")
Back to Journals » International Medical Case Reports Journal » Volume 14
A Case of Central Pontine Myelinolysis Occurred During Treatment of Hyperosmolar Hyperglycemic Syndrome
Authors Niida S, Ogawa F , Nakajima K, Sakai K, Uchiyama M, Abe T, Takeuchi I
Received 25 April 2021
Accepted for publication 4 June 2021
Published 18 June 2021 Volume 2021:14 Pages 407—412
DOI https://doi.org/10.2147/IMCRJ.S316943
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ronald Prineas
Shoko Niida, Fumihiro Ogawa, Kento Nakajima, Kazuya Sakai, Munehito Uchiyama, Takeru Abe, Ichiro Takeuchi
Department of Emergency Medicine, Yokohama City University School of Medicine, Yokohama, Kanagawa, 236-0004, Japan
Correspondence: Fumihiro Ogawa
Department of Emergency Medicine, Yokohama City University School of Medicine, 3-9, Fukuura, Kanazawa-Ku, Yokohama, Kanagawa, 236-0004, Japan
Tel +81-45 787 2800
Email [email protected]
Abstract: Central pontine myelinolysis (CPM) is a rare demyelinating condition which has been reported to occur in a variety of clinical settings, but most commonly in association with a rapid rise in plasma osmolality during correction of chronic hyponatremia. The clinical consequences can vary from a mild motor weakness that resolves completely over time to the devastating locked-in syndrome. In this presentation, we report a case of hyperosmolar hyperglycemic syndrome (HHS) with ponto-occipital disintegration. A 71-year-old female was transferred to our ER by an ambulance due to consciousness disorder and continuous fever for 10 days. We diagnosed septic shock caused by urinary tract infection (UTI), cerebral multiple infarctions, acute kidney injury (AKI) and HHS without treatment for diabetes. Then, we started therapeutic interventions for them based on the guideline with severe control for blood sugar (BS; primary 1635 mg/dl) under insulin therapy and hypernatremia (primary 153 mEq/l) under crystal infusion control in advanced care unit, apparently on routine lab data. However, the initial serum sodium value of 153 mEq/l was slowly compensated to 148 mEq/l in 60 hours under guideline on routine lab data, the initial compensated sodium value with osmolality was changed from 178 mEq/l to 150 mEq/l in the period. She recovered from her primary diagnosis and unconsciousness. After stabilized sepsis and HHS, we detected CPM on brain MRI due to following up multiple cerebral infarctions with left leg paralysis and verbal disorder. She gradually recovered over several months with intensive rehabilitation and eventually regained near normal functional capacity with stabilized BS. When we consider HHS with hypernatremia, it may be necessary to pay attention to not only to BS control and sodium control according to the guideline but also to osmolality changes to prevent CPM.
Keywords: diabetes mellites, central pontine myelinolysis, hyperosmolar hyperglycemic syndrome, hypernatremia
Introduction
Extreme fluctuations in the concentration of serum sodium and plasma osmolality may have detrimental effects on the central nervous system. Treatment of hyperosmolar hyperglycemic state (HHS) with hydration and insulin therapy diminishes blood sugar (BS) levels, and could cause major shifts in plasma osmolality and a rapid rise in plasma sodium. Failure to control BS levels and osmolality can result in the sudden development of hyponatremia, but its rapid correction can cause hypernatremia and, consequently, brain edema.1–3 The clinical symptoms of cerebral edema include dysarthria, dysphagia, quadriparesis, behavioral disturbances, lethargy, comas, and less frequent seizures.2
The excessively rapid rise in the concentration of sodium may also induce central pontine myelinolysis (CPM) or extrapontine myelinolysis.2 Moreover, the rapid normalization of hyponatremia or rise in BS are the underlying mechanisms.4 The diagnosis of either can be confirmed upon the identification of demyelinating brain lesions with magnetic resonance imaging (MRI) or computed tomography (CT); however, CT has less sensitivity than MRI.2 We present a rarely reported incident of CPM related to HHS.
Case Description
A 71-year-old female presented to our hospital with a 10-day history of consciousness disorder, continuous fever, and vomiting without abdominal pain. She had a body temperature of 37.5°C, severe dehydration, a blood pressure of 80/70 mmHg, and a heart rate of 111 bpm. Her consciousness level was E1V2M4 (7/15) on the Glasgow coma scale (GCS) with a pupil response of 3/3 mm, +/+. We suspected that the patient had developed septic shock caused by a urinary tract infection. She had never been to a clinic or hospital for a health check-up, thus she received no diagnosis or medication. Unfortunately, she also lived alone, and her detailed condition before admission was unknown. Examination of the heart, chest, and abdomen was unremarkable. Blood gas analysis showed metabolic acidosis: pH 7.268, anion gap 17.7 mmol/L, Na 154 mEq/L, K 4.8 mEq/L, Cl 115 mEq/L, and lactate 3.9 mg/dL. Blood analysis showed the following: BS 1635 mg/dL and hyperosmotic plasma 427 mOsm/L. These findings informed the diagnosis of HHS. Additionally, her family was not aware of her diabetes status, and she was never treated with insulin. This was the first time she was diagnosed with diabetes. Her white blood cell count was 13,500/μL, neutrophils 91.4%, C-reactive protein 24 mg/dL, BUN 85 mg/dL, and creatinine 3.47 mg/dL, and she had indications of severe inflammation. We treated her for HHS with sepsis caused by pyelonephritis. The brain CT suggested that her consciousness disorder was caused by metabolic encephalopathy due to uremia and multiple cerebral and cerebellar infarctions.
Under ventilation for consciousness disorder, she underwent treatment for HHS with intravenous fluids (0.9% saline) and insulin according to the international guidelines5 with antibiotic treatment for pyelonephritis. The saline infusion was initially administered at a dose of 250 mL/h, but the rate and concentration were changed when her BS levels decreased by 60 mg/dL and the serum sodium increased from 135 to 161 mEq/L in 6 hours (Figure 1). BS was administered at a dosage of 4 U/h (0.1 U/kg/h). Her BS levels decreased by 144 mg/dL (9% of her initial BS level) in the first hour and by 90–100 mg/dL per hour thereafter, totaling a drop of 1480 mg/dL within 24 hours. Although the patient’s sodium levels changed minimally, her corrected sodium decreased by 25 mEq/L within 24 hours as the infusion was modified to include sugar. The correction rate of BS was rapid, inducing a sharp decrease in sodium concentrations. Hemodynamic stability was attained gradually with fluid therapy alone, and septic shock was alleviated on the third day.
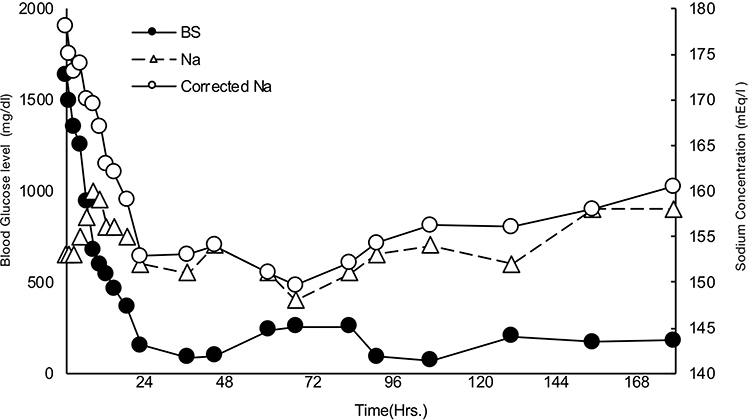 |
Figure 1 This figure shows transition of blood sugar (BS) and sodium. Corrected sodium is calculated using the formula; sodium+{(BS-100)/100*1.65}. Left bar is for BS level and right bar is for sodium concentration. |
The patient exhibited a smooth trend towards improvement under severe management for BS and sodium control. Her consciousness level had immediately improved to GCS E3VtM6 (10/15), and extubation was performed. After extubation, she could not speak, and exhibited left foot paralysis and unilateral visual impairment in her left eye (only light perception). Considering the possibility of cerebral infarction, brain MRI was performed; high signal intensity lesions were observed in the posterior central pons in T2-weighted and fluid-attenuated inversion-recovery images (Figure 2A), low signal intensity lesion on T1 weighted images (Figure 2B), and a high signal intensity lesion and a lesion without a decrease in the apparent diffusion coefficient on diffusion-weighted images (Figure 2C). These imaging findings informed a diagnosis of CPM. In addition, brain MRI showed multiple cerebral infarctions in the cerebral cortex, and a cerebellar infarction in the left cerebellar hemisphere. We consulted with an ophthalmologist regarding the cause of her visual impairment; however, no evidence of abnormalities in the fundus were found. Furthermore, multiple cerebral infarctions were not the cause owing to unilaterality. We considered further testing but did not identify the cause of visual impairment; however, preexisting unilateral visual impairment due to severe diabetes without treatment is also a possibility.
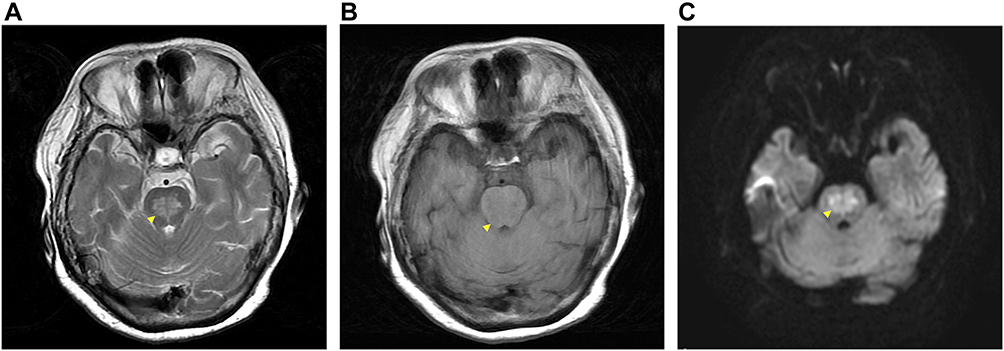 |
Figure 2 Brain magnetic resonance imaging consistent with CPM (A) high signal intensity lesions in the posterior central pons on T2WI (arrow head), (B) low signal intensity lesion on T1WI, and (C) high signal intensity lesion and a lesion without a decrease in the apparent diffusion coefficient on DWI. Abbreviations: CPM, central pontine myelinolysis; WI, weighted image; DWI, diffusion weighted image. |
Subsequently, the patient underwent intensive rehabilitation with insulin and meal management to control BS levels; she could perform more activities of daily living and gradually began to speak. The high signal area in diffusion-weighted images had shrunk after 2 weeks from last MRI. She achieved almost full functional recovery and independently performed all activities of daily living 2 months after her presentation.
Discussion
In our case report, CPM occurred during the treatment of extreme hyperglycemia with severe diabetes due to pyelonephritis and hypernatremia due to severe dehydration. CPM is complicated by renal failure, liver failure, malignant disease, severe infection, malnutrition, and chronic alcoholism. In their seminal study in 1958, Adams et al6 reported four cases of patients with alcoholism and malnutrition with varying degrees of encephalopathy, quadriplegia, and pseudobulbar palsy. Necropsy revealed significant bridge changes in the patients. It was not until 1976 that Tomlinson et al7 postulated that CPM might be caused by the rapid correction of chronic hyponatremia. This theory has been verified by animal experiments. Later, it became clear that CPM could occur in a variety of clinical situations. The most common is the rapid correction of severe hyponatremia, but it has also been described in association with other electrolyte abnormalities such as hypernatremia and hypophosphatemia,8,9 liver transplantation, and lithium and carbamate poisoning.10–12 Symptoms of CPM reportedly induce cortical bulbar signs such as impaired consciousness, spasms, dysarthria and dysphagia, and quadriplegia. If the lesion extends to the pons, it causes pupillary abnormalities and eye movement disorders. Although MRI is useful for diagnosis, CPM is not associated with any abnormal head MRI findings in 21% of CPM cases at onset.13 In such cases, MRI findings may not present until several days or weeks later.
The present report documents the case of a patient for whom the treatment of severe HHS with sepsis and pyelonephritis was complicated by CPM. Rapid and aggressive intravascular volume replacement is indicated as the first line of therapy for patients with HHS. BS is usually reduced by 75 to 100 mg/dL/h with the initial administration of isotonic sodium chloride solution for initial treatment, which restores sodium and water to address severe dehydration. Although many patients with HHS respond to fluid treatment alone, the intravenous administration of insulin in dosages similar to those used for diabetic ketoacidosis can facilitate the correction of hyperglycemia.14 Because insulin without concomitant vigorous fluid replacement increases the risk of shock, sustained insulin administration is recommended at dosages of 0.1 U/kg/h to achieve a reduction in BS of at least 10% in the first hour. The dosage of insulin can be reduced to 0.02–0.05 U/kg/h when BS levels drop below 300 mg/dL. BS levels should be maintained at 250–300 mg/dL until osmotic pressure normalizes and consciousness improves. Hypoglycemia has been reported to be as low as 100 mg/dL/h and as high as 300 mg/dL/h according to international guidelines. In our case, the correction rate of serum BS levels was maintained within the appropriate range by its 9% reduction in the first hour after therapeutic intervention and its subsequent correction to 90–100 mg/dL/h. This is recognized as factor in the onset of CPM, as previously reported.14
The exact cause of CPM is unknown. In acute hyponatremia or rapid correction of hypernatremia, an osmolar shift causes swelling of cells and cerebral edema. The most likely cause of CPM in this case was the significant shift in serum osmolality during the correction for her hyperglycemia, which was the rapid shift of plasma sodium from a baseline of 153 mEq/L to a peak of 164 mEq/L. If hyponatremia occurs for a long time (>48 h), the cells adapt to maintain a normal size. The first adaptive mechanism is the extrusion of sodium and potassium from the cell, which occurs over several hours. This is followed by organic osmolytes such as myoinisotol, taurine, and glutamate for several days.15 If the correction rate of plasma sodium is greater than the rate at which organic osmolytes can be resynthesized or returned to cells, cell contraction occurs and ultimately induces osmotic demyelination. In patients with HHS, hyperglycemia leads to a reduction in plasma sodium concentration because of translocational hyponatremia: the shift of water from intracellular to extracellular compartments. As blood sugar decreases with treatment, water returns to the intracellular space and the plasma sodium levels rise. Therefore, in patients with HHS, it is necessary to calculate the corrected plasma sodium, not the plasma sodium measured in the laboratory.16 If the concentration of the corrected serum sodium is high, then hypotonic fluids should be administered, and plasma sodium should be adjusted with careful monitoring. In addition, a decrease in BS should be gradual and carefully balanced against elevated plasma sodium rebounds. It can also be speculated that malnutrition due to HHS and a deficiency of total body potassium, sodium, phosphate, magnesium, and other osmolytes may contribute to the development of CPM in patients. Her initial plasma sodium concentration of 153 mEq/L increased to 178 mEq/L during correction of her hyperglycemia. CPM is more common in women and can affect extra-pontine areas of the brain.17 In our case, although the patient’s serum sodium level changed minimally, osmolality decreased by 50 mOsm/kg in 12 hours. According to Sterns et al,18 CPM only develops when correction of serum sodium is performed at a rate of over 12 mEq/L/day, and no reports have published rates of less than that. Hence, the correction rate for hyponatremia should be less than 12 mEq/L/day because the fluctuation of Δ24 (2 * sodium + BS/18 + BUN/2.8) should be terminated within 24 hours when considered in terms of serum osmolality. In this context, the rate at which serum osmolality was lowered in our case was rapid, possibly exacerbating the CPM that we considered to have already developed at admission. Because changing the serum osmotic pressure should be the greatest consideration when attempting to prevent CPM during the treatment of HHS, clinician should avoid changing the osmotic pressure to Δ24 (430 mg/dL/day in terms of BS levels) to prevent CPM. This approach should, however, be balanced with the treatment of hyperglycemia, which may induce organ damage and exacerbate infection when prolonged. Negotiating the two priorities requires further study.
The clinical features of CPM are diverse. The involvement of the corticospinal tract initially induces flaccid paralysis and eventually becomes spastic. Coma and confusion result from lesions in the pontine tegmentum or thalamus, and a large basis pontine lesion can cause locked-in syndrome characterized by paralysis of the lower cranial nerves and muscular tissue of the extremities. These manifestations, although rare, are important clues for clinicians to recognize to allow for correct management and treatment of CPM. Additionally, spasms, dystonia, chorea, ballismus, and paroxysmal painful tonic spasms have been associated with both CPM and extra central pontine myelinolysis.19 Although MRI has high sensitivity for detecting CPM,20 neuroimaging does not always indicate a lesion. In fact, the appearance of CPM on imaging may be delayed, thus imaging should be repeated 10–14 days later if CPM is suspected.2 The low reliability of diagnostic imaging highlights the importance of a thorough clinical examination.
No specific treatment has proven to be beneficial for patients with CPM. The best supportive care with BS management and follow-up allows for the possibility of spontaneous recovery. The previous case reports or therapeutic trials have successfully treated CPM with steroids, plasmapheresis, and intravenous immunoglobulin, but no randomized trials have been conducted to confirm the efficacy of these treatments. The prognosis of CPM is generally poor because it may cause permanent neurological sequelae or death.2 However, as the current and previous case studies show, a significant proportion of the cases can be expected to recover completely.21–23
Conclusion
Herein, we document a patient with HHS with osmotic demyelination and hypernatremia that could have induced CPM. In the treatment of HHS, BS levels should be normalized while cautiously correcting the hyperosmolar state. Additionally, the lack of free water contributed to the prevention of dilutional hyponatremia. Concurrently, serum osmolality, fluid replacement, and electrolyte imbalance should be appropriately managed to prevent CPM.
Acknowledgments
We thank other colleagues in Department of Emergency Medicine from Yokohama City University Hospital for their kind assistance. And, we would like to thank Editage for English language editing.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Adrogue HJ, Madias NE. Hypernatremia. N Engl J Med. 2000;342(20):1493–1499. doi:10.1056/NEJM200005183422006
2. Martin RJ. Central pontine and extrapontine myelinolysis: the osmotic demyelination syndromes. J Neurol Neurosurg Psychiatry. 2004;75(Suppl 3):iii22–8. doi:10.1136/jnnp.2004.045906
3. Hegazi MO, Mashankar A. Central pontine myelinolysis in the hyperosmolar hyperglycaemic state. Med Princ Pract. 2013;22(1):96–99. doi:10.1159/000341718
4. Pliquett RU, Noll A, Ibe R, et al. Hyperglycemia-related central pontine demyelinization after a binge-eating attack in a patient with type-2 diabetes: a case report. BMC Endocr Disord. 2018;18(1):18. doi:10.1186/s12902-018-0245-3
5. Karslioglu French E, Donihi AC, Korytkowski MT. Diabetic ketoacidosis and hyperosmolar hyperglycemic syndrome: review of acute decompensated diabetes in adult patients. BMJ. 2019;365:l1114. doi:10.1136/bmj.l1114
6. Adams, RD Victor, M Mancall, EL. Central pontine myelinolysis: a hitherto undescribed disease occurring in alcoholics and malnourished patients. Arch Neurol Psychiatr. 1959;81:154–172.
7. Tomlinson BE, Pierides AM, Bradley WG. Central pontine myelinolysis. Two cases with associated electrolyte disturbance. Q J Med. 1976;45(179):373–386.
8. Brown WD, Caruso JM. Extrapontine myelinolysis with involvement of the hippocampus in three children with severe hypernatremia. J Child Neurol. 1999;14(7):428–433. doi:10.1177/088307389901400704
9. Michell AW, Burn DJ, Reading PJ. Central pontine myelinolysis temporally related to hypophosphataemia. J Neurol Neurosurg Psychiatry. 2003;74(6):820. doi:10.1136/jnnp.74.6.820
10. Santinelli R, Tolone C, D’Avanzo A, Del Giudice EM, Perrone L, D’Avanzo M. Pontine myelinolysis in a child with carbamate poisoning. Clin Toxicol (Phila). 2006;44(3):327–328. doi:10.1080/15563650600584543
11. Singh N, Yu VL, Gayowski T. Central nervous system lesions in adult liver transplant recipients: clinical review with implications for management. Medicine (Baltimore). 1994;73(2):110–118. doi:10.1097/00005792-199403000-00004
12. Swartz CM. Central pontine myelinolysis and lithium toxicity. Ann Clin Psychiatry. 2003;15(2):133. doi:10.3109/10401230309085679
13. Rai GS, Rai T, Jain L, Vyas MM, Roshan R. Evaluation of CT and MRI findings among patients presented with chief complaint of headache in central India. J Clin Diagn Res. 2016;10(2):TC21–5. doi:10.7860/JCDR/2016/16852.7249
14. Kitabchi AE, Murphy MB, Spencer J, Matteri R, Karas J. Is a priming dose of insulin necessary in a low-dose insulin protocol for the treatment of diabetic ketoacidosis? Diabetes Care. 2008;31(11):2081–2085. doi:10.2337/dc08-0509
15. McCormick WF, Danneel CM. Central pontine myelinolysis. Arch Intern Med. 1967;119(5):444–478. doi:10.1001/archinte.1967.00290230082002
16. Hillier TA, Abbott RD, Barrett EJ. Hyponatremia: evaluating the correction factor for hyperglycemia. Am J Med. 1999;106(4):399–403. doi:10.1016/S0002-9343(99)00055-8
17. Wright DG, Laureno R, Victor M. Pontine and extrapontine myelinolysis. Brain. 1979;102(2):361–385. doi:10.1093/brain/102.2.361
18. Sterns RH, Silver S, Kleinschmidt-DeMasters BK, Rojiani AM. Current perspectives in the management of hyponatremia: prevention of CPM. Expert Rev Neurother. 2007;7(12):1791–1797. doi:10.1586/14737175.7.12.1791
19. Marsili L, Gallerini S, Bartalucci M, Marotti C, Marconi R. Paroxysmal painful spasms associated with central pontine myelinolisis in the context of nonketotic hyperglycemia. J Neurol Sci. 2018;388:37–39. doi:10.1016/j.jns.2018.03.005
20. Menger H, Jorg J. Outcome of central pontine and extrapontine myelinolysis (n = 44). J Neurol. 1999;246(8):700–705. doi:10.1007/s004150050435
21. Mao S, Liu Z, Ding M. Central pontine myelinolysis in a patient with epilepsia partialis continua and hyperglycaemic hyperosmolar state. Ann Clin Biochem. 2011;48(Pt 1):79–82. doi:10.1258/acb.2010.010152
22. Naik KR, Saroja AO. Seasonal postpartum hypernatremic encephalopathy with osmotic extrapontine myelinolysis and rhabdomyolysis. J Neurol Sci. 2010;291(1–2):5–11. doi:10.1016/j.jns.2010.01.014
23. van der Helm-van Mil AH, van Vugt JP, Lammers GJ, Harinck HI. Hypernatremia from a hunger strike as a cause of osmotic myelinolysis. Neurology. 2005;64(3):574–575. doi:10.1212/01.WNL.0000150730.80325.11
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.