")
Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 7
A challenging diagnosis: case report of extensive pyoderma gangrenosum at multiple sites
Authors Ye M, Ye J, Wu L, Keating C, Choi WT
Received 7 January 2014
Accepted for publication 24 January 2014
Published 26 March 2014 Volume 2014:7 Pages 105—109
DOI https://doi.org/10.2147/CCID.S60229
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Mingwei J Ye,1,2 Joshua Mingsheng Ye,2 Leonard Wu,3 Cameron P Keating,4 Wai-Ting Choi4
1Department of Dermatology, Western Hospital, Footscray, VIC, Australia; 2University of Melbourne, Department of Medicine, Dentistry and Health Sciences, Parkville, VIC, Australia; 3Department of Pathology, Western Hospital, Footscray, VIC, Australia; 4Department of Plastic Surgery, Western Hospital, Footscray, VIC, Australia
Background: Pyoderma gangrenosum (PG) is a rare dermatological condition characterized by the rapid progression of a painful, necrolytic ulcer with an irregular, undermined border and commonly affects the lower extremities, mainly in the pretibial area. The diagnosis of PG is not easy. Due to lack of diagnostic laboratory test and histopathological findings indicative of PG, it is often misdiagnosed as an infection. This results in delayed or inappropriate treatment of the condition, which leads to devastating consequences such as limb amputation and death.
Main observations: We report a rare case of a 51-year-old female who was initially diagnosed as having infected ulcers and underwent serial debridements, which resulted in extensive PG at three different sites (abdominal, left thigh, and sacral).
Conclusion: This case highlights the challenges in diagnosing PG, emphasizes the key clinical features to aid diagnosis, and the clinical consequences of delayed or misdiagnosis of this condition.
Keywords: postoperative complication, skin ulcer, surgical wound
Introduction
Pyoderma gangrenosum (PG) is a rare dermatological condition characterized by the rapid progression of a painful, necrolytic ulcer with an irregular, undermined border and commonly affects the lower extremities, mainly in the pretibial area.1 In 70% of cases, PG is associated with an underlying disease such as inflammatory bowel disease, inflammatory arthritis, and hematological malignancies.2–4 The etiology of PG is unclear but is thought to be a reactive inflammatory dermatosis and part of the spectrum of neutrophilic dermatosis. More recently, new disease entities such as PASH syndrome (PG, acne, and suppurative hidradenitis) and PAPA syndrome (pyogenic arthritis, PG, and acne) are described in the literature as part of the spectrum of autoinflammatory disorders distinct from allergic, autoimmune, and infectious processes.5 PAPA syndrome is associated with genetic mutations involved in innate immune response regulation,6 while no genetic links have been detected in patients with PASH syndrome.5
PG is a diagnosis of exclusion and can only be made after common causes of ulcers such as infection and malignancy have been ruled out. However, the management of suspected infected or malignant ulcers may require debridement or excision, which can exacerbate PG through a process called pathergy (a term used to describe an exaggerated skin injury occurring after trauma).1 We report a case of a patient who developed extensive PG at multiple sites as a result of serial debridements for suspected infected ulcers, highlighting the challenges in diagnosing PG and the clinical consequences of misdiagnosis or delayed diagnosis.
Case report
A 51-year-old female initially presented with infected abdominal, thigh, and sacral ulcers. Her ulcers developed as a result of poor self-care and hygiene. Her past medical history included type 2 diabetes, hypertension, hypercholesterolemia, and back pain. Physical examination revealed an abdominal ulcer measuring 4 cm × 2 cm in the right iliac fossa with surrounding erythema, a small ulcer on her left thigh (1 cm × 1 cm), and two sacral ulcers (7 cm × 3 cm and 4.5 cm × 2 cm, respectively). Ulcer cultures revealed skin flora. Although she did not have any systemic features of infection, blood investigations on admission showed an elevated white cell count of 14.9×109/L (reference interval, 4.0×09–11.0×109/L) and C-reactive protein of 120 mg/L (reference interval, 0–10 mg/L). She was subsequently commenced on 2 g of empirical intravenous flucloxacillin four times daily.
In the next few weeks, all her wounds worsened despite wound dressings and antibiotics. Her abdominal and thigh ulcers had increased in size. The size of her two sacral ulcers remained the same but had increased wound necrosis. One month after admission, her abdominal ulcer measured 21 cm × 7 cm, and the thigh ulcer was 4 cm × 1 cm. In view of this, surgical excisions of the necrotic areas were performed. Vacuum-assisted closure dressings were applied to the anterior abdomen and sacrum.
Three days later, the patient underwent a routine vacuum-assisted closure dressing change. It was noted that her wounds had worsened rapidly since the last debridement. The once healthy wound edges had become necrotic. Further debridement was performed. Excised tissues sent off for microscopy and culture grew methicillin-sensitive Staphylococcus aureus and Enterobacter aerogenes. Intravenous tazocin (4.5g 8 hourly) was added for suspected postoperative wound infection.
The patient’s wounds continued to deteriorate with antibiotics and further debridements. After the third debridement, the abdominal ulcer had increased to a size of 32 cm × 11 cm, and the two sacral ulcers became a huge sacral defect extending into the perianal region (Figure 1A and B). Cultures of subsequently debrided tissue were negative for bacteria and fungi. Repeated histological sections of sacral and abdominal tissues revealed neutrophilic infiltrates extending into deep dermis and no evidence of malignancy (Figure 2A and B). Antinuclear antibodies, extractable nuclear antigens, rheumatoid factor, and anti-neutrophil cytoplasmic antibodies were all negative. A diagnosis of PG was made, and our patient was commenced on 50 mg of prednisolone daily. After 1 week, her wounds showed slow improvement, with resolution of necrotic wound edges and formation of granulation tissue. Two weeks later, she developed steroid-induced hyperglycemia, and 500 mg twice daily of mycophenolate mofetil was added as a steroid sparing agent. Her prednisolone dose was gradually tapered by 5 mg every 2 weeks, and the dose of mycophenolate mofetil increased to 1 g twice daily. Four months after admission, the patient underwent successful split skin grafts and primary closure to her abdominal and sacral ulcers while on immunosuppressive therapy (Figure 3A and B). At 3-month follow-up, there was no recurrence of the disease.
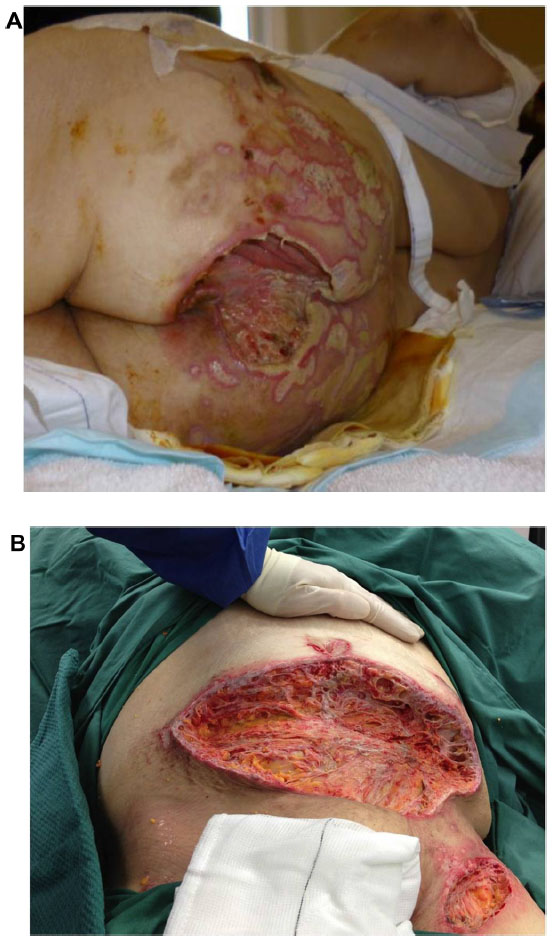 | Figure 1 (A) Sacral pyoderma gangrenosum after multiple debridements. (B) Huge abdominal and left thigh wound defect with typical violaceous borders post-debridement. |
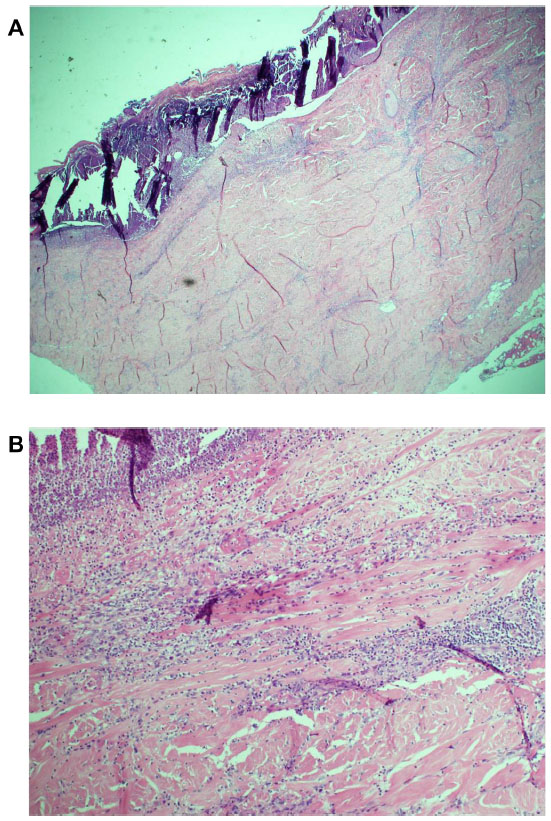 | Figure 2 Histology of incisional biopsy of abdominal ulcer. (A) Extensive skin ulceration is shown (H&E stain; original magnification ×10). (B) Higher-powered view of ulcer being covered by inflamed crust and infiltrated by a heavy collection of acute inflammatory cells (neutrophils) (H&E stain; ×20). |
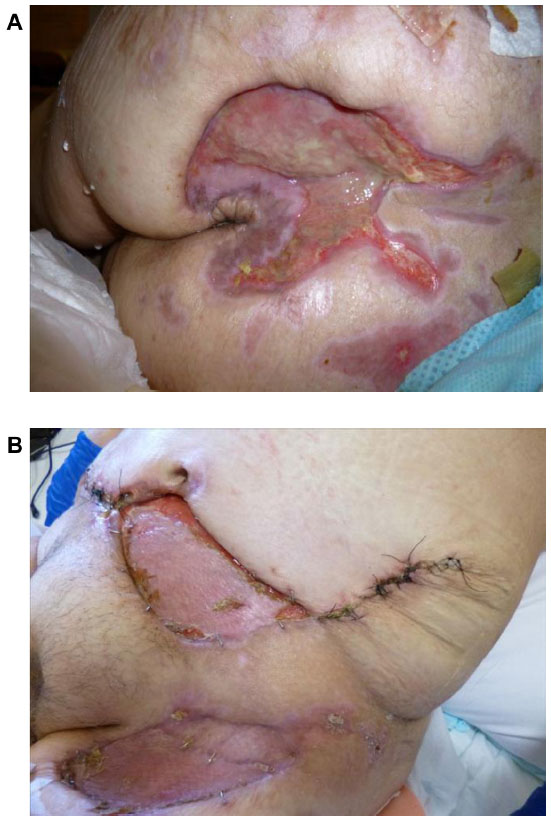 | Figure 3 After treatment with immunosuppressive medications, appearance of sacral (A) and abdominal and left thigh (B) pyoderma gangrenosum. |
Discussion
PG was first described more than a century ago by Louis Brocq, a French dermatologist.7 He reported a series of patients with typical features of PG and described the appearance of their ulcers. Today, PG still remains a clinical diagnosis. There are no diagnostic laboratory investigations. Histology findings are not specific but can serve to exclude infection, vasculitis, and malignancy.1
To aid the recognition of PG, Su et al1 proposed a set of diagnostic criteria for ulcerative PG. Diagnosis requires both major criteria and at least two minor criteria. The major criteria include the characteristic appearance of a painful, irregular ulcer with a violaceous border, and exclusion of other causes of ulceration such as malignancy, vasculitis, primary infection, and drugs. The minor criteria include history of pathergy, associated systemic illness, histology findings of dermal neutrophilia, and response to steroid treatment. Other diagnostic criteria for variants of PG, namely bullous, pustular, and vegetative PG, have also been described.4,8,9
A majority of the PG cases were often misdiagnosed as infection resulting in repeated surgical debridement.10–12 Unfortunately, surgical debridement worsens PG through a phenomenon called pathergy. Diagnosis of PG is often made after failure of initial treatment and absence of Gram stain on culture. Furthermore, the presence of secondary infections can obscure the diagnosis of this condition.
Diagnosis of PG in our patient was extremely difficult, made more complicated by the fact that her initial tissue cultures grew Staphylococcus and Enterobacter. As a result, she was placed on antibiotics and underwent repeated surgical debridement for a presumed diagnosis of a wound infection. Diagnosis of PG was considered only after repeated biopsies revealed dermal neutrophilia, failure of treatment with broad-spectrum antibiotics, and most importantly, the wound exhibited pathergy following debridement. However, multiple debridements of her ulcers and the delay in instituting appropriate treatment led to extensive ulcerations and prolonged recovery for the patient.
Currently, there are no standard guidelines for the treatment of PG. Marzano et al13 recently published a therapeutic algorithm based on the clinical extent of the lesions. However, due to its small sample size, larger controlled trials are needed to confirm the effectiveness of their algorithm. In general, the treatment of PG involves both topical and systemic approaches. Topical therapy involves the use of dressings to control exudation, improve auto-debridement, protect the surrounding skin, and provide analgesia.14 Deep wounds may benefit from vacuum sealing as it improves granulation.15 The topical use of antimicrobials is not recommended as they may irritate the ulcers and surrounding skin. Topical tacrolimus has been shown to be effective in the treatment of less severe PG.16
Systemic therapy is the mainstay of treatment for rapidly progressive PG. Corticosteroids such as prednisolone are initially used to prevent progression and arrest the inflammatory process. Combinations of steroid and cytotoxic drugs such as azathioprine, cyclophosphamide, cyclosporin, or mycophenolate mofetil are used in patients with steroid-resistant disease or as a steroid-sparing measure.14 In the presence of secondary infections, corticosteroids should be combined with antibiotics.4 Recently, tumor necrosis factor (TNF)-α blockers and other biologics have been used with some success.17,18 Infliximab, a monoclonal antibody against TNF-α, has been particularly effective in treating PG associated with inflammatory bowel disease.13 Surgical debridement is contraindicated due to pathergy.19 Surgical treatments such as skin grafts may be worthwhile but can only be performed with concomitant immunosuppression and in patients with stable disease or partial remission.15
Despite the advances in medical therapy, the outlook of PG is unpredictable. Delayed diagnosis or misdiagnosis of PG will certainly lead to exacerbation and progression of the disease. In our case, misdiagnosis of the patient’s condition resulted in multiple debridements of her ulcers and a delay in instituting appropriate treatment. The outcome was extensive ulcerations and prolonged hospital recovery. Our patient still requires long-term follow-up to monitor for recurrence of PG and further evaluation to exclude genetic mutations associated with the spectrum of autoinflammatory syndromes.
Conclusion
PG is a rare occurrence and often proves to be a diagnostic challenge. The presence of a rapidly progressing, necrotizing ulcer and a history suggestive of pathergy are key features of PG. It is also important to reconsider a diagnosis when the disease process worsens or does not respond to treatment. A dermatology opinion should be obtained if there is any uncertainty regarding the diagnosis.
Disclosure
The authors report no conflicts of interest in this work.
References
Su WPD, Davis MDP, Weenig RH, et al. Pyoderma gangrenosum: clinicopathologic correlation and proposed diagnostic criteria. Int J Dermatol. 2004;43(11):790–800. | |
Greenstein AJ, Janowitz HD, Sachar DB. The extraintestinal complications of Crohn’s disease and ulcerative colitis: a study of 700 patients. Medicine (Baltimore). 1976;55:401–412. | |
Stolman LP, Rosenthal D, Yaworsky R, et al. Pyoderma gangrenosum and rheumatoid arthritis. Arch Dermatol. 1975;111:1020–1023. | |
Perry HO, Winkelmann RK. Bullous pyoderma gangrenosum and leukemia. Arch Dermatol. 1972;106:901–905. | |
Braun-Falco M, Kovnerystyy O, Lohse P, Ruzicka T. Pyoderma gangrenosum, acne, and suppuratives hidradenitis (PASH) – a new autoinflammatory syndrome distint from PAPA syndrome. J Am Acad Dermatol. 2012;66:408–415. | |
Wise CA, Gillum JD, Seidman CE, et al. Mutations in CD2BP1 disrupt binding to PTP PEST and are responsible for PAPA syndrome, an autoinflammatory disorder. Hum Mol Genet. 2002;11:961–969. | |
Brocq L. A new contribution to the study of geometric phagedenism. Ann Dermatol Syphiligr. 1916;9:1–39. | |
O’Loughlin S, Perry HO. A diffuse pustular eruption associated with ulcerative colitis. Arch Dermatol. 1978;114:1061–1064. | |
Wilson-Jones E, Winkelmann RK. Superficial granulomatous pyoderma: a localized vegetative form of pyoderma gangrenosum. J Am Acad Dermatol. 1988;18:511–521. | |
Bennett CR, Brage ME, Mass DP. Pyoderma gangrenosum mimicking postoperative infection in the extremities. A report of two cases. J Bone Joint Surg Am. 1999;81:1013–1018. | |
Greidanus TG, Honl BA. More than a hand infection. Pyoderma gangrenosum. Postgrad Med. 2002;112:107–109. | |
Barańska-Rybak W, Kakol M, Naesstrom M, Komorowska O, Sokołowska-Wojdyło M, Roszkiewicz J. A retrospective study of 12 cases of pyoderma gangrenosum: why we should avoid surgical intervention and what therapy to apply. Am Surg. 2011;77(12):1644–1649. | |
Marzano AV, Trevisan V, Lazzari R, Crosti C. Pyoderma gangrenosum: study of 21 patients and proposal of a ‘clinicotherapeutic classification’. J Dermatolog Treat. 2011;22(5):254–260. | |
Wollina U. Clinical management of pyoderma gangrenosum. Am J Clin Dermatol. 2002;3(3):149–158. | |
Fulbright RK, Wolf JE, Tschen JA. Pyoderma gangrenosum at surgery sites. J Dermatol Surg Oncol. 1985;11:883–886. | |
Marzano AV, Trevisan V, Lazzari R, Crosti C. Topical tacrolimus for the treatment of localized, idiopathic, newly diagnosed pyoderma gangrenosum. J Dermatolog Treat. 2010;21(3):140–143. | |
Regueiro M, Valentine J, Plevy S, Fleisher MR, Lichtenstein GR. Infliximab for treatment of pyoderma gangrenosum associated with inflammatory bowel disease. Am J Gastroenterol. 2003;98(8):1821–1826. | |
Hubbard VG, Friedmann AC, Goldsmith P. Systemic pyoderma gangrenosum responding to infliximab and adalimumab. Br J Dermatol. 2005;152:1059–1061. | |
Bennett ML, Jackson JM, Jorizzo JL, et al. Pyoderma gangrenosum. A comparison of typical and atypical forms with an emphasis on time to remission. Case review of 86 patients from 2 institutions. Medicine (Baltimore). 2000;79:37–46. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.