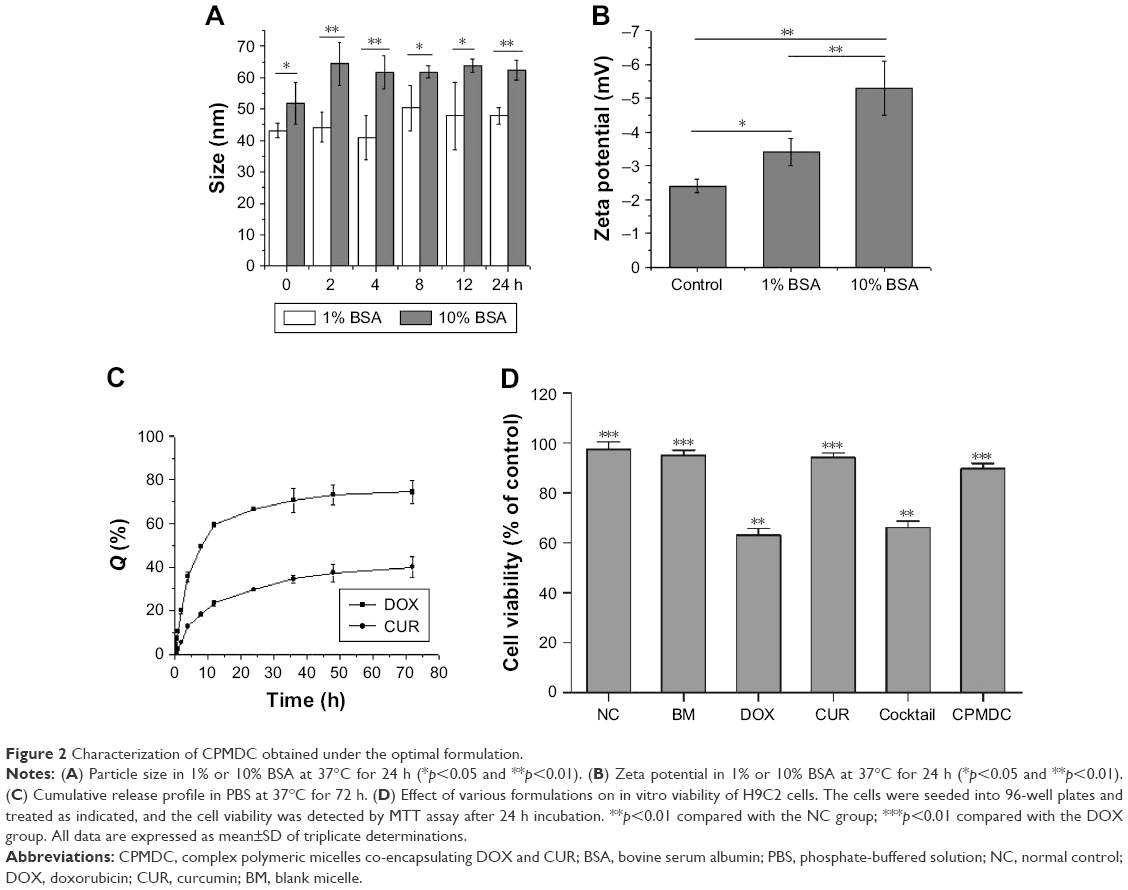 | Figure 2 Characterization of CPMDC obtained under the optimal formulation. |
The release profile of CPMDC was investigated for both drugs using the PBS containing 0.5% Tween 80 at pH 3.5 as the medium to maintain the chemical stability for quantification. As illustrated in Figure 2C, DOX showed much higher release rate in contrast to CUR that obtained a cumulative release of <40% within 72 h, while the value was up to 75% for DOX. The release kinetics could be well fitted with the Higuchi model for both drugs. The equations about the change in cumulative release percentage at some time point (Qt) with release time (t) were Qt=−0.21t1/2+4.63 (r=0.979) for DOX and Qt=−0.064t1/2+4.62 (r=0.978) for CUR, indicating that the drug release from CPMDC was controlled by diffusion through the polymeric matrix for both DOX and CUR.24
The effect on viability of H9C2 cells in vitro was further assessed to gain an insight into the safety of this formulation. In contrast to the vehicle, both CUR (5 μM) and the blank micelles without loading drugs showed no effect on the cell proliferation, whereas free DOX at 1 μM significantly inhibited H9C2 cell growth after 24 h incubation (p<0.01). It was worth noting that co-treatment using the cocktail formulation displayed no significant improvement in cell viability compared with free DOX alone (p>0.05). On the contrary, CPMDC at same dosages increased viability of H9C2 cells to near normal (Figure 2D), suggesting a favorable biocompatibility of mPEG–PCL–BP that may be responsible for the good safety and unique cellular uptake profile of the CPMDC formulation.
Effects on tumor growth in xenograft mice model
The tumor-bearing mouse model was used to evaluate how the CPMDC affected the antitumor activity of DOX. As shown in Figure 3A, DOX significantly inhibited the growth of xenograft tumor, whereas no obvious antitumor effect of CUR alone was observed. By contrast with the mice only treated with vehicle, the tumor growth inhibition was calculated as 65.9%, 61.4% and 68.2% for the DOX, cocktail and CPMDC groups, respectively, and no significant difference was found among these groups (p>0.05; Figure 3B). This result indicated that co-treatment with CUR by the CPMDC formulation did not attenuate but rather seemed to augment the tumor growth inhibiting properties of DOX to a certain degree.
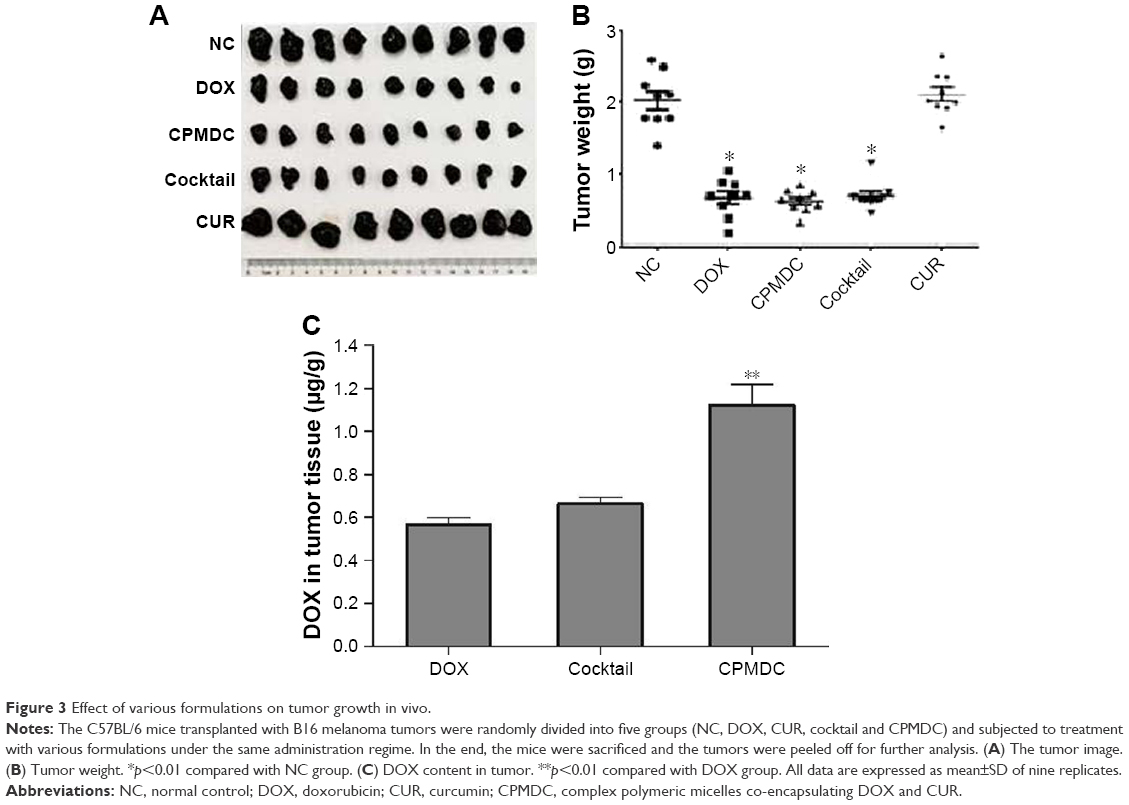 | Figure 3 Effect of various formulations on tumor growth in vivo. |
Biodistribution profile of DOX in tumor was further investigated (Figure 3C). One prominent feature of CPMDC was the ability to maintain the highest level of DOX in the tumor. By contrast, both the free DOX alone and the cocktail formulation showed a significantly lower DOX content (p<0.01), indicating the CPMDC formulation with a distinctive biodistribution profile of DOX that may contribute to its advantage as a chemotherapeutic agent.
Effects of CPMDC on DOX-induced cardiotoxicity in rats
The effects of CPMDC on DOX-induced cardiotoxicity were evaluated on rats treated with different formulations by comparing some principal physiological and biochemical markers concerned with cardiotoxicity in these animals. First of all, body weight (BW) and heart weight (HW) of each rat were measured to calculate body weight change (BWC) and cardiac weight index (CWI; the ratio of HW to BW). As shown in Figure 4A, the group administered CUR alone displayed no significant difference in the BWC or CWI level (p>0.05), while a single i.p. injection of free DOX caused significant decrease in both indices in contrast to the NC group treated with vehicle – (3.9±0.9)% vs (4.9±1.4)% for BWC and (2.3±0.6)% vs (3.1±1.3)% for CWI, indicating it is not CUR or the copolymer but DOX that caused remarkable toxicity in rats. Moreover, significant differences were observed in both indices between the cocktail and CPMDC groups that co-administered CUR and DOX at same dosages (p<0.01), suggesting the differentiated effects of various co-treatment formulations on DOX-induced toxicity and the effectiveness of CPMDC formulation that could promote BWC and CWI to near normal levels.
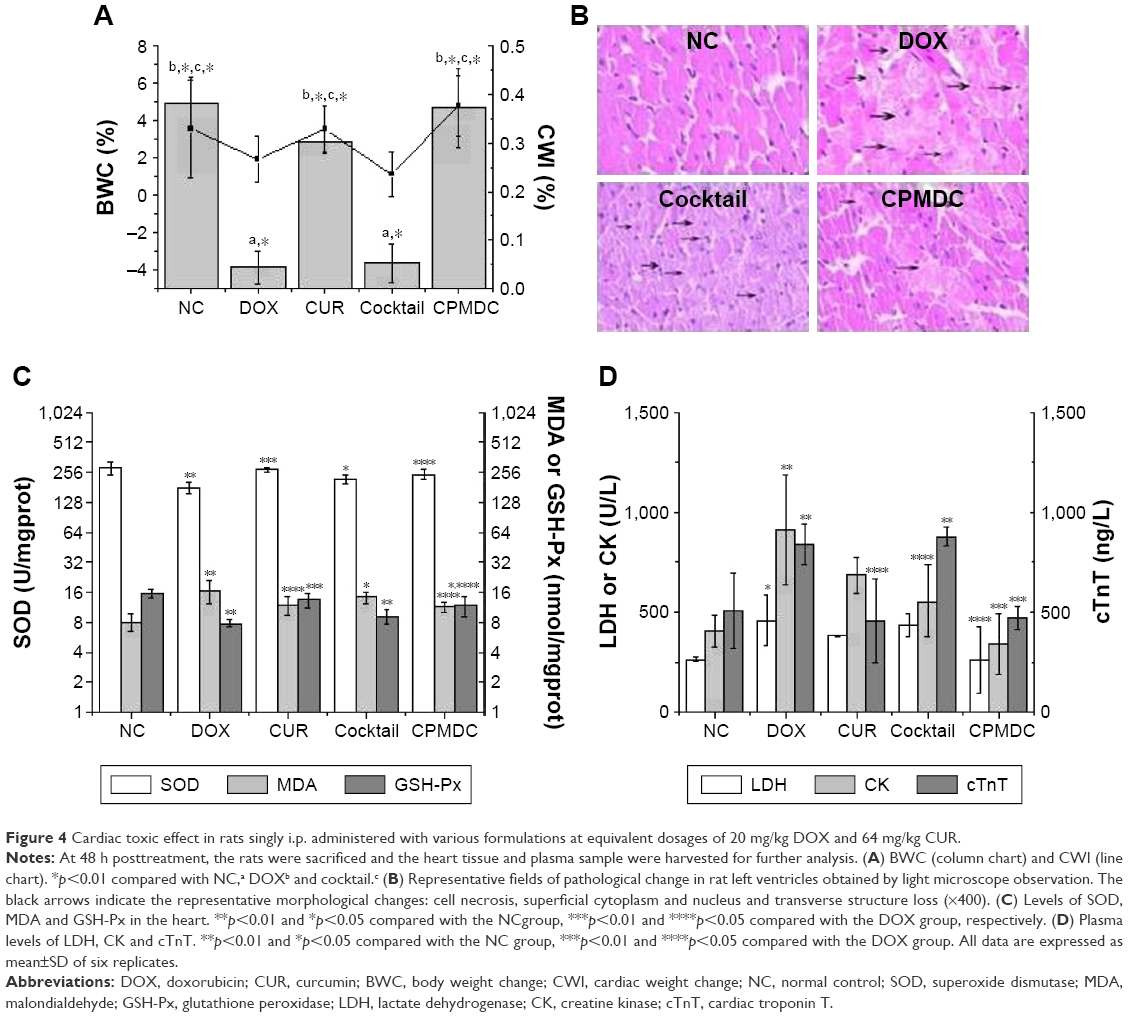 | Figure 4 Cardiac toxic effect in rats singly i.p. administered with various formulations at equivalent dosages of 20 mg/kg DOX and 64 mg/kg CUR. |
Considerable differences among various formulations were further observed by histological examination of cardiac tissues. The rats in the NC group showed normal morphology of cardiac myocyte in the left ventricle, whereas those only administered the DOX solution clearly exhibited myocardial injuries, such as cross-striation, myocardial endochylema puffing and sarcoplasmic matrix partly resorbed, myocardial cells derangement and necrotic cells flakily connected. For the two groups both co-treated with CUR and DOX at same dosages, fewer histopathological changes were observed in the CPMDC group than in the cocktail group (Figure 4B). These results from independent observations confirmed each other and demonstrated fairly high potency of CUR against cardiotoxic effect of DOX by the complex micelles co-loading both drugs.
Oxidative stress is generally recognized as one of the main causes of DOX-induced cardiotoxicity;4,6 biochemical analysis thus was performed to measure those well-known markers of oxidative injury involved in cardiac toxicity. As shown in Figure 4C, the rats administered free DOX alone displayed significantly decreased levels of SOD and GSH-Px and elevated MDA content in contrast to the control group (p<0.01). By contrast, co-administration by CPMDC formulation led to a significant decrease in the MDA level and alleviated the reduction of SOD and GSH-Px (p<0.05). Meanwhile, a significant increase in the plasma levels of CK, LDH and cTnT could be observed in the rats only treated with free DOX, which confirmed the occurrence of cardiac injury induced by this cardiotoxic agent (Figure 4D). Comparison of these biomarkers among the DOX, cocktail and CPMDC groups further revealed that co-treatment with CUR by the CPMDC, but not the cocktail formulation, could have a significant reduction in all these markers.
PK and biodistribution profiles in rats
The plasma drug concentrations of DOX and/or CUR were measured for the rats following a single i.p. administration of DOX or CUR alone or their combination by cocktail or CPMDC. As shown in Figure 5A, the PK profile of DOX was comparable between free DOX alone and the cocktail group, whereas it was remarkably altered by the CPMDC. The concentrations of DOX delivered by CPMDC were decreased bi-exponentially over 48 h with a long elimination phase, and the CPMDC formulation led to significant changes in all the PK parameters, including a substantial increase in plasma area under curve (AUC) and apparent volume of distribution (Vd) along with a notably extended half-life (t1/2) and a significant decrease in the total body clearance (CL) of DOX (p<0.01) in contrast to the group treated with free DOX alone or the cocktail.
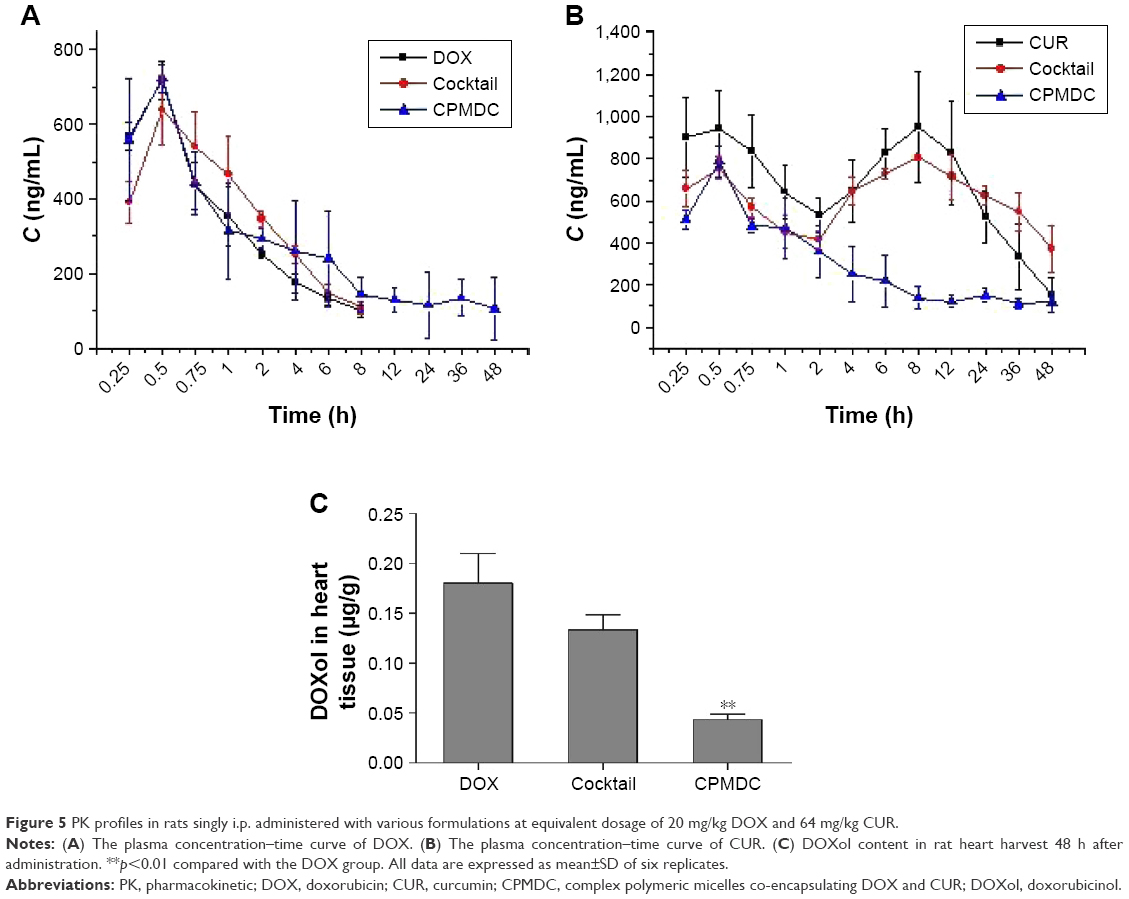 | Figure 5 PK profiles in rats singly i.p. administered with various formulations at equivalent dosage of 20 mg/kg DOX and 64 mg/kg CUR. |
CPMDC is a kind of complex formulation of CUR and DOX. However, this formulation altered the pharmacokinetic profile of the two active pharmaceutical ingredients in different way. As illustrated in Figure 5B and Table 2, a significant difference in all the PK parameters of CUR was observed among the three groups (p<0.01). By contrast with the group only administered CUR micelles, co-treatment with DOX using the cocktail formulation caused a more than twofold increase in plasma AUC and t1/2 of CUR along with more than 50% reduction in the CL value, whereas the CPMDC group displayed ~36% increase in the CL and a more than fivefold increase in t1/2 and Vd of CUR. Furthermore, the content of the main toxic metabolite DOXol in rat heart was measured. As shown in Figure 5C, administration of the cocktail formulation did not result in any significant difference in the DOXol level by contrast to free DOX alone, whereas the rats administered CPMDC displayed notably lower DOXol content in heart than those treated with free DOX alone or the cocktail formulation (p<0.01). These results demonstrated that the CPMDC formulation co-encapsulating CUR and DOX into polymeric micelles could bring about distinctive PK and metabolic characteristics by interaction among both drugs and the copolymers and then enable their retention within nanocarriers and significantly extend the circulation times.
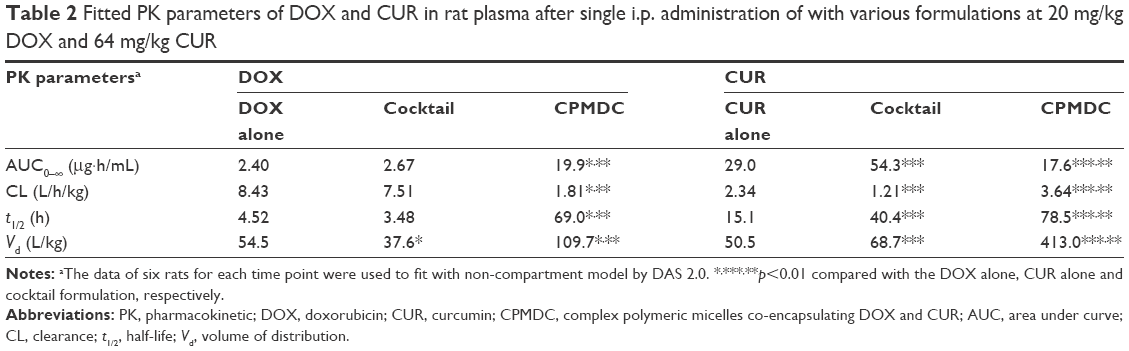 | Table 2 Fitted PK parameters of DOX and CUR in rat plasma after single i.p. administration of with various formulations at 20 mg/kg DOX and 64 mg/kg CUR |
Discussion
Following the recent development of highly bioavailable polymeric micellar formulations, we sought to develop complex micelles co-loading CUR and DOX, namely CPMDC, which would be expected to have improved PK and biodistribution profiles and make the possibility of increased safety at fairly high cumulative doses. The optimal formulation was first investigated by orthogonal-design experiments. Both direct and variance analysis revealed that the diblock copolymer mPEG–PCL–BP could co-encapsulate DOX and CUR at a molar ratio of 1:5 with the total drugs loading up to 1:8 and encapsulation efficiency >90%. This outcome coincided with that from our previous studies on CUR and its derivative.18,19 Molecular dynamics simulation provided a reasonable explanation for this formulation. When the complex micelles formed by self-assembly, both the copolymer and the small molecules adjusted conformations and the distance between each other to obtain energetically favorable binding sites and the interaction mode, and then, the stable complex micelles developed with the ball-shaped copolymer binding DOX on the surface and CUR into the cavity.
Characterization of the CPMDC reproduced under the optimal formulation revealed favorable physico-pharmaceutical properties, including fairly high encapsulation efficacy and drug loadings and desirable particle size for passive tumor targeting through the EPR effect.6,12 The measurement of particle size change in BSA solution indicated that CPMDC could maintain good colloidal stability in blood circulation by its appropriate zeta potential and the coating effect of blood albumin. Moreover, the two drugs in CPMDC displayed similar sustained-release profiles that could be well fitted with the Higuchi model, and marked difference was observed in the release rate, suggesting the different degrees of DOX and CUR binding with mPEG–PCL–BP despite that they were released from CPMDC by the same diffusion-controlled mechanism.
MTT assay of in vitro viability of H9C2 cells further demonstrated direct myocardial cytotoxicity of free DOX, as well as the safety of CPMDC that could be ascribed to favorable biocompatibility of mPEG–PCL–BP. Significantly different effects of the two co-treatment formulations further indicated that CPMDC may provide both DOX and CUR with unique PK characteristics that are closely related to the drug release and cellular uptake profiles and thus result in remarkable changes in pharmacodynamics of DOX in vivo.
The therapeutic potential of CPMDC as a new formulation for DOX therapy was investigated first since the mechanism of DOX-induced cardiotoxicity is independent of that acting in tumors.4 The mice bearing melanoma B16 treated with CUR alone exhibited no obvious antitumor effect. By contrast, those administered equivalent dosages of free DOX alone, cocktail or CPMDC formulation showed definite signs of decrease in xenograft tumor growth, and the CPMDC formulation displayed the highest DOX level in tumor. This finding provided an evidence base for the postulation that co-treatment with CUR by CPMDC would selectively block the cardiotoxicity of DOX without affecting its therapeutic benefit to tumor growth. It also indicated the passive accumulation of CPMDC in tumor via the EPR effect due to the leaky tumor blood vessels and malfunctioning lymphatic vessels,32 which would lead to the local bioavailability of DOX for the antitumor efficacy afforded.
CPMDC was next subjected to a series of in vivo measurements to address the advantage of protecting from cardiac toxicity. DOX-induced toxicity in the rat model, especially in the heart tissue, was manifested as a significant BW reduction and CWI decrease of the rats singly i.p. administered free DOX in contrast to the control group (p<0.01). Such differences further indicated that co-treatment by CPMDC, but not the cocktail at same dosages, could significantly ameliorate DOX-induced toxicities in rats. The histopathological evaluation of cardiac tissues strengthened the evidence for severe myocardial injuries caused by DOX and the minimum heart toxicity of CPMDC compared with the cocktail formulation or free DOX alone.
It has been reported that the protective effect of CUR on acute DOX-induced toxicity is related to its antioxidant capacity.33 Measurements of those biomarkers in the rat model further demonstrated the involvement of free radical over-formation and oxidative stress in DOX-induced cardiotoxicity. In contrast with the control, free DOX alone treatment caused a remarkable elevation of MDA and escalating levels of SOD and GSH-Px in rat heart tissue, which could be significantly improved by CPDMC but not the cocktail formulation at same dosages. Comparable effects were found in those markers of cardiac injury in plasma, such as CK, cTnT and LDH. Free DOX alone led to the highest levels of these markers, while significant decreases in all these markers were observed in rats administered CPMDC (p<0.01). These findings from various independent observations confirmed each other and manifested the effectiveness of CPMDC against DOX-induced cardiotoxicity.
Combination chemotherapy is prone to PK drug–drug interactions. As to the CPMDC formulation for co-administration of CUR with DOX, the PK and biodistribution profiles further revealed the occurrence of drug–drug interactions. DOX is mainly metabolized in rodents by the liver and biliary system with a 7–10 h elimination half-life, whereas CUR exhibits a different elimination profile with much shorter half-life.34 When they were administered concomitantly in rats by CPMDC, drug–drug interactions occurred, and significant changes could be observed in plasma PK parameters (Table 2). The elevated value of Vd may be ascribed to the competition between DOX and CUR for red blood cells and plasma proteins since they both have strong binding affinity and the competition and displacement from binding would lead to increased free drug and unbound fractions.35 Meanwhile co-encapsulation of both drugs into CPMDC could enable DOX retention within nanocarriers and extend the circulation time accompanied with the increased plasma AUC and decreased CL level of DOX. DOX is known as one of the most common substrates of CYP3A and P-gp that could be inhibited by CUR in a concentration-dependent manner.36 Such changes in plasma PK parameters of DOX therefore could suggest that the two drugs in CPMDC may both undergo metabolic elimination and the co-existing CUR would decrease DOX elimination by inhibiting metabolism. On the contrary, the cocktail formulation at equivalent dosages displayed obvious decrease in Vd and t1/2 along with comparable levels of AUC and CL of DOX, which further demonstrated that drug–drug interaction highly depends on the drug delivery system.
It has been found that DOX is prone to be metabolized to DOXol through reduction of the carbonyl group by aldo-keto reductase (AKR) in the cytosol, and then the incidence of cardiotoxicity increases with the formation of DOXol.37,38 Determination of DOXol in rat heart provided an additional evidence for the occurrence of metabolic drug–drug interaction of CPMDC and its close relation to the advantage of therapeutic efficacy. High levels of DOXol in the heart tissues of rats treated with free DOX alone or the cocktail were associated with prominent acute cardiotoxicity, and the dually loaded micellar system exhibited significantly decreased DOXol content in the heart (Figure 5C), suggesting that CPMDC may shield DOX from metabolism in rat heart, thus decreasing the DOX-to-DOXol conversion. It was also probably due to the extended half-life and the limited cellular uptake of DOX released from the dually loaded micelles. The underlying mechanism needs further investigation.
Conclusion
A dually loaded polymeric micellar system was developed for co-administration of DOX and CUR in the present study to overcome the dose-dependent cardiac toxicity associated with cancer chemotherapy. The premium natural antioxidant compound CUR was used for combination, and the amphiphilic diblock copolymer mPEG–PCL–BP with good biocompatibility was used as a nanocarrier for drug loading. The complex polymeric micelles CPMDC with favorable physico-pharmaceutical properties could be obtained by colloidal self-assembly with CUR encapsulated within the hydrophobic cavity and DOX bound to its ball-shaped surface. As demonstrated in multiple in vitro and in vivo models, such complex micellar formulation does produce the effect of killing two birds with one stone by maintaining the antitumor efficacy of free DOX alone while overcoming its associated cardiac toxicity. The findings from the PK study further revealed that such distinctive therapeutic efficacy of CPMDC may at least partially be ascribed to drug–drug interactions between DOX and CUR in vivo. Along with favorable physicochemical and PK characteristics, the superior safety and efficacy would provide CPMDC a great promise for clinical translation as an improved nanomedicine. Further investigations concerning this dually loaded micellar system will be focused on the mechanisms of drug combination and drug–drug interactions that play a pivotal role in combination therapy.
Acknowledgment
This work was supported by the Key Research and Development Project of Shandong Province (grant number 2018GSF119010), Natural Science Foundation of Shandong Province (grant number ZR2017MH061) and the Innovation Fund of Science and Technology for Graduate Students of Yantai University, China (grant number YDYB1725).
Disclosure
The authors report no conflicts of interest in this work.
References
Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev. 2004;56(2):185–229. | ||
Muggia FM, Speyer JL. Doxorubicin-induced cardiomyopathy. N Engl J Med. 1999;340(8):654–655. | ||
Swain SM, Whaley FS, Ewer MS. Congestive heart failure in patients treated with doxorubicin: a retrospective analysis of three trials. Cancer. 2003;97(11):2869–2879. | ||
Chatterjee K, Zhang J, Honbo N, Karliner JS. Doxorubicin cardiomyopathy. Cardiology. 2010;115(2):155–162. | ||
Ewer MS, Ewer SM. Cardiotoxicity of anticancer treatments: what the cardiologist needs to know. Nat Rev Cardiol. 2010;7(10):564–575. | ||
Mohajeri M, Sahebka A. Protective effects of curcumin against doxorubicin-induced toxicity and resistance: a review. Crit Rev Oncol Hematol. 2018;122:30–51. | ||
Olson RD, Mushlin PS, Brenner DE, et al. Doxorubicin cardiotoxicity may be caused by its metabolite, doxorubicinol. Proc Natl Acad Sci U S A. 1988;85(10):3585–3589. | ||
Pereira GC, Silva AM, Diogo CV, Carvalho FS, Monteiro P, Oliveira PJ. Drug-induced cardiac mitochondrial toxicity and protection: from doxorubicin to carvedilol. Curr Pharm Des. 2011;17(20):2113–2129. | ||
Ojha S, Taee HA, Goyal S, et al. Cardioprotective potentials of plant-derived small molecules against doxorubicin associated cardiotoxicity. Oxid Med Cell Longev. 2016;2016:5724973. | ||
Aggarwal BB, Sung B. Pharmacological basis for the role of curcumin in chronic diseases: an age-old spice with modern targets. Trends Pharmacol Sci. 2008;30(2):85–94. | ||
Benzer F, Kandemir FM, Ozkaraca M, Kucukler S, Caglayan C. Curcumin ameliorates doxorubicin-induced cardiotoxicity by abrogation of inflammation, apoptosis, oxidative DNA damage, and protein oxidation in rats. J Biochem Mol Toxic. 2018;32(2):e22030. | ||
Yallapu MM, Jaggi M, Chauhan SC. Curcumin nanoformulations: a future nanomedicine for cancer. Drug Discov Today. 2012;17(1–2):71–80. | ||
Fan X, Zhang C, Liu DB, Yan J, Liang HP. The clinical applications of curcumin: current state and the future. Curr Pharm Des. 2013;19(11):2011–2031. | ||
Ma W, Guo Q, Li Y, et al. Co-assembly of doxorubicin and curcumin targeted micelles for synergistic delivery and improving anti-tumor efficacy. Eur J Pharm Biopharm. 2016;112:209–223. | ||
Gu Y, Li J, Li Y, et al. Nanomicelles loaded with doxorubicin and curcumin for alleviating multidrug resistance in lung cancer. Int J Nanomed. 2016;11:5757–5770. | ||
Wang J, Ma W, Tu P. Synergistically improved anti-tumor efficacy by co-delivery doxorubicin and curcumin polymeric micelles. Macromol Biosci. 2015;15(9):1252–1261. | ||
Ramasamy T, Ruttala HB, Chitrapriya N, et al. Engineering of cell microenvironment-responsive polypeptide nanovehicle co-encapsulating a synergistic combination of small molecules for effective chemotherapy in solid tumors. Acta Biomater. 2017;48:131–143. | ||
Ramasamy T, Ruttala HB, Kanu BG, et al. Smart chemistry-based nanosized drug delivery systems for systemic applications: a comprehensive review. J Control Release. 2017;258:226–253. | ||
Sundaramoorthy P, Ramasamy T, Mishra SK, et al. Engineering of caveolae-specific self-micellizing anticancer lipid nanoparticles to enhance the chemotherapeutic efficacy of oxaliplatin in colorectal cancer cells. Acta Biomater. 2016;42:220–231. | ||
Ramasamy T, Haidar ZS, Tran TH, et al. Layer-by-layer assembly of liposomal nanoparticles with PEGylated polyelectrolytes enhances systemic delivery of multiple anticancer drugs. Acta Biomater. 2014;10(12):5116–5127. | ||
Ruttala HB, Ramasamy T, Madeshwaran T, et al. Emerging potential of stimulus-responsive nanosized anticancer drug delivery systems for systemic applications. Arch Pharm Res. 2018;41(2):111–129. | ||
Liu H, Xu H, Jiang YX, et al. Preparation, characterization, in vivo pharmacokinetics, and biodistribution of polymeric micellar dimethoxycurcumin for tumor targeting. Int J Nanomedicine. 2015;10:6395–6410. | ||
Wang BL, Shen Y, Zhang Q, et al. Codelivery of curcumin and doxorubicin by MPEG-PCL results in improved efficacy of systemically administered chemotherapy in mice with lung cancer. Int J Nanomedicine. 2013;8:3521–3531. | ||
Jiang Y, Wang F, Xu H, Liu H, Meng Q, Liu W. Development of andrographolide loaded PLGA microspheres: optimization, characterization and in vitro-in vivo correlation. Int J Pharm. 2014;475(1–2):475–484. | ||
Wang HB, Yu PF, Gou HT, et al. Cardioprotective effects of 20(S)-ginsenoside Rh2 against doxorubicin-induced cardiotoxicity in vitro and in vivo. Evid Based Complement Alternat Med. 2012;2012:506214. | ||
Lv GY, Sun DJ, Zhang JW, et al. Lx2-32c, a novel semi-synthetic taxane, exerts antitumor activity against prostate cancer cells in vitro and in vivo. Acta Pharm Sin B. 2017;7(1):52–58. | ||
Manju S, Sreenivasan K. Synthesis and characterization of a cytotoxic cationic polyvinylpyrrolidone-curcumin conjugate. J Pharm Sci. 2011;100(2):504–511. | ||
Zhang D, Xu Q, Liu K, Xu H. Comparison of pharmacokinetics of curcumin in rats administered with two kinds of polymeric micelles. Zhongguo Zhong Yao Za Zhi. 2016;41(19):3668–3673. | ||
Jaequet JM, Bressolle F, Galtier M, et al. Doxorubicin and doxorubicinol: intra- and inter-individual variations of pharmacokinetic parameters. Cancer Chemother Pharmacol. 1990;27(3):219–225. | ||
Ruggiero A, De RG, Rizzo D, et al. Myocardial performance index and biochemical markers for early detection of doxorubicin-induced cardiotoxicity in children with acute lymphoblastic leukaemia. Int J Clin Oncol. 2013;18:927–933. | ||
Jorgensen WL, Maxwell DS, Tiradorives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc. 1996;118:11225–11236. | ||
Torchilin V. Tumor delivery of macromolecular drugs based on the EPR effect. Adv Drug Deliv Rev. 2011;63(3):131–135. | ||
Namdari M, Eatemadi A. Cardioprotective effects of curcumin-loaded magnetic hydrogel nanocomposite (nanocurcumin) against doxorubicin-induced cardiac toxicity in rat cardiomyocyte cell lines. Artif Cells Nanomed Biotechnol. 2017;45(4):731–739. | ||
Gustafson DL, Rastatter JC, Colombo T, Long ME. Doxorubicin pharmacokinetics: macromolecule binding, metabolism, and excretion in the context of a physiologic model. J Pharm Sci. 2002;91(6):1488–1501. | ||
Schrijvers D. Role of red blood cells in pharmacokinetics of chemotherapeutic agents. Clin Pharmacokinet. 2003;42(9):779–791. | ||
Li C, Choi BC, Kim DK, et al. Effects of curcumin on the pharmacokinetics of loratadine in rats: possible role of CYP3A4 and p-glycoprotein inhibition by curcumin. Biomol Ther. 2011;19:364–370. | ||
Menna P, Recalcati S, Cairo G, Minotti G. An introduction to the metabolic determinants of anthracycline cardiotoxicity. Cardiovasc Toxicol. 2007;7(2):80–85. | ||
Heibein AD, Guo B, Sprowl JA, et al. Role of aldo-keto reductases and other doxorubicin pharmacokinetic genes in doxorubicin resistance, DNA binding, and subcellular localization. BMC Cancer. 2012;12:381–395. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.