")
Back to Journals » OncoTargets and Therapy » Volume 8
A case of EGFR mutant lung adenocarcinoma that acquired resistance to EGFR-tyrosine kinase inhibitors with MET amplification and epithelial-to-mesenchymal transition
Authors Miyoshi S , Kato T, Katayama H, Ito R, Mizuno Y, Okura T, Higaki J
Received 8 December 2014
Accepted for publication 19 March 2015
Published 9 April 2015 Volume 2015:8 Pages 783—787
DOI https://doi.org/10.2147/OTT.S78911
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Daniele Santini
Seigo Miyoshi,1 Takahide Kato,1 Hitoshi Katayama,1 Ryoji Ito,1 Yosuke Mizuno,2 Takafumi Okura,1 Jitsuo Higaki1
1Department of Cardiology, Pulmonology, Hypertension and Nephrology, Ehime University Graduate School of Medicine, Ehime, Japan; 2Department of Molecular Pathology, Ehime University Graduate School of Medicine, Ehime, Japan
Abstract: EGFR mutant lung cancer responds to EGFR tyrosine kinase inhibitors (TKIs), but all patients eventually develop resistance to EGFR-TKIs. Herein we report a case of EGFR mutant lung adenocarcinoma that acquired resistance to EGFR-TKI with MET amplification and epithelial-to-mesenchymal transition (EMT). A 73-year-old woman was diagnosed with adenocarcinoma harboring an EGFR exon 19 deletion. She received gefitinib as second-line therapy. Tumors were reduced 1 month after gefitinib therapy. However, only a few months later, chest computed tomography results indicated cancer progression. Gefitinib therapy was stopped and docetaxel therapy started. However, she died 13 days after admission. Microscopic examination of postmortem specimens revealed a diffuse proliferation of atypical giant cells in primary and metastatic lesions, but no adenocarcinomatous components as in the antemortem specimens. Immunohistochemical analyses showed that antemortem tumor specimens were positive for CDH1 but negative for VIM. In contrast, postmortem tumor specimens were positive for VIM but negative for CDH1. Genetic analyses revealed MET amplification. We concluded that resistance to EGFR-TKI might be caused by MET amplification and EMT. To our knowledge, no clinical studies have reported that MET amplification and EMT together may be associated with acquired resistance to EGFR-TKI. Second biopsy after the development of EGFR-TKI resistance may be recommended to determine the best therapeutic strategy.
Keywords: epidermal growth factor receptor tyrosine kinase inhibitor, MET amplification, epithelial-to-mesenchymal transition
Background
Patients with EGFR mutant lung cancer derive significant therapeutic benefit from treatment with EGFR tyrosine kinase inhibitors (TKIs). However, acquired resistance is an inevitable consequence of this treatment strategy, with a broad variety of resistance mechanisms.1,2 Herein we report a case of potential acquired resistance to EGFR-TKI in primary lung adenocarcinoma with both MET amplification and epithelial-to-mesenchymal transition (EMT).
Case report
A 73-year-old woman was admitted to our hospital due to progressive dyspnea. She had been diagnosed with T2bN1M1a adenocarcinoma (stage IV with visceral pleural nodules) harboring an EGFR exon 19 deletion by computed tomography (CT)-guided lung tumor biopsy (biopsy was performed twice) and by visceral pleural nodule biopsy using video-assisted thoracoscopy (biopsy was performed once) (Figure 1). She had a performance status of 1 and was a never smoker. As first-line chemotherapy, she received carboplatin and pemetrexed, because there have been no reports that using EGFR-TKI, compared with cytotoxic agents, as first-line chemotherapy significantly prolongs the overall survival of patients with EGFR mutant lung cancer. In addition, our patient had a good performance status that withstood cytotoxic chemotherapy at the time of disease diagnosis. After a five-course regimen, progressive disease was observed, and gefitinib was administered as second-line therapy. Chest CT showed that the tumor and right hilar lymphadenopathy were reduced 1 month after gefitinib therapy was initiated (the longest dimension of the tumor decreased by 62.1% after gefitinib therapy) (Figure 2A–D). However, only a few months after gefitinib therapy (on the present admission), CT showed atelectasis in the right middle and lower lobes (Figure 2E and F). Upon suspicion of recurrence, gefitinib therapy was stopped and docetaxel therapy started as third-line chemotherapy according to the Japanese Clinical Practice Guideline for Lung Cancer.3 However, she died 13 days after admission.
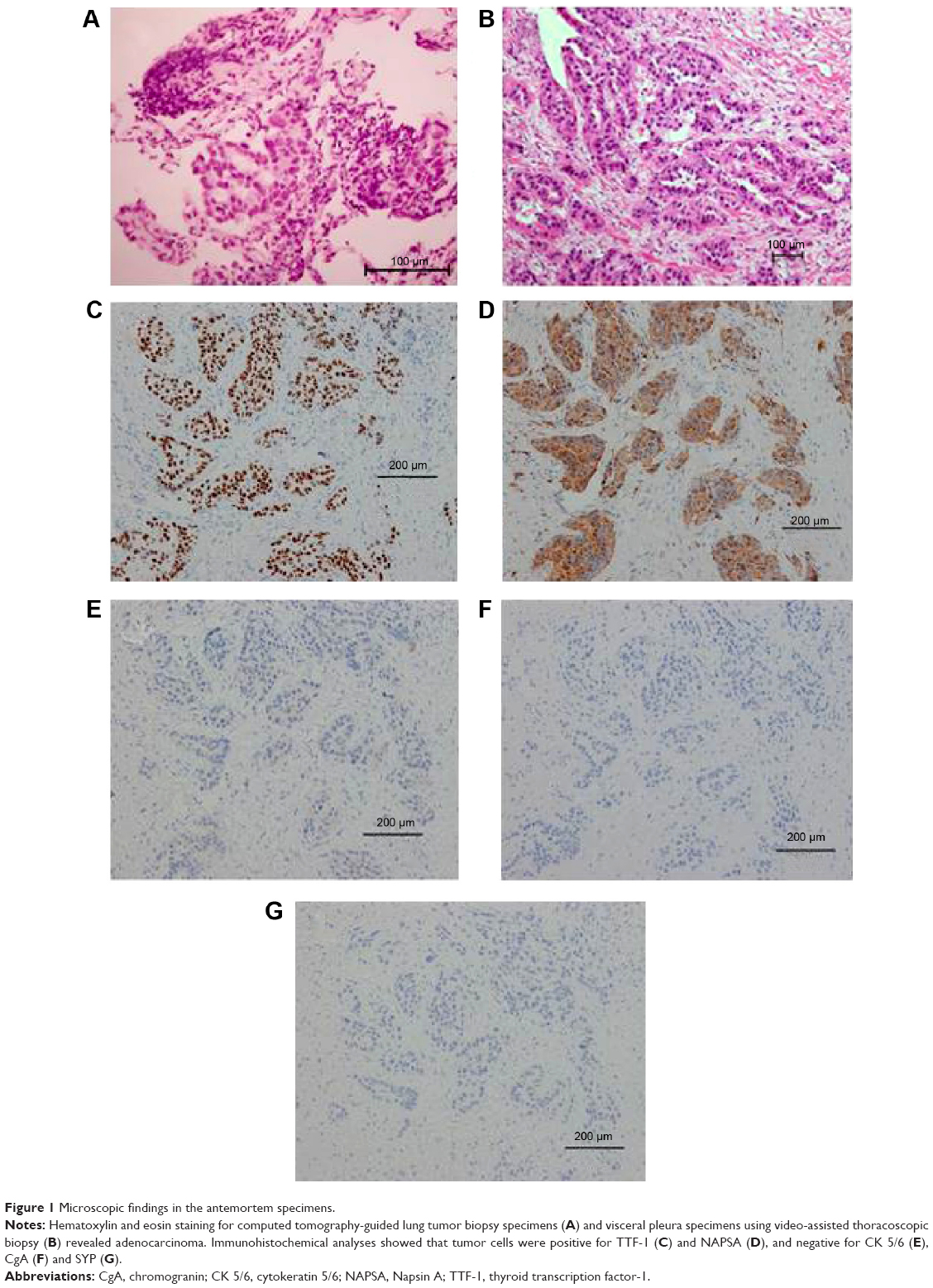 | Figure 1 Microscopic findings in the antemortem specimens. |
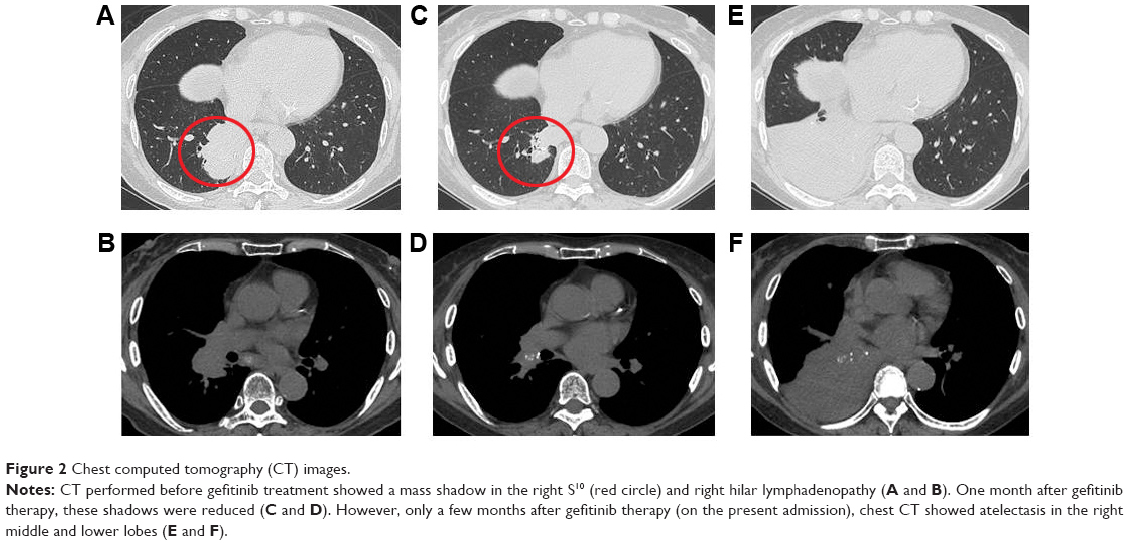 | Figure 2 Chest computed tomography (CT) images. |
We performed an autopsy on the patient with her son’s consent. All organs within the chest were resected. Postmortem macroscopic examination showed a tumor in the right lower lobe and right hilar and mediastinal lymphadenopathy. Tumor invasion was also observed in the esophagus and trachea. Surprisingly, microscopic examination revealed a diffuse proliferation of atypical giant cells in the primary and metastatic lesions, but no adenocarcinomatous components such as those seen in the antemortem specimens were observed (Figure 3). We assessed CDH1 and VIM expression using immunohistochemistry in both antemortem and postmortem specimens. The antemortem pretreatment specimens showed staining for CDH1, but not for VIM. In contrast, postmortem specimens stained for VIM, but not for CDH1. These results suggested that the resistant tumor had undergone an EMT, which caused a phenotypic transformation to giant cell carcinoma.
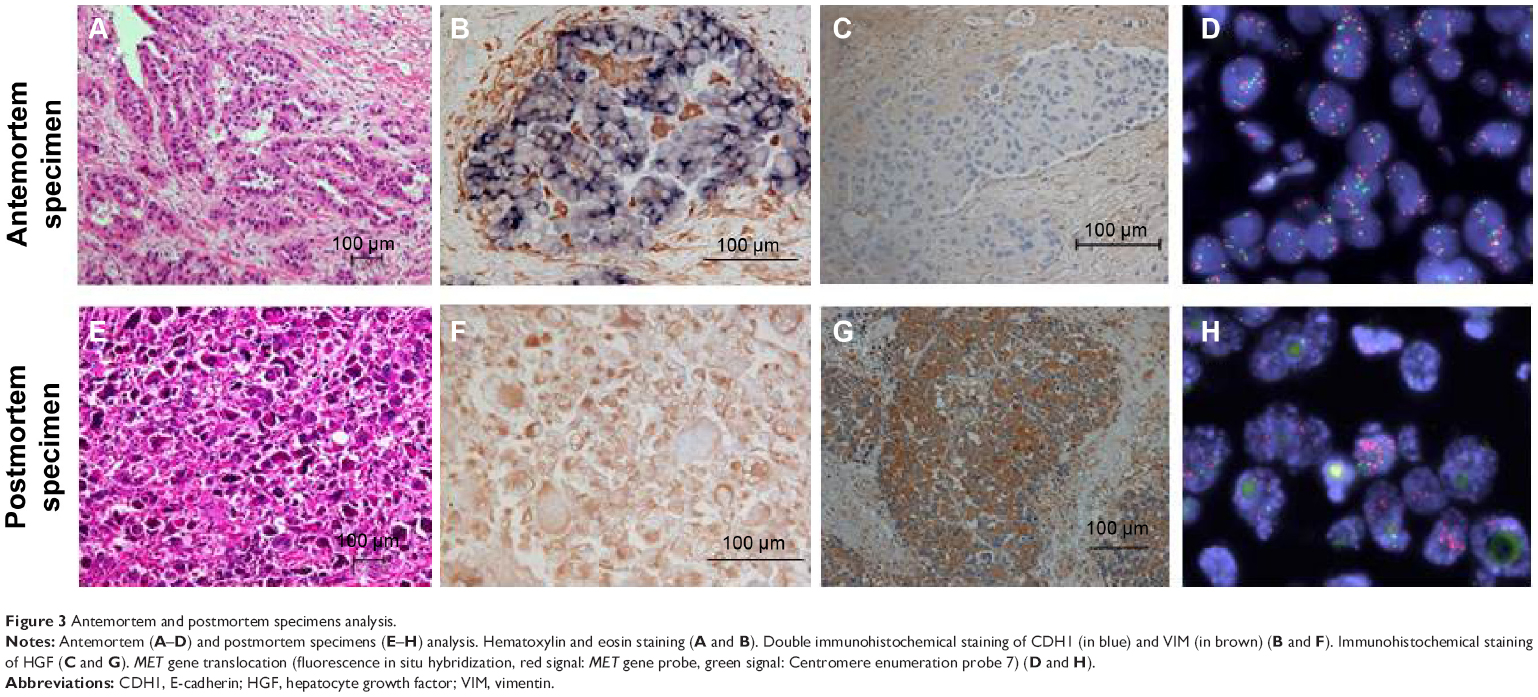 | Figure 3 Antemortem and postmortem specimens analysis. |
Using the antemortem and postmortem specimens, we also evaluated EGFR mutations, PIK3CA mutations, and MET gene amplification (Table 1 and Figure 3). Genetic analyses of the EGFR gene and PIK3CA gene were performed using direct sequencing. MET amplification was analyzed by fluorescent in situ hybridization. Signals were enumerated in at least 60 tumor nuclei per core. We considered MET-positive cases to have a mean ≥5 copies per cell according to a previous report.4 EGFR exon 19 deletion was observed in both specimens. MET amplification was also observed in both specimens, but MET gene copy numbers of postmortem tumor cells were higher than those of antemortem tumor cells. In addition, the expression of HGF, which is a ligand of MET, increased in postmortem, compared with antemortem, tumor cells. We concluded that resistance to EGFR-TKI might be caused by MET amplification and EMT.
 | Table 1 Analyses of genetic mutations or amplification in antemortem and postmortem specimens |
Discussion
The reported mechanisms underlying acquired EGFR-TKI resistance include EGFR T790M mutation, PIK3CA mutation, MET amplification, small cell lung cancer (SCLC) transformation, and EMT.1,2 In the present case, we considered that MET amplification and EMT together may cause resistance to EGFR-TKI. To our knowledge, no clinical studies have reported both MET amplification and EMT to be associated with acquired resistance to EGFR-TKI.
The association between MET amplification and EMT is unclear. However, one in vitro study showed that EGFR-TKI resistance in a non-SCLC cell line harboring MET amplification induced EMT following gefitinib removal.5 The present case also showed that EGFR mutant adenocarcinoma harboring MET amplification had undergone an EMT, suggesting these mechanisms may lead to rapid progression of tumors.
Second biopsy after the development of EGFR-TKI resistance may be recommended to determine the best therapeutic strategy. The characterization of the mechanisms underlying acquired resistance to EGFR-TKIs has led to the design of several clinical trials.2 In addition, Sequist et al reported sensitivity to standard SCLC treatments in SCLC tumors transformed from non-SCLC.1
Conclusion
We describe EGFR-TKI resistance acquired in non-SCLC with MET amplification and EMT. When EGFR mutant tumors acquire resistance to EGFR-TKI, second biopsy may be recommended to select the best therapeutic strategy.
Disclosure
Jitsuo Higaki received research grants from Takeda Pharm Ltd., Mochida Pharm Ltd., Novartis Pharm Ltd., Pfizer Ltd., MSD Ltd, Astellas Pharm Ltd., Japan Boehringer-Ingelheim Ltd., Daiichi-Sankyo Ltd., and Dainihon-Sumitomo Pharm Ltd. Jitsuo Higaki received honoraria from Takeda Pharm Ltd., Inter-Science Ltd., Mochida Pharm Ltd., Novartis Pharm Ltd., Pfizer Ltd., MSD Ltd., Astellas Pharm Ltd., Japan Boehringer-Ingelheim Ltd., Daiichi-Sankyo Ltd., Dainihon-Sumitomo Pharm Ltd., and Teijin Pharm Ltd. However, all authors declare no conflicts of interest associated with this work.
References
Sequist LV, Waltman BA, Dias-Santagata D, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3(75):75ra26. | ||
Yu HA, Riely GJ, Lovly CM. Therapeutic Strategies Utilized in the Setting of Acquired Resistance to EGFR Tyrosine Kinase Inhibitors. Clin Cancer Res. 2014;20(23):5898–5907. | ||
The Japan Lung Cancer Society [homepage on the Internet]. Japan: Japanese Clinical Practice Guideline for Lung Cancer. Available from: http://www.haigan.gr.jp/modules/guideline/index.php?content_id=3. Accessed August 1, 2013. | ||
Cappuzzo F, Marchetti A, Skokan M, et al. Increased MET gene copy number negatively affects survival of surgically resected non-small-cell lung cancer patients. J Clin Oncol. 2009;27(10):1667–1674. | ||
La Monica S, Caffarra C, Saccani F, et al. Gefitinib inhibits invasive phenotype and epithelial-mesenchymal transition in drug-resistant NSCLC cells with MET amplification. PLoS One. 2013;8(10):e78656. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.