")
Back to Journals » Clinical and Experimental Gastroenterology » Volume 7
A clinical perspective on the role of chronic inflammation in gastrointestinal cancer
Received 28 February 2014
Accepted for publication 30 May 2014
Published 11 August 2014 Volume 2014:7 Pages 261—272
DOI https://doi.org/10.2147/CEG.S43457
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Sebastian Rasch, Hana Algül
II Medizinische Klinik und Poliklinik, Klinikum rechts der Isar, Technische Universität München, Munich, Germany
Abstract: Chronic inflammation has been identified as an important risk factor for the development of malignancy, and knowledge about its molecular and cellular mechanisms is increasing. Several chronic inflammatory diseases of the gastrointestinal tract are important as risk factors for malignancy and have been studied in detail. In this review, we summarize important molecular mechanisms in chronic inflammation and highlight established and potential links between chronic inflammation and gastrointestinal cancer. In addition, we present the role of chronic inflammation in numerous tumors within the gastrointestinal tract as well as the relevant pathways or epidemiologic observations linking the pathogenesis of these tumors to inflammation.
Keywords: inflammation, immune response, carcinogenesis, gastrointestinal cancer, risk factor
Different entities of gastrointestinal cancer
Despite advances in cancer therapy, including targeted therapies, many malignancies are still associated with high mortality. Several cancers with a particularly poor prognosis, such as pancreatic ductal adenocarcinoma (PDAC) or gastric carcinoma, are located within the gastrointestinal (GI) tract. According to the GLOBOCAN database, neoplastic disorders of the GI tract account for more than 37% of cancer-related deaths worldwide, and the incidence of some GI malignancies, such as esophageal adenocarcinoma (EAC), has even been increasing over the last decade.1,2
GI cancers are a very heterogeneous group, as they can derive from various cell types. There are lymphoproliferative disorders like mucosa-associated lymphoid tissue (MALT) lymphoma and neuroendocrine tumors, predominantly located in the small intestine and pancreas. In addition to hepatocellular carcinoma (HCC), cholangiocarcinomas also occur in the liver, and stromal neoplasias such as GI stromal tumors (GIST) and squamous cell carcinomas can be found in the stomach and esophagus. However, most GI cancers are adenocarcinomas.
As a barrier to the environment on the one hand, and as the main organ system for digestion and absorption on the other, the GI tract is exposed to many substances, microorganisms, and irritants. Some of these, such as alcohol, Helicobacter pylori, and arsenic are well established risk factors for GI cancers.3–5 The common link between most of these carcinogens is chronic inflammation (Table 1).
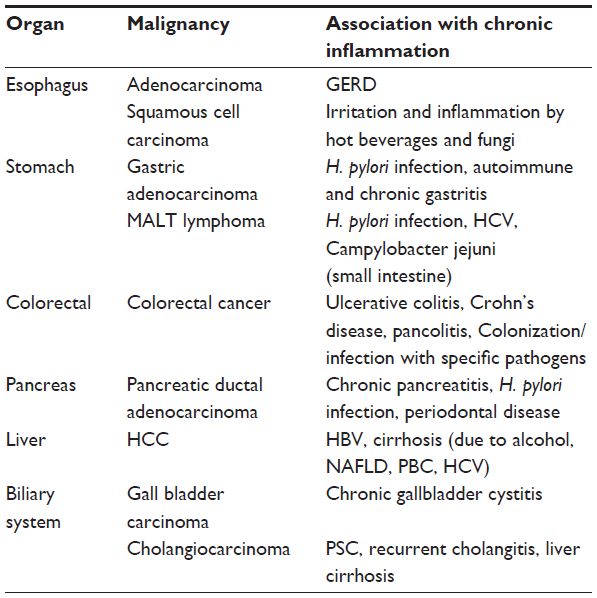 | Table 1 Gastrointestinal tumors that have been associated with chronic inflammation |
Virchow6 first described an infiltration of leukocytes in cancer in 1881. In the meantime, inflammation is considered to be one of the hallmarks of carcinogenesis and has become a main focus of research.
Processes of chronic inflammation
The immune system acts as a defense against bacterial, viral, or parasitic infections. It is also responsible for identification, destruction, and phagocytosis of damaged, apoptotic, or neoplastic cells. Local activation of the immune system results in inflammation. The presentation of antigens from pathogens, mechanical or chemical cell irritation, necrotic cells, or autoimmune processes can induce an immune response and inflammation.
In general, the immune system can be distinguished into an innate and an adaptive immune system. Natural killer cells, mast cells, monocytes, macrophages, dendritic cells, and granulocytes usually evoke the first immune reaction and initiate inflammation. They activate specialized T- and B-lymphocytes as part of the adaptive immune system that play an important role in the regulation of inflammation and in the pathogen-specific immune response. However, the latest research has revealed a group of innate lymphoid cells that resemble T-helper (TH) cells in function, and are presumably involved in the initiation of the immune response and inflammation.7,8
Accordingly, acute inflammation is characterized by activation and infiltration of innate immune cells. After interaction with pathogens or through attraction by chemokines, such as transforming growth factor (TGF)-β or platelet-derived growth factor (PDGF) from damaged epithelial cells, neutrophils, macrophages, and mast cells secrete proinflammatory cytokines as well as more chemokines. This signaling results in invasion and activation of additional immune cells. Infiltrating granulocytes produce cytotoxic oxygen and nitrogen radicals to fight pathogens.
Interleukin (IL)-1 from mononuclear phagocytes induces an autocrine activation. Together with tumor necrosis factor (TNF), it enhances the presentation of adhesion molecules and stimulates macrophages to segregate metalloproteinases (MMPs) and collagenases, leading to connective tissue damage. In addition TNF, histamine, and prostaglandins synergistically increase endothelial permeability, which facilitates leukocyte infiltration.
TNF also induces fibroblast growth and differentiation as well as apoptosis of affected epithelial cells, but it also stimulates IL-1 and IL-6 release. It can act as a pro-angiogenic at the site of inflammation, but can also act as an anti-angiogenic at high concentrations.9
After phagocytosis of damaged cells or pathogens, the adaptive immune response is mediated by antigen-presenting cells (APCs) such as dendritic cells. An infiltration with T- and B-lymphocytes is typical for sites of chronic inflammation. According to their IL secretion, TH cells are divided into TH1, TH2, and TH17 cells.10,11 TH1 cells produce interferon (IFN)-γ, TNF-α, and IL-12 and thus control cellular immunity. TH2 cells produce IL-4, IL-5, and IL-13, mediating B-cell-associated humoral immunity, and TH17 cells produce IL-17, IL-6, and IL-23, regulating chronic inflammation via regulatory T-cells (Treg).12
The extent of cytokine release by innate, TH, and Treg cells and the cytokine composition is crucial for the degree and duration of inflammation.13
The role of inflammation in cancer development and maintenance
Genetic instability and cell death resistance
In more than 15% of cancer patients worldwide, the cancer is believed to be caused by infection. Persistent infections, like some other causes, can be a reason for chronic inflammation.14,15 At sites of acute and chronic inflammation, cells are exposed to oxidative cell stress as well as to aggressive oxygen and nitrogen radicals from mononuclear phagocytes and leukocytes. Release of reactive oxygen and nitrogen species is enhanced by proinflammatory cytokines such as TNF, IL-1β, and IFN-α. Particularly in proliferating cells, these radicals can cause DNA damage by nitration and strand breakage.16 Thus, chronic inflammation creates an environment of genomic instability.
Cells can accumulate several mutations as DNA control and repair mechanisms are suppressed in inflammatory tissue. Nitric oxide itself can inhibit DNA mismatch repair (MMR) directly at the protein level and also by induction of hypermethylation.17,18 In this fashion, it inactivates the promoter of the MMR protein hMLH1.19 Another mechanism to alter MMR activity is the induction of hypoxia-inducible factor (HIF)-1α by TNF, IL-1β, and prostaglandin E2. Furthermore, HIF-1α removes c-MYC from the promoter of the MMR genes MSH2 and MSH6.20,21
Besides downregulation of MMR activity, it has been shown that DNA damage-induced apoptosis via the p53 pathway is impaired in chronic inflammation. TNF-α and the TH2 cytokines IL-4 and IL-13, for example, can lead to mutations in the TP53 gene by amplification of activation-induced cytidine deaminase expression.22 Furthermore, migration inhibitory factor (MIF) from macrophages can repress p53 function.23 The dimension of genomic instability together with impaired MMR and cell cycle control in chronic inflammation is emphasized by a high frequency of p53 mutations in chronic inflammatory diseases such as rheumatoid arthritis.24
In addition to p53, inflammation alters a wide range of other cell-cycle control mechanisms. TNF induces fibroblast proliferation (compare page 2), and IL-1 advances the growth of gastric carcinoma.25 Proliferation is stimulated by the activation of toll-like receptor (TLR)-4 by bacteria. This results in increased prostaglandin E2 production by induction of cyclooxygenase (COX)-2 and activation of epidermal growth factor receptor (EGFR) signaling.26
Another mechanism leading to activation of signal transducer and activator of transcription (STAT)-3 is the secretion of IL-6 from cells of the innate immune system and IL-22 from TH17 cells. Apart from various other genes, STAT3 is involved in the transcriptional regulation of cyclin B, cyclin D1, and cyclin D2, which play important roles in cell-cycle control and proliferation.27 Similarly, upregulation of the expression of BCL2 and BCL2-like1 genes by STAT3 results in prolonged cell survival by avoiding apoptosis.28,29
In summary, inflammation leads to DNA damage by oxygen and nitrogen radicals, especially in cells that are prone because of diminished capacity for DNA repair, apoptosis, and/or cell-cycle control.
Persistent proliferation and perpetual tumorigenic signaling in chronic inflammation
Persistence of this mutagenic environment in chronic inflammation enables cells to accumulate several mutations that are required for neoplastic transformation. For example, Kras and p53 gene mutations are very common in patients with GI malignancies. Tumor suppressor p53 is often designated as ‘guardian of the genome’ as it prompts cell-cycle arrest and apoptosis in defective cells via Mdm2, p21, mammalian target of rapamycin (mTOR), and PUMA signaling. In contrast, Kras is an oncogene, which is activated in many different tumors. It belongs to the Ras family of guanosine triphosphates (GTPases) and is involved in many cellular processes, such as cell proliferation, differentiation, apoptosis, and senescence. Phosphatidylinositol 3-kinase (PI-3-K), Raf, Rac, Rho, and TGF-β belong to its key downstream effector pathway.30–32
IL-6/STAT3 signaling can maintain a mutagenic state. Upregulation of sphingosine-1-phosphate (S1P) activates nuclear factor (NF)-κB, which in turn induces IL-6 transcription.33 NF-κB can also be activated by TNF and IL-1.34 A self-augmenting loop maintaining chronic inflammation is described after the activation of the proto-oncogene SRC. SRC activation as the triggering event initiates NF-κB/STAT3-mediated cell growth and malignant transformation. Concurrently, NF-κB rapidly reduces let7-microRNA via Lin28 activation, which acts as a direct inhibitor of IL-6. High IL-6 concentrations in return activate NF-κB signaling and by these means start a positive feedback loop and link signaling in chronic inflammation to malignant transformation.35 NF-κB directly, but also via WNT-β-catenin activation, has been shown to provoke dedifferentiation, proliferation, and resistance to cell death in many tumors.36,37 NF-κB as the signal pathway relating carcinogenesis to inflammation was first described by Greten et al38 in colorectal cancer (CRC). In fact, there is evidence for a role of NF-κB in the carcinogenesis of various GI tumors. NF-κB can be activated by TNF and is involved in the transcriptional regulation of many cellular functions. Amongst these are a plethora of tumorigenic functions such as proliferation, invasion, and suppression of apoptosis. Beyond that, NF-κB influences the inflammatory response by controlling the secretion of cytokines such as TNF-α and IL-1.39
Angiogenesis, invasion, and migration
Genomic instability and evasion of cell death are considered to be key steps in carcinogenesis and metastasis formation. But inflammatory cytokines are involved in several of these processes.40 For example, TNF, IL-1, and IL-6 from neutrophils, mast cells, and macrophages increase transcription of vascular endothelial growth factor (VEGF) – an important stimulating factor for angiogenesis.41,42 Another example is the pro-angiogenic effect of IL-8. IL-8 can also be induced by TNF and IL-1.43,44
In accordance with Hanahan and Weinberg’s Hallmarks of Cancer,40 infiltration and migration are crucial features of malignant cells. Altered endothelial permeability by TNF and degradation of extracellular matrix by MMP and cathepsins not only fosters leukocyte infiltration during inflammation, but also paves the way for invasive growth, migration, and intravasation of tumor cells.45,46 Inhibition of IL-1 in a metastatic mouse model actually revealed a distinct decrease in the rate of metastasis.47 To detach epithelial cells and enable these cells to migrate, an epithelial mesenchymal transition (EMT) is assumed to be decisive. STAT3 transcriptional regulation also includes transcription of E-cadherin, which is characteristic for epithelial cells and transcription of MMP. TNF, IL-1, and IL-6-associated NF-κB and STAT3 activation leads to downregulation of E-cadherin and increased MMP transcription, constituting a mesenchymal cell phenotype. A switch from E-cadherin to N-cadherin is typical of EMT. As the switch from E- to N-cadherin depends on the transcription factor Snail, Snail is an important regulator of EMT.48,49 In CRC, active NF-κB has been shown to inhibit Snail ubiquitination and degradation downstream, thereby highlighting the significance of inflammatory cytokine signaling for metastasis.50–52 Through Wnt and Ras signaling, TGF-β can also induce EMT.53
Through these mechanisms, chronic inflammation is a decisive player in early carcinogenesis. It generates a mutagenic environment and may enable susceptible cells with inherited or spontaneously altered genes to accumulate enough mutations for malignant transformation. This is supported by studies that show that treatment with nonsteroidal anti-inflammatory drugs (NSAIDs) consistently decreases the risk of cancer, most likely by direct inhibition of the NF-κB/STAT3 signaling pathway. NF-κB/STAT3 signaling increases expression of COX-2 and in turn is activated itself by COX-2. COX-2 is significantly overexpressed in malignancies, and NSAIDs also increase cell death in existing tumors.27,54–58 The anti-neoplastic activity of NSAIDs seems to be mediated by several effectors. NSAIDs without COX inhibitory activity, for example, have similar anti-tumorigenic efficacy. Among others, induction of apoptosis as well as suppression of cyclic guanosine monophosphate (cGMP)- and β-catenin-dependent transcription were identified as alternative NSAID properties. This leads to the assumption that inflammatory cytokines have an influence on tumor progression.59,60
Inflammation in tumor progression
Tumor interactions with the immune system have recently been the focus of extensive research, and a comprehensive description would be beyond the scope of this article. However, as tumors induce a peritumoral chronic state of inflammation, major aspects of tumor-immune cell interactions will be illustrated in this article. To maintain carcinogenic signaling from immune cells, tumors actively attract macrophages and T-cells by chemokines such as IL-4 and TGF-β, bridging the gap to Virchow’s description of leukocyte infiltration of tumors. Cytokines, MMPs, and cathepsins from the so-called tumor-associated macrophages (TAMs) are pivotal for further growth, angiogenesis, and invasion. In addition, macrophage-derived EGF forms a paracrine signaling loop, with tumor-derived colony-stimulating factor (CSF)-1 driving neoplastic invasion.61,62 These experimental findings have been confirmed by demonstrating an association of macrophage infiltration with a poor prognosis in human cancer patients.63 As necrotic tissue fosters macrophage invasion, tumors might even benefit from limited intra-tumoral necrosis.62 In contrast, the immune system poses a threat to malignant cells and it is essential for tumors to escape a cytotoxic immune response. Gabrilovich et al.64 discovered tumor-derived VEGF-A, which has the potential to suppress dendritic cell antigen presentation. APC function is also directly impaired by TGF-β and IL-10 secretion of tumor cells.62,65 Another mechanism to avoid natural killer cells is to attract myeloid-derived suppressor cells (MDSCs). Amongst others, the release of chemokines such as CC-chemokine ligands (CCL) -2, -5, and -12, prostaglandins, and granulocyte colony-stimulating factor (G-CSF) can evoke MDSC accumulation in tumor tissue. As a consequence, MDSCs suppress T-cell function by production of IL-10 and TGF-β.42,62,66,67 Only the immune-suppressive function of Treg cells is maintained by TGF-β, and elevated levels of MDSC and Treg correlate with tumor burden.68
In conclusion, inflammatory signaling can both support and suppress the growth of established tumors. By partial immune suppression with concomitant selective activation of myeloid cells, tumors influence the inflammatory response to their favor.
Specific role of chronic inflammation in gastrointestinal cancer
Esophagus
Barrett’s esophagus and esophageal adenocarcinoma
Gastroesophageal reflux disease (GERD), smoking, body mass index, and a low fruit consumption are well established risk factors for EAC and account for the majority of cases. Although the interaction of several of these risk factors is crucial for the development of this cancer, GERD appears to be the most important individual risk factor.69 The reflux of gastric acid causes mucosal damage of the distal esophagus, leading to chronic esophagitis. In some patients, metaplasia from a squamous to a columnar epithelium occurs as a consequence of chronic inflammation during mucosal regeneration – the so-called Barrett’s mucosa. Whether Barrett’s mucosa is caused by a translocation or migration of gastric cells, by bone marrow stem cells, or by true metaplasia of esophageal cells is controversial. However, proinflammatory cytokines such as IL-6, IL-8, and IL-1β appear to be involved in the development of Barrett’s mucosa.70–73 Consistently, inhibition of COX-2 inhibits proliferation of Barrett’s mucosal cells.74 As a result, the incidence of EAC is 30 times higher in people with Barrett’s esophagus than in the general population.74 Nevertheless, the annual risk of patients with Barrett’s mucosa for developing EAC is 0.12%–0.4% and is relatively low overall.75,76
Squamous cell carcinoma
Besides the major risk factors of alcohol, smoking, and genetic polymorphisms, a carcinogenesis-promoting role of chronic inflammation in squamous cell carcinoma is assumed. Hot beverages and fungal invasion are suspected to cause chronic irritation and inflammation of the squamous cell mucosa. The influence of human papilloma virus is still under debate, but its pathogenetic influence on carcinogenesis might be by way of genomic integration rather than inflammation.77–79
Stomach
Carcinomas other than adenocarcinomas are very rare in the stomach. Gastric adenocarcinomas are classified into an intestinal and a diffuse type. A total of 80% of gastric cancer cases are associated with H. pylori infections, and H. pylori-associated gastric cancer is one of the best elucidated relationships between chronic inflammation and cancer overall. While being protected by antioxidants itself, H. pylori can expose host cells to oxidative stress, impair MMR, and alter DNA methylation.80,81 In response to H. pylori, the gastric mucosa releases the inflammatory cytokine IL-8, an overexpression of which correlates with a poor prognosis.82 Virulence factor cytotoxin-associated gene A (cagA)-positive H. pylori strains were identified as particularly carcinogenic. After delivering cagA into gastric epithelial cells, H. pylori can initiate signaling that activates growth factor receptors that increase proliferation, invasion, and angiogenesis and also inhibit apoptosis.83 Additionally, chronic inflammation caused by infection with H. pylori contributes to neoplastic transformation by establishing a positive feedback loop via STAT3-dependent COX-2 induction, which in turn influences STAT3 regulation via IL-6.84
However, gastric carcinogenesis is considered a multistep process, and other inflammatory risk factors have been identified that either act independently of H. pylori infections or further enhance their effects. For example, chronic gastritis caused by bile reflux can cause intestinal metaplasia as a neoplastic precursor lesion. Also, T-cell-mediated autoimmune gastritis fosters the development of intestinal-type gastric cancer.85,86
All of these risk factors lead to a state of chronic inflammation. Activation of signaling mediators that are typical of chronic inflammation such as TNF, IL-6, and prostaglandin E2 has been identified as a promoter of gastric tumorigenesis. For example, TNF-α fosters carcinogenesis by upregulating nicotinamide adenine dinucleotide phosphate (NADPH) oxidase organizer 1 (Noxo1) and Gna14, resulting in dedifferentiation of gastric epithelial cells.87–89 Furthermore, tumor suppressor microRNA miR-7, which inhibits proliferation in vitro, is downregulated in gastric tumors in an inflammation-dependent manner.90
Colorectal cancer
Inflammatory bowel disease (IBD) has long been identified as risk factor for CRC. As CRC in IBD arises from flat dysplastic tissue or dysplasia-associated lesions or masses, carcinogenesis seems to differ from the well described adenoma–carcinoma sequence. Concomitantly, IBD-associated CRC has a worse prognosis. The risk of CRC in patients with colonic manifestation of Crohn’s disease and in patients with ulcerative colitis is similar. Recent population-based studies have shown an overall standardized incidence ratio of CRC in patients with IBD of 1.7, with a significantly lower age of CRC manifestation (7–12 years earlier). However, this ratio is lower than those reported for years before 2000, which might be a result of aging cohorts.91,92 The risk of malignancy correlates with the duration of IBD (10 years after IBD diagnosis: increase of approximately 1% per year). Several clinical symptoms of IBD, such as anatomic extent of disease, disease activity, and primary sclerosing cholangitis (PSC) characterize patients with a comparatively higher risk for malignancy.93,94 Inflammation-driven carcinogenesis in patients with CRC is based on the above-illustrated conditions in chronic inflammation, with oxidative stress and impaired MMR combined with proliferation, invasion, and angiogenesis promoting signaling. In contrast with sporadic CRC, p53 mutations occur rather early in disease development, and APC mutations rather late.95 Downregulation of tumor-suppressor genes by hypermethylation was often found at sites of chronic inflammation, and recently the role of intestinal pathogens in carcinogenesis has attracted increasing attention. The DNA repair gene MUTYH is a homologue to the Escherichia coli gene mutY, and colonization by exclusively intracellular E. coli was detected at tumor sites.95,96 Pathogen antigen binding to TLRs, such as TLR9, can initiate inflammation and could provide a link between pathogens such as E. coli and neoplastic transformation.97 As Treg concentration in CRC correlates with morbidity, activation of Tregs in IBD might also contribute to cancer initiation and progression.98
Pancreas
Chronic pancreatitis is a well established risk factor for the development of PDAC, and the risk correlates with the duration of chronic pancreatitis. As mutations found in hereditary pancreatitis differ from those found in tumors, this epidemiologic observation has to be based on enhanced carcinogenesis due to inflammation.99,100 In fact, chemokine receptor 2 (CXCR2) ligands such as IL-6 and IL-8 have been identified as autocrine promoters of tumor growth in pancreatic cancer.101 Another inflammatory chemokine, the so-called macrophage pro-inflammatory chemokine-3a (MIP-3a/CCL20), is overexpressed in pancreatic cancer cells and TAMs. It stimulates growth and migration of neoplastic cells and activates further macrophages.102 Furthermore, patients with homozygous allele 2 of IL-1β, which is associated with higher IL-1β concentrations in pancreatic cancer, had a significantly shorter survival, which once again points to the importance of crosstalk of pancreatic cancer and immune cells.103 TGF-β signaling is enhanced in chronic inflammation and plays a prominent role in its regulation (compare pages 2 and 4). Concomitantly, inhibition of TGF-β signaling in pancreatic cancer resulted in increased survival.104 In 90% of pancreatic cancers, Kras-activating mutations are responsible for continuous proliferative and anti-apoptotic signaling. Interestingly, activation of NF-κB signaling in chronic pancreatitis can ultimately lead to Kras activation and neoplastic transformation. Also, permanently mutated Kras has been detected in chronic pancreatitis.105–108 Only recently has a pivotal role of the IL-6/STAT3 pathway in pancreatic cancer been described. Oncogenic Ras (KrasG12D) induces the secretion of IL-6 from different cell types, and genetic inactivation of IL-6 impedes Ras-driven tumorigenesis.109 The activation of mutant Kras (KrasG12D) in pancreatic ductal epithelial cells reprograms the tumor microenvironment and is associated with a robust inflammatory response characterized by release of inflammatory cytokines, such as granulocyte macrophage (GM)-CSF. KrasG12D-dependent GM-CSF production promotes infiltration of myeloid cells into the surrounding stroma.110,111 TAMs secrete IL-6 that directly activates the Janus kinases (JAKs), thus influencing pancreatic intraepithelial neoplasia (PanIN) progression and pancreatic cancer development.112,113 The activated JAK in turn phosphorylates several downstream targets, including cytoplasmic STAT3, which after dimerization rapidly translocates into the nucleus and promotes PanIN progression through transcriptional regulation of anti-apoptotic and pro-proliferative genes. Pancreatic epithelial STAT3 deletion and inactivation of IL-6 trans-signaling impairs PanIN formation and inhibits PDAC development.113 These studies, together with numerous other observations, suggest that Kras serves as a link between chronic inflammation and carcinogenesis. In combination with other risk factors, H. pylori infection and periodontal disease also appear to contribute to carcinogenesis in the pancreas.114
Hepatobiliary
Hepatocellular carcinoma
In recent decades, incidence of HCC has been increasing in the USA, and a growing number of hepatitis C virus (HCV) and hepatitis B virus (HBV) infections have been identified as the underlying cause. Other risk factors for HCC have not increased in incidence during this time.115 HBV is a DNA-integrating virus. Accordingly, activation or suppression of oncogenes and tumor suppressor genes by random integration of viral DNA is suspected to be the key effect in HBV-associated carcinogenesis. However, HBV infection-induced inflammation with activation of NF-κB signaling and DNA damage by oxidative stress appear to be important pathogenetic mechanisms.116 In contrast, no specific virally induced oncogenes have been identified in HCV infection. Carcinogenesis in patients with HCV (an RNA virus) infection is considered to be an inflammation-mediated process (eg, TGF-β signaling) with a high cell turnover, fibrosis, and cirrhosis. This hypothesis is supported by the fact that HCV-related HCC correlates with the degree of inflammation, whereas HCC can emerge in HBV-infected patients even without evidence of cirrhosis.117–119 This hypothesis was further supported by the finding that antiviral therapy was beneficial in select patients with HBV/HCV-related HCC.120
However, as these lesions develop secondary to hepatic cirrhosis in most patients with HCC, this is considered to be the major risk factor. While hepatocytes are more susceptible to mutagenesis because of the high cell turnover during regeneration, recurrent necrosis of liver cells induces chronic inflammation. This process leads to hepatic fibrosis, which ultimately culminates in cirrhosis.121–123 Hepatocellular necrosis can be caused by toxins such as alcohol, other environmental factors such as diet, or by chronic inflammation itself. Accordingly, non-alcoholic fatty liver disease and primary biliary cirrhosis have also been identified as risk factors of HCC that are based on chronic inflammation.124–126
During inflammation, macrophages (ie, Kupffer cells) activate fibroblasts and stellate cells by release of TNF and PDGF. Activated fibroblasts produce numerous cytokines, including EGF, hepatocyte growth factor, fibroblast growth factor, IL-6, and MMP-9. This combined signaling causes fibroblastic differentiation of stellate cells, which plays a central role in the pathogenesis of fibrosis. After initiation of the tumor, infiltrating Treg cells that are attracted by Kupffer cells and TAMs modify the immune response and stimulate angiogenesis. Taken together, accumulation of mutations during inflammation-driven fibrosis or cirrhosis is a prerequisite for HCC development.127,128
Cholangiocarcinoma
The risk factor that accounts for the most cases of cholangiocarcinoma worldwide is infection with parasites such as Opisthorchis viverrini, Clonorchis sinensis, or Ascaris lumbricoides. These parasites are endemic in several Asian regions, cause chronic inflammation of the biliary tree, and expose the biliary epithelium to reactive oxygen and nitrogen species.129,130
In Western nations, almost one-third of the patients with cholangiocarcinoma suffer from PSC and, on average, develop the malignancy at a significantly lower age.131 Interestingly, the duration of PSC does not correlate with the incidence of cholangiocarcinoma. DNA damage and proliferative and mutagenic signaling in chronic inflammation of PSC are considered key steps in the development of cholangiocarcinoma.132
Similar to HCC, chronic infection with HBV or HCV, hepatic cirrhosis, and non-alcoholic fatty liver disease also increase the risk of cholangiocarcinoma.133,134
Recurrent bacterial infections due to cholestasis by biliary stones, congenital abnormalities such as fibropolycystic liver disease, or due to choledocho-enteric anastomoses are also associated with a higher incidence of cholangiocarcinoma. Chronic infections of the biliary tree are presumably not solitary triggers of carcinogenesis and can provide the necessary second hit for malignant transformation in genetically predisposed patients.135
Several causative signaling pathways linking inflammation to carcinogenesis in biliary disease have been identified. Human immune response to parasitic biliary infections is mediated by metastasis-associated protein 1 (MTA1) with its adjunctive cytokeratins K-18 and 19. Besides regulating TH1 and TH2 immune responses, MTA1 has a central role in carcinogenesis and is overexpressed in malignant cells.136 Auto- and paracrine stimulation of proliferation mediated by IL-6, IL-8, TGF-β, and TNF-α can be found in cholangiocytes. This stimulation is mediated by IL-6-related activation of p38 mitogen-activated protein kinase and overexpression of the anti-apoptotic myeloid cell leukemia (Mcl)-1.137–139
A similar correlation of recurrent cholecystitis and chronic inflammation of the bile ducts with malignancy is evident for the gallbladder.140
Mucosa-associated lymphoid tissue lymphoma
MALT lymphoma is an extra-nodal B-cell non-Hodgkin lymphoma that can manifest itself in epithelial tissues throughout the body. Typical GI organs of manifestation are the stomach and the small intestine. Although several chromosomal translocations have been identified in MALT lymphomas, recent research provides augmenting evidence for an inflammatory genesis. Chronic inflammation by infections with Campylobacter jejuni, HCV, or, predominantly, H. pylori precede many cases of MALT lymphoma and an association of extra-intestinal MALT and autoimmune disease has been described.141–143 The release of a proliferation-inducing ligand (APRIL), which belongs to the TNF family, by macrophages as response to infection is a potential link between inflammation and neoplastic transformation. APRIL is important for B-cell maturation and survival. Also, NF-κB is frequently activated in MALT lymphoma. This signaling pathway can be induced in inflammation or might be permanently mutated by oxygen radicals during chronic inflammation.141,144 H. pylori virulence factor cagA is found in approximately 90% of patients with gastric MALT lymphoma. Also, eradication of H. pylori leads to regression of GI MALT lymphomas in about 90% of the cases, and H. pylori eradication is the standard therapy for patients affected.145,146
Clinical perspective
There is accumulating evidence for the involvement of chronic inflammation in the carcinogenesis of most GI malignancies, and the underlying cellular and molecular processes are increasingly understood. Chronic inflammation as a potent player in tumor initiation and progression has extended our concept of the multivariate process of neoplastic transformation (Figure 1). This knowledge has already been implemented in clinical routine. For example, in patients with Barrett’s esophagus, H. pylori is eradicated to prevent esophageal or gastric cancer. In patients with MALT lymphoma, eradication of H. pylori is used to induce remission in these patients. In many inflammatory diseases such as IBD, anti-inflammatory drugs are the mainstay of therapy. However, while mucosal healing in IBD patients results in a decreasing rate of dysplasia, a similar effect could not be proven for single agents such as 5-aminosalicylates or thiopurines.147,148 In contrast, many studies showed a chemoprophylactic property of NSAIDs in other inflammation-associated malignancies, such as gastric carcinoma and CRC. Nevertheless, unselective COX inhibitors carry a notable risk of GI bleeding, and selective COX-2 inhibitors also carry a risk of cardiovascular complications that outweigh the anti-neoplastic benefits.149 Thus, the identification of COX-independent anti-neoplastic activity of NSAIDs and the development of NSAIDs that are more selective might offer future treatment options for defined patient populations that carry a high risk of cancer, such as patients with a history of familial adenomatous polyposis.
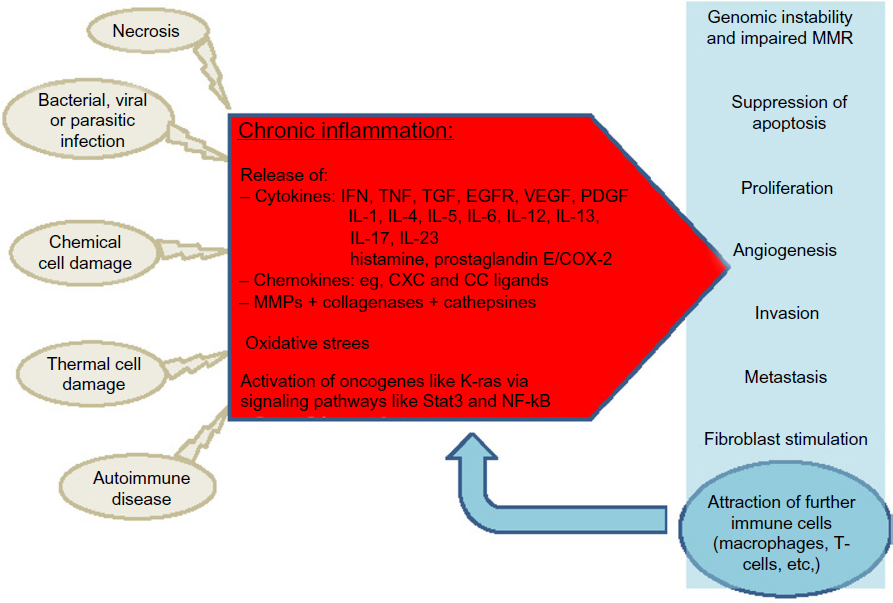 | Figure 1 Chronic inflammation in carcinogenesis. |
Nowadays, immune-suppressive drugs not only restrict chronic inflammation but are also associated with an increased risk of developing malignancies. More specific agents that interfere with selected immune functions might actually be equally effective as an anti-inflammatory and as an anti-neoplastic medication. Inhibition of IL-6 signaling via IL-6 receptor antibodies (Tocilizumab®; Hoffmann-La Roche AG, Basel, Swiss) represents an effective treatment for patients with rheumatoid arthritis, but also showed promising results in a mouse model of Crohn’s disease.150
However, similar to TNF, with its pro- and anti-angiogenic effects, many mediators of the immune response have a dual role in carcinogenesis; further research is required for clarification.
Also complicating is the fact that many cytokines and signaling pathways have multiple functions throughout the human body. For example, Tocilizumab-induced inhibition of IL-6 – a decisive cytokine linking inflammation and carcinogenesis – is associated with severe side effects such as gastric perforation.151
More importantly, it is crucial to obtain further insight into the molecular pathways that drive carcinogenesis in inflammation. Identification of such signaling pathways is therefore of utmost importance. For example, blocking the soluble IL-6 receptor that is predominantly activated during stress situations such as chronic inflammation and cancer might evolve into a future treatment option.152 Another potentially interesting target is the JAK2/STAT3 pathway. In fact, several studies are currently underway that address the pharmacological inhibition of the IL-6-dependent JAK2/STAT3 pathway in patients with various solid GI tumors.
Furthermore, there is already evidence of NK cells being capable of restoring the antigen-presenting function of dendritic cells and MDSC.68 Detailed understanding of the tumor–immune system interaction would provide the possibility to redirect the immune response against neoplastic cells. Such immune-modulatory drugs could be beneficial for cancer prevention as well as for limiting cancer progression or improving response to existing therapies.
Conclusion
Chronic inflammation is a major risk factor for the development of GI malignancies. As ‘wounds that do not heal’, tumors benefit from inflammatory signaling while escaping damage through the immune system by modifying the immune response in their favor. Further insight into the function of the mediators and the regulation of the immune response could help to identify more specific future treatment targets. In our opinion, understanding of the tumor microenvironment and tumor–immune system interactions will be crucial, as these aspects have not yet been implemented in therapeutic regimens. Altering a tumor-susceptible micro environment, or even avoiding formation of the so-called metastatic niche, might reduce metastasis. In addition, re-establishing an immune response against neoplastic cells might improve the efficacy of existing therapies. This estimation is supported by the finding that assessment of the immunologic tumor environment allows better conclusions regarding prognosis and response to therapy than the traditional anatomic International Union Against Cancer ‘Tumor/Node/Metastasis’ (UICC-TNM) classification system for tumor staging.153 As assay availability and protocols vary among laboratories, an international task force has proposed the ‘Immunoscore’ as a standardized prognostic tool.154 The ongoing clinical evaluation of this score will reveal its relevance for clinical practice.
Disclosure
The authors do not have any conflicts of interest to disclose.
References
Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008:GLOBOCAN 2008. Int J Cancer. 2010;127(12):2893–2917. | |
Cook MB, Chow WH, Devesa SS. Oesophageal cancer incidence in the United States by race, sex, and histologic type, 1977–2005. Br J Cancer. 2009;101(5):855–859. | |
Liu J, Waalkes MP. Liver is a target of arsenic carcinogenesis. Toxicol Sci. 2008;105(1):24–32. | |
Peek RM Jr, Crabtree JE. Helicobacter infection and gastric neoplasia. J Pathol. 2006;208(2):233–248. | |
Varela-Rey M, Woodhoo A, Martinez-Chantar ML, Mato JM, Lu SC. Alcohol, DNA methylation, and cancer. Alcohol Res. 2013; 35(1):25–35. | |
Virchow R. An address on the value of pathological experiments. Br Med J. 1881;2(1075):198–203. | |
Spits H, Cupedo T. Innate lymphoid cells: emerging insights in development, lineage relationships, and function. Annu Rev Immunol. 2012;30:647–675. | |
Spits H, Artis D, Colonna M, et al. Innate lymphoid cells – a proposal for uniform nomenclature. Nat Rev Immunol. 2013;13(2):145–149. | |
Lejeune FJ, Rüegg C, Liénard D. Clinical applications of TNF-alpha in cancer. Curr Opin Immunol. 1998;10(5):573–580. | |
Dong C. Diversification of T-helper-cell lineages: finding the family root of IL-17-producing cells. Nat Rev Immunol. 2006;6(4):329–333. | |
Romagnani S. Lymphokine production by human T cells in disease states. Annu Rev Immunol. 1994;12:227–257. | |
Miossec P. IL-17 and Th17 cells in human inflammatory diseases. Microbes Infect. 2009;11(5):625–630. | |
Gallin JI, Snyderman R, Fearon DT, Haynes BF, Nathan C, editors. Inflammation: Basic Principles and Clinical Correlates. 3rd ed. Philadelphia: Lippincott Williams & Wilkins; 1999. | |
Kuper H, Adami HO, Trichopoulos D. Infections as a major preventable cause of human cancer. J Intern Med. 2000;248(3):171–183. | |
de Martel C, Ferlay J, Franceschi S, et al. Global burden of cancers attributable to infections in 2008: a review and synthetic analysis. Lancet Oncol. 2012;13(6):607–615. | |
Maeda H, Akaike T. Nitric oxide and oxygen radicals in infection, inflammation, and cancer. Biochemistry (Mosc). 1998;63(7):854–865. | |
Jaiswal M, LaRusso NF, Burgart LJ, Gores GJ. Inflammatory cytokines induce DNA damage and inhibit DNA repair in cholangiocarcinoma cells by a nitric oxide-dependent mechanism. Cancer Res. 2000;60(1):184–190. | |
Chang CL, Marra G, Chauhan DP, et al. Oxidative stress inactivates the human DNA mismatch repair system. Am J Physiol Cell Physiol. 2002;283(1):C148–C154. | |
Fleisher AS, Esteller M, Harpaz N, et al. Microsatellite instability in inflammatory bowel disease-associated neoplastic lesions is associated with hypermethylation and diminished expression of the DNA mismatch repair gene, hMLH1. Cancer Res. 2000;60(17):4864–4868. | |
Koshiji M, To KK, Hammer S, et al. HIF-1alpha induces genetic instability by transcriptionally downregulating MutSalpha expression. Mol Cell. 2005;17(6):793–803. | |
Jung YJ, Isaacs JS, Lee S, Trepel J, Neckers L. IL-1beta-mediated up-regulation of HIF-1alpha via an NFkappaB/COX-2 pathway identifies HIF-1 as a critical link between inflammation and oncogenesis. FASEB J. 2003;17(14):2115–2117. | |
Endo Y, Marusawa H, Kou T, et al. Activation-induced cytidine deaminase links between inflammation and the development of colitis-associated colorectal cancers. Gastroenterology. 2008;135(3):889–898, 898. e1–e3. | |
Hudson JD, Shoaibi MA, Maestro R, Carnero A, Hannon GJ, Beach DH. A proinflammatory cytokine inhibits p53 tumor suppressor activity. J Exp Med. 1999;190(10):1375–1382. | |
Yamanishi Y, Boyle DL, Rosengren S, Green DR, Zvaifler NJ, Firestein GS. Regional analysis of p53 mutations in rheumatoid arthritis synovium. Proc Natl Acad Sci U S A. 2002;99(15):10025–10030. | |
El-Omar EM, Carrington M, Chow WH, et al. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature. 2000;404(6776):398–402. | |
Fukata M, Chen A, Vamadevan AS, et al. Toll-like receptor-4 promotes the development of colitis-associated colorectal tumors. Gastroenterology. 2007;133(6):1869–1881. | |
Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9(11):798–809. | |
Bollrath J, Phesse TJ, von Burstin VA, et al. gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell. 2009;15(2):91–102. | |
Sonnenberg GF, Fouser LA, Artis D. Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat Immunol. 2011;12(5):383–390. | |
Castellano E, Downward J. RAS interaction with PI3K: more than just another effector pathway. Genes Cancer. 2011;2(3):261–274. | |
Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer. 2011;11(11):761–774. | |
Suppiah A, Greenman J. Clinical utility of anti-p53 auto-antibody: systematic review and focus on colorectal cancer. World J Gastroenterol. 2013;19(29):4651–4670. | |
Liang J, Nagahashi M, Kim EY, et al. Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell. 2013;23(1):107–120. | |
Faustman D, Davis M. TNF receptor 2 pathway: drug target for autoimmune diseases. Nat Rev Drug Discov. 2010;9(6):482–493. | |
Iliopoulos D, Hirsch HA, Struhl K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 microRNA, and IL6 links inflammation to cell transformation. Cell. 2009;139(4):693–706. | |
Ben-Neriah Y, Karin M. Inflammation meets cancer, with NF-kappaB as the matchmaker. Nat Immunol. 2011;12(8):715–723. | |
Schwitalla S, Fingerle AA, Cammareri P, et al. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell. 2013;152(1–2):25–38. | |
Greten FR, Eckmann L, Greten TF, et al. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118(3):285–296. | |
Kim HJ, Hawke N, Baldwin AS. NF-kappaB and IKK as therapeutic targets in cancer. Cell Death Differ. 2006;13(5):738–747. | |
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. | |
Ferrara N. Vascular endothelial growth factor: basic science and clinical progress. Endocr Rev. 2004;25(4):581–611. | |
Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer. 2008;8(8):618–631. | |
Keane MP, Strieter RM. The role of CXC chemokines in the regulation of angiogenesis. Chem Immunol. 1999;72:86–101. | |
Torisu H, Ono M, Kiryu H, et al. Macrophage infiltration correlates with tumor stage and angiogenesis in human malignant melanoma: possible involvement of TNFalpha and IL-1alpha. Int J Cancer. 2000;85(2):182–188. | |
Peinado H, Lavotshkin S, Lyden D. The secreted factors responsible for pre-metastatic niche formation: old sayings and new thoughts. Semin Cancer Biol. 2011;21(2):139–146. | |
Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141(1):39–51. | |
Vidal-Vanaclocha F, Fantuzzi G, Mendoza L, et al. IL-18 regulates IL-1beta-dependent hepatic melanoma metastasis via vascular cell adhesion molecule-1. Proc Natl Acad Sci U S A. 2000;97(2):734–739. | |
Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119(6):1420–1428. | |
Wu Y, Zhou BP. TNF-alpha/NF-kappaB/Snail pathway in cancer cell migration and invasion. Br J Cancer. 2010;102(4):639–644. | |
Sullivan NJ, Sasser AK, Axel AE, et al. Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene. 2009;28(33):2940–2947. | |
Wang H, Wang HS, Zhou BH, et al. Epithelial-mesenchymal transition (EMT) induced by TNF-α requires AKT/GSK-3β-mediated stabilization of snail in colorectal cancer. PLoS One. 2013;8(2):e56664. | |
Wu Y, Deng J, Rychahou PG, Qiu S, Evers BM, Zhou BP. Stabilization of snail by NF-kappaB is required for inflammation-induced cell migration and invasion. Cancer Cell. 2009;15(5):416–428. | |
Fuxe J, Karlsson MC. TGF-β-induced epithelial-mesenchymal transition: a link between cancer and inflammation. Semin Cancer Biol. 2012;22(5–6):455–461. | |
Elder DJ, Halton DE, Hague A, Paraskeva C. Induction of apoptotic cell death in human colorectal carcinoma cell lines by a cyclooxygenase-2 (COX-2)-selective nonsteroidal anti-inflammatory drug: independence from COX-2 protein expression. Clin Cancer Res. 1997;3(10):1679–1683. | |
García-Rodríguez LA, Huerta-Alvarez C. Reduced risk of colorectal cancer among long-term users of aspirin and nonaspirin nonsteroidal antiinflammatory drugs. Epidemiology. 2001;12(1):88–93. | |
Gupta RA, Dubois RN. Colorectal cancer prevention and treatment by inhibition of cyclooxygenase-2. Nat Rev Cancer. 2001;1(1):11–21. | |
Yin MJ, Yamamoto Y, Gaynor RB. The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature. 1998;396(6706):77–80. | |
Roelofs HM, Te Morsche RH, van Heumen BW, Nagengast FM, Peters WH. Over-expression of COX-2 mRNA in colorectal cancer. BMC Gastroenterol. 2014;14:1. | |
Gurpinar E, Grizzle WE, Piazza GA. COX-independent mechanisms of cancer chemoprevention by anti-inflammatory drugs. Front Oncol. 2013;3:181. | |
Gurpinar E, Grizzle WE, Piazza GA. NSAIDs inhibit tumorigenesis, but how? Clin Cancer Res. 2014;20(5):1104–1113. | |
Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19(11):1423–1437. | |
Smith HA, Kang Y. The metastasis-promoting roles of tumor-associated immune cells. J Mol Med (Berl). 2013;91(4):411–429. | |
Leek RD, Landers RJ, Harris AL, Lewis CE. Necrosis correlates with high vascular density and focal macrophage infiltration in invasive carcinoma of the breast. Br J Cancer. 1999;79(5–6):991–995. | |
Gabrilovich DI, Chen HL, Girgis KR, et al. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat Med. 1996;2(10):1096–1103. | |
Geissmann F, Revy P, Regnault A, et al. TGF-beta 1 prevents the noncognate maturation of human dendritic Langerhans cells. J Immunol. 1999;162(8):4567–4575. | |
Huang B, Lei Z, Zhao J, et al. CCL2/CCR2 pathway mediates recruitment of myeloid suppressor cells to cancers. Cancer Lett. 2007; 252(1):86–92. | |
Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9(3):162–174. | |
Lindau D, Gielen P, Kroesen M, Wesseling P, Adema GJ. The immunosuppressive tumour network: myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology. 2013;138(2):105–115. | |
Zhai R, Chen F, Liu G, et al. Interactions among genetic variants in apoptosis pathway genes, reflux symptoms, body mass index, and smoking indicate two distinct etiologic patterns of esophageal adenocarcinoma. J Clin Oncol. 2010;28(14):2445–2451. | |
Quante M, Bhagat G, Abrams JA, et al. Bile acid and inflammation activate gastric cardia stem cells in a mouse model of Barrett-like metaplasia. Cancer Cell. 2012;21(1):36–51. | |
Sarosi G, Brown G, Jaiswal K, et al. Bone marrow progenitor cells contribute to esophageal regeneration and metaplasia in a rat model of Barrett’s esophagus. Dis Esophagus. 2008;21(1):43–50. | |
Wang X, Ouyang H, Yamamoto Y, et al. Residual embryonic cells as precursors of a Barrett’s-like metaplasia. Cell. 2011;145(7):1023–1035. | |
Fitzgerald RC, Abdalla S, Onwuegbusi BA, et al. Inflammatory gradient in Barrett’s oesophagus: implications for disease complications. Gut. 2002;51(3):316–322. | |
Van der Veen AH, Dees J, Blankensteijn JD, Van Blankenstein M. Adenocarcinoma in Barrett’s oesophagus: an overrated risk. Gut. 1989;30(1):14–18. | |
de Jonge PJ, van Blankenstein M, Looman CW, Casparie MK, Meijer GA, Kuipers EJ. Risk of malignant progression in patients with Barrett’s oesophagus: a Dutch nationwide cohort study. Gut. 2010;59(8):1030–1036. | |
Hvid-Jensen F, Pedersen L, Drewes AM, Sørensen HT, Funch-Jensen P. Incidence of adenocarcinoma among patients with Barrett’s esophagus. N Engl J Med. 2011;365(15):1375–1383. | |
Stoner GD, Gupta A. Etiology and chemoprevention of esophageal squamous cell carcinoma. Carcinogenesis. 2001;22(11):1737–1746. | |
Sun L, Yu S. Meta-analysis: non-steroidal anti-inflammatory drug use and the risk of esophageal squamous cell carcinoma. Dis Esophagus. 2011;24(8):544–549. | |
Zhang Y. Epidemiology of esophageal cancer. World J Gastroenterol. 2013;19(34):5598–5606. | |
Kim SS, Ruiz VE, Carroll JD, Moss SF. Helicobacter pylori in the pathogenesis of gastric cancer and gastric lymphoma. Cancer Lett. 2011;305(2):228–238. | |
Nagini S. Carcinoma of the stomach: a review of epidemiology, pathogenesis, molecular genetics and chemoprevention. World J Gastrointest Oncol. 2012;4(7):156–169. | |
Lee KE, Khoi PN, Xia Y, et al. Helicobacter pylori and interleukin-8 in gastric cancer. World J Gastroenterol. 2013;19(45):8192–8202. | |
Hatakeyama M. Oncogenic mechanisms of the Helicobacter pylori CagA protein. Nat Rev Cancer. 2004;4(9):688–694. | |
Xiong H, Du W, Sun TT, et al. A positive feedback loop between STAT3 and cyclooxygenase-2 gene may contribute to Helicobacter pylori-associated human gastric tumorigenesis. Int J Cancer. 2014;134(9):2030–2040. | |
Sobala GM, O’Connor HJ, Dewar EP, King RF, Axon AT, Dixon MF. Bile reflux and intestinal metaplasia in gastric mucosa. J Clin Pathol. 1993;46(3):235–240. | |
Nguyen TL, Khurana SS, Bellone CJ, et al. Autoimmune gastritis mediated by CD4+ T cells promotes the development of gastric cancer. Cancer Res. 2013;73(7):2117–2126. | |
Oshima H, Oshima M. The role of PGE2-associated inflammatory responses in gastric cancer development. Semin Immunopathol. 2013;35(2):139–150. | |
Oshima H, Ishikawa T, Yoshida GJ, et al. TNF-α/TNFR1 signaling promotes gastric tumorigenesis through induction of Noxo1 and Gna14 in tumor cells. Oncogene. Epub August 26, 2013. | |
Kinoshita H, Hirata Y, Nakagawa H, et al. Interleukin-6 mediates epithelial-stromal interactions and promotes gastric tumorigenesis. PLoS One. 2013;8(4):e60914. | |
Kong D, Piao YS, Yamashita S, et al. Inflammation-induced repression of tumor suppressor miR-7 in gastric tumor cells. Oncogene. 2012;31(35):3949–3960. | |
Lutgens MW, van Oijen MG, van der Heijden GJ, Vleggaar FP, Siersema PD, Oldenburg B. Declining risk of colorectal cancer in inflammatory bowel disease: an updated meta-analysis of population-based cohort studies. Inflammatory Bowel Diseases. 2013;19(4):789–799. | |
Peyrin-Biroulet L, Lepage C, Jooste V, Guéant JL, Faivre J, Bouvier AM. Colorectal cancer in inflammatory bowel diseases: a population-based study (1976-2008). Inflamm Bowel Dis. 2012;18(12):2247–2251. | |
Herszenyi L, Miheller P, Tulassay Z. Carcinogenesis in inflammatory bowel disease. Dig Dis. 2007;25(3):267–269. | |
Dyson JK, Rutter MD. Colorectal cancer in inflammatory bowel disease: what is the real magnitude of the risk? World J Gastroenterol. 2012;18(29):3839–3848. | |
Rogler G. Chronic ulcerative colitis and colorectal cancer. Cancer Lett. 2014;345(2):235–241. | |
Khan AA, Cash P. E. coli and colon cancer: is mutY a culprit? Cancer Lett. 2013;341(2):127–131. | |
Füri I, Sipos F, Germann TM, et al. Epithelial toll-like receptor 9 signaling in colorectal inflammation and cancer: clinico-pathogenic aspects. World J Gastroenterol. 2013;19(26):4119–4126. | |
Müzes G, Molnár B, Sipos F. Regulatory T cells in inflammatory bowel diseases and colorectal cancer. World J Gastroenterol. 2012;18(40):5688–5694. | |
Whitcomb DC. Inflammation and cancer V. Chronic pancreatitis and pancreatic cancer. Am J Physiol Gastrointest Liver Physiol. 2004;287(2):G315–G319. | |
Wörmann SM, Diakopoulos KN, Lesina M, Algül H. The immune network in pancreatic cancer development and progression. Oncogene. 2014;33(23):2956–2967. | |
Hertzer KM, Donald GW, Hines OJ. CXCR2: a target for pancreatic cancer treatment? Expert Opin Ther Targets. 2013;17(6):667–680. | |
Kleeff J, Kusama T, Rossi DL, et al. Detection and localization of Mip-3alpha/LARC/Exodus, a macrophage proinflammatory chemokine, and its CCR6 receptor in human pancreatic cancer. Int J Cancer. 1999;81(4):650–657. | |
Barber MD, Powell JJ, Lynch SF, Fearon KC, Ross JA. A polymorphism of the interleukin-1 beta gene influences survival in pancreatic cancer. Br J Cancer. 2000;83(11):1443–1447. | |
Wörmann SM, Algül H. Risk factors and therapeutic targets in pancreatic cancer. Front Oncol. 2013;3:282. | |
Kolodecik T, Shugrue C, Ashat M, Thrower EC. Risk factors for pancreatic cancer: underlying mechanisms and potential targets. Front Physiol. 2013;4:415. | |
Farrow B, Evers BM. Inflammation and the development of pancreatic cancer. Surg Oncol. 2002;10(4):153–169. | |
Rivera JA, Rall CJ, Graeme-Cook F, et al. Analysis of K-ras oncogene mutations in chronic pancreatitis with ductal hyperplasia. Surgery. 1997;121(1):42–49. | |
Hidalgo M. Pancreatic cancer. N Engl J Med. 2010;362(17):1605–1617. | |
Ancrile B, Lim KH, Counter CM. Oncogenic Ras-induced secretion of IL6 is required for tumorigenesis. Genes Dev. 2007;21(14):1714–1719. | |
Bayne LJ, Beatty GL, Jhala N, et al. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell. 2012;21(6):822–835. | |
Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, Bar-Sagi D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell. 2012;21(6):836–847. | |
Beatty GL, Chiorean EG, Fishman MP, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331(6024):1612–1616. | |
Lesina M, Kurkowski MU, Ludes K, et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell. 2011;19(4):456–469. | |
Michaud DS. Role of bacterial infections in pancreatic cancer. Carcinogenesis. 2013;34(10):2193–2197. | |
Davila JA, Morgan RO, Shaib Y, McGlynn KA, El-Serag HB. Hepatitis C infection and the increasing incidence of hepatocellular carcinoma: a population-based study. Gastroenterology. 2004;127(5):1372–1380. | |
Ayub A, Ashfaq UA, Haque A. HBV induced HCC: major risk factors from genetic to molecular level. Biomed Res Int. 2013; 2013:810461. | |
Budhu A, Wang XW. The role of cytokines in hepatocellular carcinoma. J Leukoc Biol. 2006;80(6):1197–1213. | |
Kim do Y, Han KH. Epidemiology and surveillance of hepatocellular carcinoma. Liver Cancer. 2012;1(1):2–14. | |
Matsuzaki K, Murata M, Yoshida K, et al. Chronic inflammation associated with hepatitis C virus infection perturbs hepatic transforming growth factor beta signaling, promoting cirrhosis and hepatocellular carcinoma. Hepatology. 2007;46(1):48–57. | |
Lok AS. Does antiviral therapy for hepatitis B and C prevent hepatocellular carcinoma? J Gastroenterol Hepatol. 2011;26(2):221–227. | |
Aravalli RN, Cressman EN, Steer CJ. Cellular and molecular mechanisms of hepatocellular carcinoma: an update. Arch Toxicol. 2013;87(2):227–247. | |
Severi T, van Malenstein H, Verslype C, van Pelt JF. Tumor initiation and progression in hepatocellular carcinoma: risk factors, classification, and therapeutic targets. Acta Pharmacol Sin. 2010;31(11):1409–1420. | |
Ramakrishna G, Rastogi A, Trehanpati N, Sen B, Khosla R, Sarin SK. From cirrhosis to hepatocellular carcinoma: new molecular insights on inflammation and cellular senescence. Liver Cancer. 2013;2(3–4):367–383. | |
White DL, Kanwal F, El-Serag HB. Association between nonalcoholic fatty liver disease and risk for hepatocellular cancer, based on systematic review. Clin Gastroenterol Hepatol. 2012;10(12):1342–1359. e2. | |
Tacke F, Yoneyama H. From NAFLD to NASH to fibrosis to HCC: role of dendritic cell populations in the liver. Hepatology. 2013;58(2):494–496. | |
Liang Y, Yang Z, Zhong R. Primary biliary cirrhosis and cancer risk: a systematic review and meta-analysis. Hepatology. 2012;56(4):1409–1417. | |
Aravalli RN. Role of innate immunity in the development of hepatocellular carcinoma. World J Gastroenterol. 2013;19(43):7500–7514. | |
Yang JD, Nakamura I, Roberts LR. The tumor microenvironment in hepatocellular carcinoma: current status and therapeutic targets. Semin Cancer Biol. 2011;21(1):35–43. | |
Pinlaor S, Ma N, Hiraku Y, et al. Repeated infection with Opisthorchis viverrini induces accumulation of 8-nitroguanine and 8-oxo-7,8-dihydro-2′-deoxyguanine in the bile duct of hamsters via inducible nitric oxide synthase. Carcinogenesis. 2004;25(8):1535–1542. | |
Shin HR, Oh JK, Masuyer E, et al. Epidemiology of cholangiocarcinoma: an update focusing on risk factors. Cancer Sci. 2010;101(3):579–585. | |
Claessen MMH, Vleggaar FP, Tytgat KM, Siersema PD, van Buuren HR. High lifetime risk of cancer in primary sclerosing cholangitis. J Hepatol. 2009;50(1):158–164. | |
Ehlken H, Schramm C. Primary sclerosing cholangitis and cholangiocarcinoma: pathogenesis and modes of diagnostics. Dig Dis. 2013;31(1):118–125. | |
Al-Bahrani R, Abuetabh Y, Zeitouni N, Sergi C. Cholangiocarcinoma: risk factors, environmental influences and oncogenesis. Ann Clin Lab Sci. 2013;43(2):195–210. | |
Chang JS, Tsai CR, Chen LT. Medical risk factors associated with cholangiocarcinoma in taiwan: a population-based case-control study. PLoS One. 2013;8(7):e69981. | |
Khan SA, Thomas HC, Davidson BR, Taylor-Robinson SD. Cholangiocarcinoma. Lancet. 2005;366(9493):1303–1314. | |
Nair SS, Bommana A, Pakala SB, et al. Inflammatory response to liver fluke Opisthorchis viverrini depends on host master coregulator MTA1, a marker for parasite-induced cholangiocarcinoma in humans. Hepatology. 2011;54(4):1388–1397. | |
Berthiaume EP, Wands J. The molecular pathogenesis of cholangiocarcinoma. Semin Liver Dis. 2004;24(2):127–137. | |
Park J, Tadlock L, Gores GJ, Patel T. Inhibition of interleukin 6-mediated mitogen-activated protein kinase activation attenuates growth of a cholangiocarcinoma cell line. Hepatology. 1999;30(5):1128–1133. | |
Isomoto H, Kobayashi S, Werneburg NW, et al. Interleukin 6 upregulates myeloid cell leukemia-1 expression through a STAT3 pathway in cholangiocarcinoma cells. Hepatology. 2005;42(6):1329–1338. | |
Kumar S, Kumar S, Kumar S. Infection as a risk factor for gallbladder cancer. J Surg Oncol. 2006;93(8):633–639. | |
Witkowska M, Smolewski P. Helicobacter pylori infection, chronic inflammation, and genomic transformations in gastric MALT lymphoma. Mediators Inflamm. 2013;2013:523170. | |
Libra M, Gloghini A, Malaponte G, et al. Association of t(14;18) translocation with HCV infection in gastrointestinal MALT lymphomas. J Hepatol. 2008;49(2):170–174. | |
Lecuit M, Abachin E, Martin A, et al. Immunoproliferative small intestinal disease associated with Campylobacter jejuni. N Engl J Med. 2004;350(3):239–248. | |
Thieblemont C, Bertoni F, Copie-Bergman C, Ferreri AJ, Ponzoni M. Chronic inflammation and extra-nodal marginal-zone lymphomas of MALT-type. Semin Cancer Biol. 2014;24:33–42. | |
Wang HP, Zhu YL, Shao W. Role of Helicobacter pylori virulence factor cytotoxin-associated gene A in gastric mucosa-associated lymphoid tissue lymphoma. World J Gastroenterol. 2013;19(45):8219–8226. | |
Zullo A, Hassan C, Cristofari F, et al. Effects of Helicobacter pylori eradication on early stage gastric mucosa-associated lymphoid tissue lymphoma. Clin Gastroenterol Hepatol 2010. 2010;8(2):105–110. | |
Lichtenstein GR, Rutgeerts P. Importance of mucosal healing in ulcerative colitis. Inflamm Bowel Dis. 2010;16(2):338–346. | |
Nguyen GC, Gulamhusein A, Bernstein CN. 5-aminosalicylic acid is not protective against colorectal cancer in inflammatory bowel disease: a meta-analysis of non-referral populations. Am J Gastroenterol. 2012;107(9):1298–1304; quiz 1297, 1305. | |
Sahin IH, Hassan MM, Garrett CR. Impact of non-steroidal anti-inflammatory drugs on gastrointestinal cancers: current state-of-the science. Cancer Lett. 2014;345(2):249–257. | |
Yao X, Huang J, Zhong H, et al. Targeting interleukin-6 in inflammatory autoimmune diseases and cancers. Pharmacol Ther. 2014;141(2):125–139. | |
Ruderman EM. Overview of safety of non-biologic and biologic DMARDs. Rheumatology (Oxford). 2012;51 Suppl 6:vi37–vi43. | |
Rose-John S, Waetzig GH, Scheller J, Grötzinger J, Seegert D. The IL-6/sIL-6R complex as a novel target for therapeutic approaches. Expert Opin Ther Targets. 2007;11(5):613–624. | |
Galon J, Costes A, Sanchez-Cabo F, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313(5795):1960–1964. | |
Angell H, Galon J. From the immune contexture to the Immunoscore: the role of prognostic and predictive immune markers in cancer. Curr Opin Immunol. 2013;25(2):261–267. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.