")
Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 14
A Chinese Boy with Mowat–Wilson Syndrome Caused by a 10 bp Deletion in the ZEB2 Gene
Authors Wei L, Han X, Li X, Han B , Nie W
Received 18 May 2021
Accepted for publication 9 August 2021
Published 23 August 2021 Volume 2021:14 Pages 1041—1045
DOI https://doi.org/10.2147/PGPM.S320128
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Video abstract of "A Chinese boy with Mowat-Wilson syndrome" [ID 320128].
Views: 1295
Lin Wei,1 Xiao Han,2 Xue Li,1 Bingjuan Han,1 Wenying Nie1
1Jinan Maternity and Child Care Hospital Affiliated to Shandong First Medical University, Jinan Maternal and Child Care Hospital, Jinan, 250001, Shandong Province, People’s Republic of China; 2Jinan Central Hospital, Cheeloo College of Medicine, Shandong University, Jinan, 250012, Shandong Province, People’s Republic of China
Correspondence: Bingjuan Han; Wenying Nie No. 2, Jianguo Xiaojing 3rd Road, Shizhong District, Jinan, Shandong Province, People’s Republic of China
Tel/Fax +86 531-82060983
; +86 531-8902957
Email [email protected]; [email protected]
Purpose: Mowat–Wilson syndrome (MWS) is a rare complex malformation syndrome which is characterized by typical facial dysmorphism, moderate to severe intellectual disability, global developmental delay, and multiple congenital anomalies. Here, we summarize the clinical characteristics and gene mutation analysis of a Chinese boy with MWS.
Patients and Methods: The clinical features of the patient were monitored. DNA extracted from peripheral blood was subjected to sequencing analysis. Then, the whole-exome sequencing was performed.
Results: A novel deletion mutation (c.1137_1146del TAGTATGTCT) was identified in exon 8 of the ZEB2 gene. The deletion mutation was predicted to produce a truncated protein (p.S380Nfs*13), resulting in haploinsufficiency. The patient presented with short stature, microcephaly, congenital heart defects, cryptorchidism, corpus callosum agenesis, global developmental delay, and intellectual disability. Furthermore, he demonstrated bilateral sensorineural hearing loss. This manifestation is less common in MWS. It is first reported in Chinese patients with MWS. Clinical follow-up showed that the facial features of MWS developed with time. The facial features of the patient were not obvious except for the uplifted ear lobes at the age of 3 months. At the age of 22 months, the facial characteristics of the patient included ocular hypertelorism, frontal bossing, rounded nasal tip, sparse eyebrows, prominent chin, widely spaced teeth, and uplifted ear lobes with a central depression.
Conclusion: A novel deletion mutation of the ZEB2 gene was identified. This work contributes to expanding the mutation spectra of MWS. Our results may reflect the variability of the phenotype in MWS.
Keywords: Mowat–Wilson syndrome, ZEB2, gene mutation, phenotype
Introduction
Mowat–Wilson syndrome (MWS) is a rare complex malformation syndrome which is characterized by typical facial dysmorphism, moderate to severe intellectual disability, global developmental delay, and multiple congenital anomalies.1,2 Facial features include high forehead with frontal bossing, hypertelorism, strabismus, deep-set eyes, open mouth with M-shaped upper lip, prominent nasal tip, prominent chin, and uplifted earlobes. Other congenital anomalies are variable, but may include short stature, microcephaly, Hirschsprung disease (HSCR), corpus callosum agenesis, malformations of the brain, genitourinary anomalies (most commonly hypospadias), and congenital heart defects.1,2 The syndrome is caused by de novo mutations in one allele of zinc-finger E-box-binding homeobox 2 gene (ZEB2), which is also called SIP1 (for Smad-interacting protein 1) and ZHFX1B.3,4 The ZEB2 gene is located on the long arm of chromosome 2q22-23 and consists of 10 exons (exon 1 is a noncoding exon), which encodes the zinc-finger E-box binding homeobox 2 protein consisting of 1214 amino acids.5 ZEB2 is a member of the two-handed zinc-finger/homeodomain transcription factor family and functions as a transcriptional repressor and interacts with Smad1 proteins in the transforming growth factor-β signaling pathway.5,6 ZEB2 is evolutionarily highly conserved and plays important roles in development of the neural crest during fetal development.7,8 It is widely expressed in neural crest-derived cells during embryological development of central and enteric nervous systems, craniofacial mesoderm, heart, and some other midline structures, such as corpus callosum and genitalia.7
MWS was first described by Mowat et al.9 The incidence of the syndrome is estimated in the range of 1 per 50,000~70,000 live births in Europe and USA.4 To date, more than 200 different mutations in the ZEB2 gene have been reported in various populations (Human Gene Mutation Database, http://www.hgmd.cf.ac.uk). In this study, we summarized the clinical presentation, genetic mutations and follow-up of a Chinese boy with MWS. Furthermore, a novel de novo mutation of the ZEB2 gene was identified in this patient.
Patients and Methods
The patient was from Jinan Maternity and Child Care Hospital Affiliated to Shandong First Medical University. All tests were performed as routine clinical investigations in accordance with the ethical principles of the Declaration of Helsinki. Written informed consent was obtained from the guardians of the patient. This study was approved by the ethical committee of Jinan Maternity and Child Care Hospital Affiliated to Shandong First Medical University.
Genomic DNA was extracted from peripheral blood leukocytes of using a commercial kit (Qiagen). Then, the whole-exome sequencing was performed. The genetic analysis was approved by the ethical committee of Jinan Maternity and Child Care Hospital Affiliated to Shandong First Medical University.
Results
The patient was the second child of healthy nonconsanguineous couple and his older brother is healthy. He was born by cesarean section at 37 weeks of gestation. His birth weight was 2.65 kg and birth length was 48 cm. Head circumference at birth was not measured. There was no history of birth asphyxia. Although he failed his newborn hearing screening, his initial newborn period was otherwise uncomplicated, and he was discharged home at 5 days of life. At the age of 3 months, he was referred to our hospital because he could not raise his head and cried easily. On physical examination, his weight was 5.8 kg, length was 59.5 cm, and head circumference was 37.2 cm. He has marked microcephaly (<1%). No specific facial characteristics were found except for uplifted ear lobes with a central depression. Furthermore, he had a cardiac murmur and echocardiography revealed interventricular septal defects and patent ductus arteriosus. Cryptorchidism was detected. Brain magnetic resonance imaging (MRI) showed corpus callosum agenesis (Figure 1). Because he failed his newborn hearing screening, he was retested using transient evoked otoacoustic emission and automated auditory brainstem response (ABR). However, he still failed to pass the tests. Then, the patient was further tested using clinical auditory brainstem response and tympanometry at the age of six months. As a result, the minimum stimulation intensity that can evoke wave V is generally considered as the clinical ABR threshold. In click evoked ABR test, the latencies of waves I, III, and V were delayed. The threshold was 50 dB nHL in both ears. In tone burst ABR test, the thresholds were as follows: 60 dB nHL at 500 Hz, 70 dB nHL at 1000 Hz, 60 dB nHL at 2000 Hz, and 50 dB nHL at 4000 Hz (left ear); 60 dB nHL at 500 Hz, 70 dB nHL at 1000 Hz, 70 dB nHL at 2000 Hz, and 60 dB nHL at 4000 Hz (right ear). The tympanogram result was type A. Thus, he demonstrated bilateral sensorineural hearing loss.
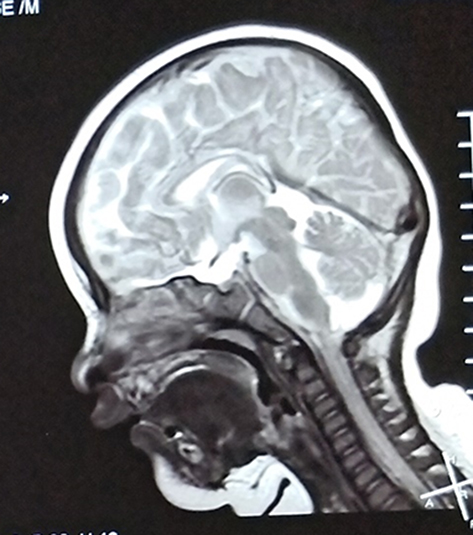 |
Figure 1 Brain MRI showed agenesis of the corpus callosum. |
Clinical follow-up showed that facial features developed with time. At the age of 22 months, the patient had facial dysmorphism including ocular hypertelorism, frontal bossing, rounded nasal tip, sparse eyebrows, prominent chin, widely spaced teeth, and uplifted ear lobes with a central depression (Figure 2, Supplementary Figure 1). Furthermore, he presented with failure to thrive (length 79.8 cm, <3%; weight 9.7 kg, <3%), microcephaly (head circumference 43 cm, <1%), developmental delay and intellectual disability. All developmental milestones were delayed. He raised head at 6 months, sat alone at 12 months, stood without assistance at 18 months, walked on a wide base with support at 22 months, and walked alone at 3 years. Surprisingly, he never presented with epileptic seizures during follow-up. The bilateral sensorineural hearing loss did not improve with growth.
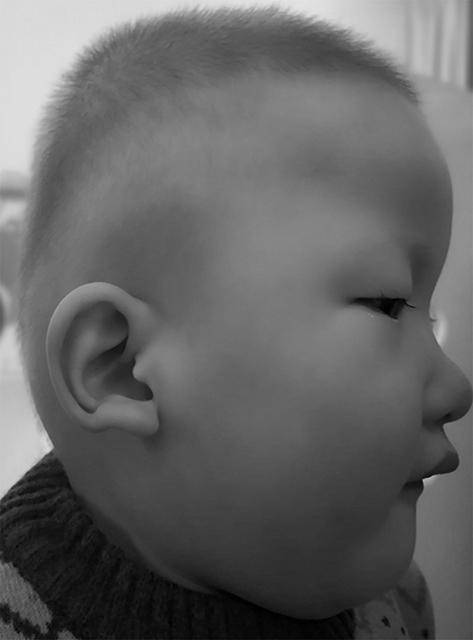 |
Figure 2 Facial appearance of the patient with MWS at the age of 22 months showing ocular hypertelorism, frontal bossing, rounded nasal tip, sparse eyebrows, and uplifted ear lobes with a central depression. We obtained the permission to use the photographs from guardians of the patient. |
A deletion mutation (c.1137_1146del TAGTATGTCT) was identified in exon 8 of the ZEB2 gene. The mutation was predicted to produce a truncated protein (p.S380Nfs*13), resulting in haploinsufficiency. The sequencing analyses of parents did not reveal any difference with respect to normal controls. Thus, the mutation of the patient was de novo.
Discussion
Here we describe the clinical features, gene mutation, and follow-up of a patient with MWS. Mowat–Wilson syndrome is a rare well-described congenital malformation syndrome. The diagnosis of MWS may be clinically difficult because of the variability of the phenotype and the widespread nature of the phenotypic features.10,11 Several groups have suggested that MWS is likely underdiagnosed.2,12,13 Even though the patients have distinctive facial appearance and intellectual disability, other clinical signs appear in various spectra for each patient. Initially, MWS was described in patients who had HSCR and severe intellectual disability.9 At present, it is clear that the phenotype is highly variable. While microcephaly, seizures, and congenital anomalies are common, none is obligatory, including HSCR. Overall, only half of all patients have HSCR.7,14
With more than 300 patients reported to date, MWS has become a well-known entity in clinical dysmorphology.15 The most frequent features include typical facial dysmorphism, intelligent disability, seizures, HSCR, constipation, and congenital heart defects.15 Most clinical features of our patient, including global developmental delay, intellectual disability, facial features, microcephaly, short stature, congenital heart defects, cryptorchidism and corpus callosum agenesis, were consistent with those described in previous literatures.2,7,15 One of the most specific clinical signs of MWS is a distinctive facial appearance. In our study, facial features of the patient were not obvious, except for the uplifted ear lobes at 3 months. It is worth noting that facial characteristics of children with MWS become more prominent as they grow up. At 22 months, the patient had ocular hypertelorism, frontal bossing, rounded nasal tip, sparse eyebrows, prominent chin, widely spaced teeth, and uplifted ear lobes with a central depression. The uplifted earlobes did not change significantly over time. Achievement of motor milestones was also delayed. He was able to walk on a wide base with support at 22 months and walk alone at 3 years. Nevertheless, several major clinical manifestations were not present such as HSCR, constipation, seizures and eye anomalies. On the other hand, this patient demonstrated bilateral sensorineural hearing loss. This manifestation is less common and has been described only rarely.14–16 It was first reported in Chinese patients with MWS. The manifestation of bilateral sensorineural hearing loss may reflect the variability of the phenotype in MWS. Seizures are very common manifestations and founded in approximately 70–75% of patients.13 We need to maintain follow-up of this patient to observe whether seizures develop in the future. The most consistent finding from brain MRI in patients with MWS in the literature is hypoplasia or corpus callosum agenesis.14,17 The utility of brain MRI in patients with suspected MWS is primarily to aid in diagnosis, as the finding of corpus callosum agenesis is a significant positive predictor.17 In our patient, brain MRI also showed corpus callosum agenesis, which was consistent with previous reports.17
Since MWS is often misdiagnosed during early infancy, it is very important to perform genetic testing. The haploinsufficiency of the ZEB2 gene is responsible for all typical MWS patients. Therefore, molecular confirmation of ZEB2 gene variation is required for diagnosis. In this study, a deletion mutation (c.1137_1146del TAGTATGTCT) was identified in the ZEB2 gene. In order to confirm whether the mutation was novel, the sequencing data were compared with those from the Human Gene Mutation Database (www.hgmd.cf.ac.uk) and gnomAD database (http://gnomad.broadinstitute.org). This deletion mutation was not found in the Human Gene Mutation Database and gnomAD database, which suggested that it was a novel pathogenic variant. To study the mutation spectra of disease-causing genes associated with MWS in the Chinese population, a large-scale investigation is necessary.
Conclusion
A novel mutation (c.1137_1146del TAGTATGTCT) in the ZEB2 gene was identified. The novel mutation results in the clinical manifestation of MWS. This work contributes to expanding the knowledge of the genetic basis of MWS. Furthermore, our results may reflect the variability of the phenotype in MWS.
Abbreviations
MWS, Mowat–Wilson syndrome; HSCR, Hirschsprung disease; ZEB2, zinc finger E-box-binding homeobox 2 gene; MRI, magnetic resonance imaging; ABR, auditory brainstem response.
Compliance with Ethical Standards
All tests were performed as routine clinical investigations in accordance with the ethical principles of the Declaration of Helsinki. Written informed consent has been provided by the guardians of the patient to have the case details and accompanying images published. This study was approved by the ethical committee of Jinan Maternity and Child Care Hospital Affiliated to Shandong First Medical University.
Acknowledgments
We are grateful to the patient and his family for making this study possible.
Disclosure
The authors declare no conflicts of interest for this work.
References
1. Zweier C, Thiel CT, Dufke A, et al. Clinical and mutational spectrum of Mowat-Wilson syndrome. Eur J Med Genet. 2005;48(2):97–111. doi:10.1016/j.ejmg.2005.01.003
2. Wenger TL, Harr M, Ricciardi S, et al. CHARGE-like presentation, craniosynostosis and mild Mowat-Wilson syndrome diagnosed by recognition of the distinctive facial gestalt in a cohort of 28 new cases. Am J Med Genet A. 2014;164A(10):2557–2566. doi:10.1002/ajmg.a.36696
3. Cacheux V, Dastot-le Moal F, Kääriäinen H, et al. Loss-of-function mutations in SIP1 Smad interacting protein 1 result in a syndromic hirschsprung disease. Hum Mol Genet. 2001;10(14):1503–1510. doi:10.1093/hmg/10.14.1503
4. Evans E, Einfeld S, Mowat D, Taffe J, Tonge B, Wilson M. The behavioral phenotype of Mowat-Wilson syndrome. Am J Med Genet A. 2012;158A(2):358–366. doi:10.1002/ajmg.a.34405
5. Saunders CJ, Zhao W, Ardinger HH. Comprehensive ZEB2 gene analysis for Mowat-Wilson syndrome in a North American cohort: a suggested approach to molecular diagnostics. Am J Med Genet A. 2009;149A(11):2527–2531. doi:10.1002/ajmg.a.33067
6. Dastot-le Moal F, Wilson M, Mowat D, Collot N, Niel F, Goossens M. ZFHX1B mutations in patients with Mowat-Wilson syndrome. Hum Mutat. 2007;28(4):313–321. doi:10.1002/humu.20452
7. Kilic E, Cetinkaya A, Utine GE, Boduroğlu K. A diagnosis to consider in intellectual disability: Mowat-Wilson syndrome. J Child Neurol. 2016;31(7):913–917. doi:10.1177/0883073815627884
8. Van de Putte T, Maruhashi M, Francis A, et al. Mice lacking ZFHX1B, the gene that codes for Smad-interacting protein-1, reveal a role for multiple neural crest cell defects in the etiology of hirschsprung disease-mental retardation syndrome. Am J Hum Genet. 2003;72(2):465–470. doi:10.1086/346092
9. Mowat DR, Croaker GD, Cass DT, et al. Hirschsprung disease, microcephaly, mental retardation, and characteristic facial features: delineation of a new syndrome and identification of a locus at chromosome 2q22-q23. J Med Genet. 1998;35(8):617–623. doi:10.1136/jmg.35.8.617
10. Mowat DR, Wilson MJ, Goossens M. Mowat-Wilson syndrome. J Med Genet. 2003;40(5):305–310. doi:10.1136/jmg.40.5.305
11. Moore SW, Fieggen K, Honey E, Zaahl M. Novel Zeb2 gene variation in the Mowat Wilson syndrome (MWS). J Pediatr Surg. 2016;51(2):268–271. doi:10.1016/j.jpedsurg.2015.10.070
12. Adam MP, Schelley S, Gallagher R, et al. Clinical features and management issues in Mowat-Wilson syndrome. Am J Med Genet A. 2006;140(24):2730–2741. doi:10.1002/ajmg.a.31530
13. Paz JA, Kim CA, Goossens M, Giurgea I, Marques-Dias MJ. Mowat-Wilson syndrome: neurological and molecular study in seven patients. Arq Neuropsiquiatr. 2015;73(1):12–17. doi:10.1590/0004-282X20140182
14. Garavelli L, Zollino M, Mainardi PC, et al. Mowat-Wilson syndrome: facial phenotype changing with age: study of 19 Italian patients and review of the literature. Am J Med Genet A. 2009;149A(3):417–426. doi:10.1002/ajmg.a.32693
15. Ivanovski I, Djuric O, Caraffi SG, et al. Phenotype and genotype of 87 patients with Mowat-Wilson syndrome and recommendations for care. Genet Med. 2018;20(9):965–975. doi:10.1038/gim.2017.221
16. Abdalla EM, Zayed LH. Mowat-Wilson syndrome: deafness in the first Egyptian case who was conceived by intracytoplasmic sperm injection. J Child Neurol. 2014;29(12):NP168–NP170. doi:10.1177/0883073813509120
17. Garavelli L, Ivanovski I, Caraffi SG, et al. Neuroimaging findings in Mowat-Wilson syndrome: a study of 54 patients. Genet Med. 2017;19(6):691–700. doi:10.1038/gim.2016.176
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.