")
Back to Journals » International Journal of General Medicine » Volume 11
A case of a patient with severe epidermolysis bullosa surviving to adulthood
Authors Hubail AR , Belkharoeva RK, Tepluk NP, Grabovskaya OV
Received 17 July 2018
Accepted for publication 13 September 2018
Published 15 November 2018 Volume 2018:11 Pages 413—421
DOI https://doi.org/10.2147/IJGM.S180464
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Amal R Hubail, Roza K Belkharoeva, Natalya P Tepluk, Olga V Grabovskaya
Department of Skin and Venereal Diseases, I.M. Sechenov First Moscow State Medical University, Moscow, Russia
Purpose: The aim of this study was to evaluate the progression of a case of a patient with epidermolysis bullosa (EB) since early age who survived to adulthood, presenting with recurrent skin blisters and disfiguring scars and disabling musculoskeletal deformities.
Background: EB is a rare group of inherited diseases that affect the skin fragility causing it to blister in response to even minor trauma. Established novel treatments are limited in the literature due to its rarity, and more research is needed to set a global management approach. Clinical manifestations range widely from localized to generalized blistering.
Methods: A rare case of EB surviving to adulthood despite the complications, which has been evaluated, treated during a relapse, and followed up.
Conclusion: The described case is of considerable clinical interest due to its rarity and severity. Optimal management requires a multidisciplinary approach and revolves around the protection of the skin against slightest injury, use of careful wound care dressings, aggressive nutritional support, and early medical or surgical interventions if needed to manage any complications. Prognosis varies considerably depending on each case.
Keywords: newborn epidermolysis bullosa, defect of type VII collagen, blistering skin diseases, skin fragility
Introduction
Epidermolysis bullosa (EB) is a group of rare inherited connective tissue diseases that result in blister formation on the skin. The incidence rate is 20 per million births in the United States. It occurs in all ethnic groups.1 The lesions, which can initially appear after birth or anytime until early adulthood, often present as dense blisters that may eventually rupture and lead to scars. Blisters usually occur at the sites of trauma or pressure, which are primarily the hands, feet, and diaper area in children, but may also present in the mouth, gastrointestinal tract, or genitalia.2 Generally, EB is a chronic condition that is disfiguring and painful. Patients with EB may have co-occurring conditions such as fused digits and clubbed hands or feet.3
EB is generally classified into four types as follows: 1) simplex EB with blisters within the epidermis, 2) dystrophic epidermolysis bullosa (DEB), 3) junctional EB with blisters in the epidermal dermal junction, and 4) Kindler syndrome with blisters at multiple levels of the skin.3
Case
Written informed consent has been provided and signed by the patient to have the case details and any accompanying images published.
A 23-year-old man presented with a diagnosis of DEB and secondary pyoderma. His family history was unremarkable.
Review of medical history revealed that vesicles first started to appear on the patient’s feet when he was beginning to walk at 11 months of age. Later, at the age of 20 months, the vesicles spread to the hands, and subsequently, lesions have always been associated with injury. As his growth progressed, skin lesions continuously progressed and occupied a large area of skin. Permanent formation of vesicles on the skin was noted, with nonhealing erosive surfaces with a bright red base at the age of 4 years. By the age of 8 years, bullous and erosive lesions were localized to the skin of the abdomen, back, and upper and lower extremities. On erosion sites, scar tissue had formed. At the same age, he had also developed contractures and pseudosyndactyly of the toes and hands.
Later in his life, periodic exacerbations of the disease occurred approximately every 2 months leading to the formation of new vesicles with purulent exudate and erosions, sometimes accompanied by fever up to 39°C. During exacerbations, the patient was given oral antibiotics, immunoglobulin, antihistamines, and topical agents that are mentioned in more detail below. The effect of treatment was insignificant. In addition, in the last 5 years, the patient experienced an increased frequency of allergic reactions, particularly to food, which were accompanied by pronounced redness and pruritus. More recently from 2016 to 2017, the patient used bandages comprising coarse nonwoven material impregnated with anhydrous ointment and silicone bandages. Zinc-containing creams and pastes were applied during dressing changes. As a result of this treatment, the patient noted a decrease in purulent exudate from the lesions and scar tissue formation.
In November 2012, the patient was examined at The Epidermolysis Bullosa Center Freiburg. Histological examination of skin biopsy showed no signs of squamous cell carcinoma, and X-ray contrast study of the esophagus revealed second-degree stenosis of the upper esophagus. Indirect immunofluorescence (IF) using antibodies to numerous proteins of the skin (desmoplakin, placefiles-1, citace-5, cytokeratin-14, plectin, integrin-α6, integrin-β4, collagen type XVII/BP 180, silvery-nu-332, collagen type IV, collagen type VII, and kind-Lin-1) showed decreased expression of collagen type VII on the surface of the vesicle. Based on these data, the diagnosis of recessive DEB was established. The acute generalized form was unlikely in this case because of the presence of a distinct residual glow of collagen type VII in biopsy material of the skin.
To clarify the subgroup of recessive EB, molecular genetic analysis of mutations in COL7A1 was suggested, but the patient declined.
Clinical findings on examination
On admission, the patient complained of rashes on the skin of the trunk and upper and lower extremities, accompanied by difficulty in swallowing, intense itching, and pain, which were aggravated by movements. Examination of the skin revealed that 70% of the body surface was covered with lesions. Rashes were localized to the skin of the trunk and upper and lower limbs. The scalp and face were free of lesions. The patient’s height and weight were 166 cm and 39.7 kg, respectively.
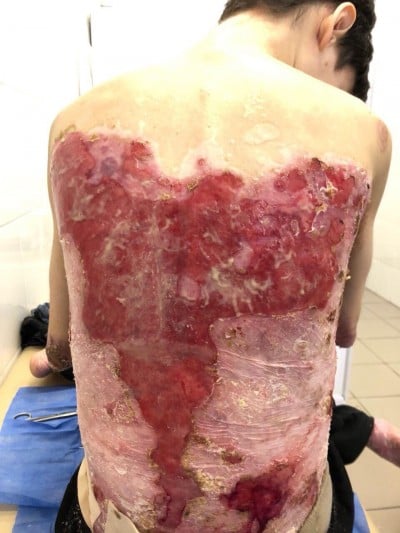
There were multiple erosions, which were bright red and irregular in shape, on the skin of the upper extremities, mainly in the shoulders, armpits, projection of the clavicle, and elbow and wrist joints. The surface of the erosions was marked by multiple-layered dirty yellow crust that secreted purulent discharge when pressed. The skin of the trunk, back, shoulder girdle, buttocks, inguinal areas, thighs, and knee joints showed extensive lesions of bright erythema, with erosive defect areas of atrophy of the skin, and multiple yellow crusts. He had pronounced flexion contractures of his hand joints with minimal finger range of motion. Although there existed finger deformities on the left hand, range of motion was preserved (Figures 1–6).
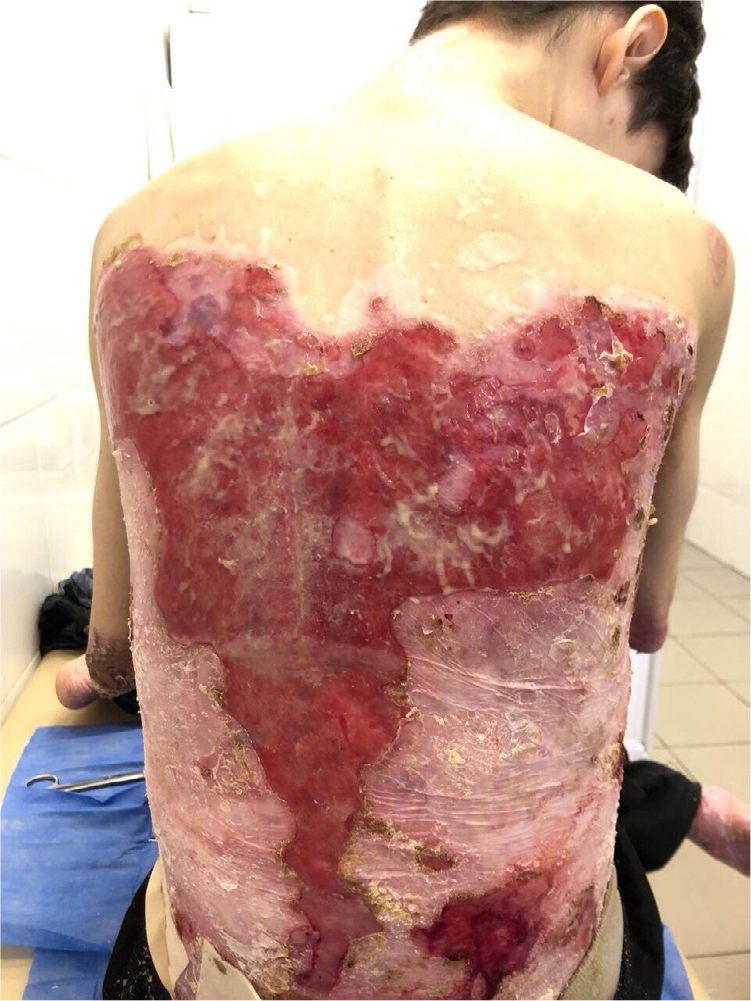 | Figure 1 Appearance of lesions on the patient’s back on presentation. |
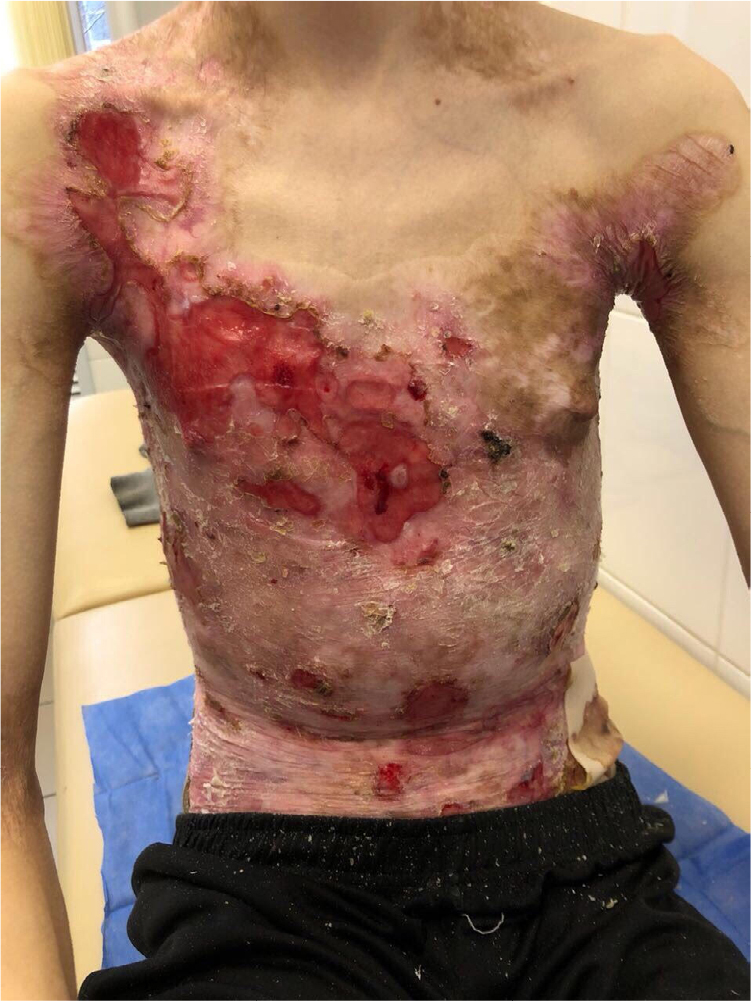 | Figure 2 Appearance of lesions on the patient’s chest and abdomen on presentation. |
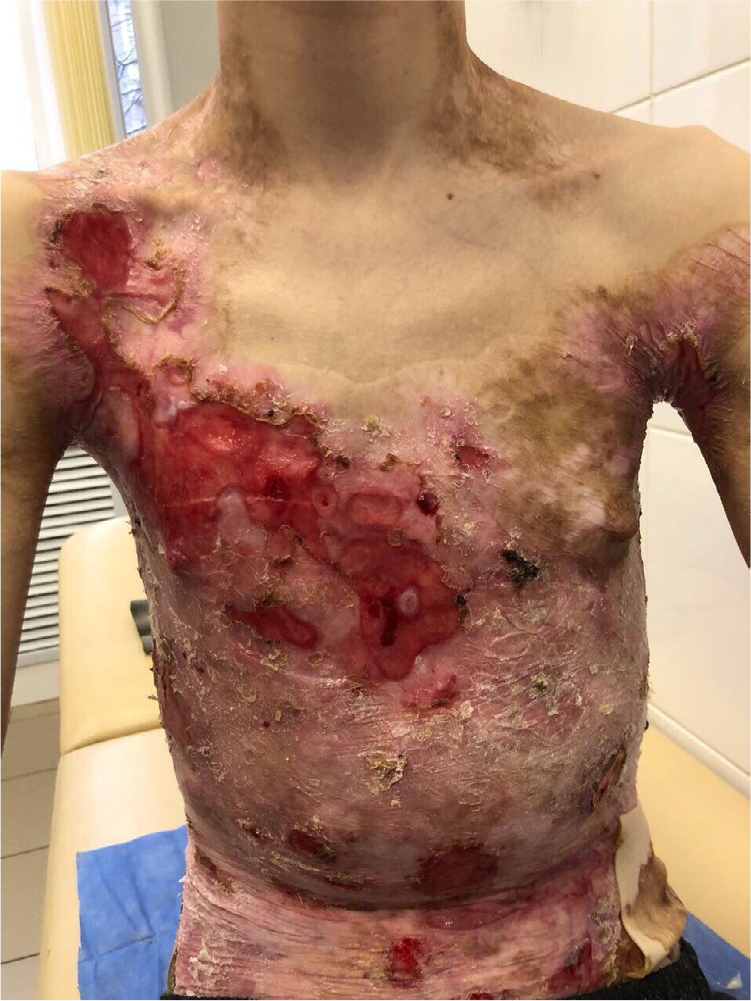 | Figure 3 Appearance of lesions on the patient’s chest and abdomen on presentation. |
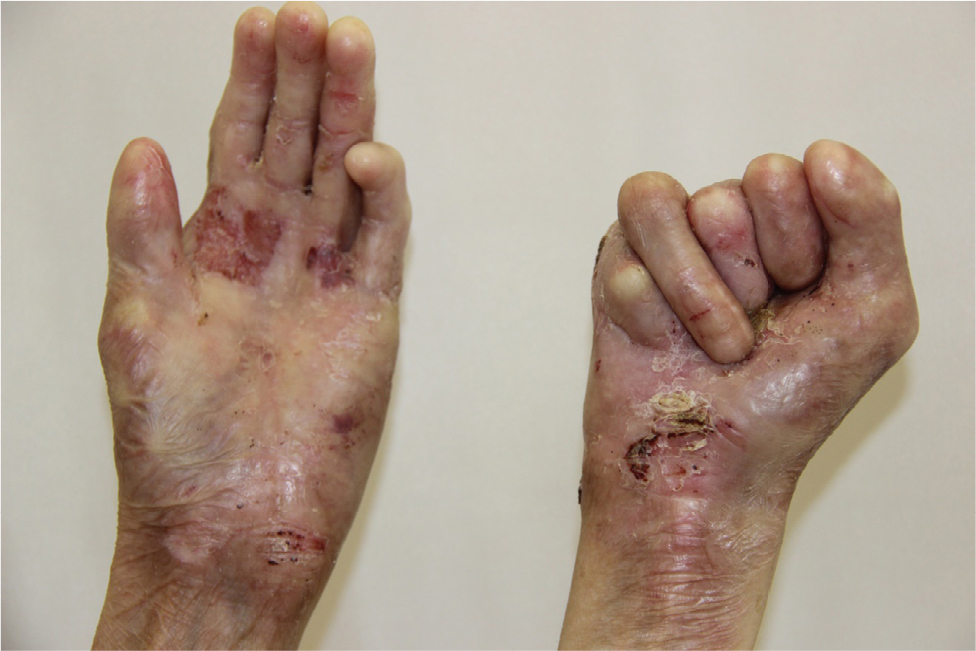 | Figure 4 Appearance of deformities on the patient’s hands on presentation. |
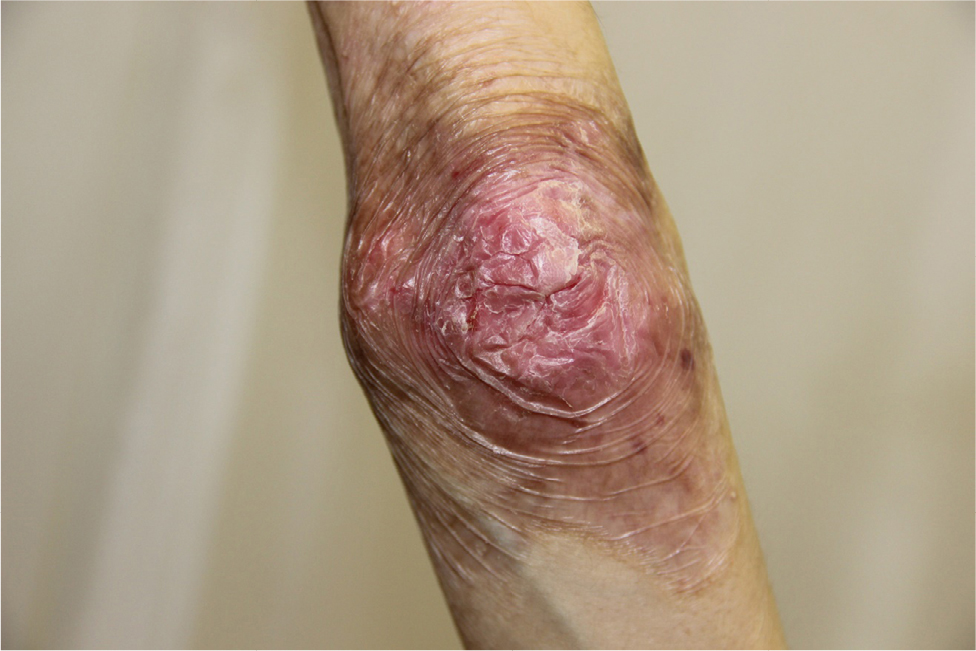 | Figure 5 Appearance of lesions on the patient’s elbow on presentation. |
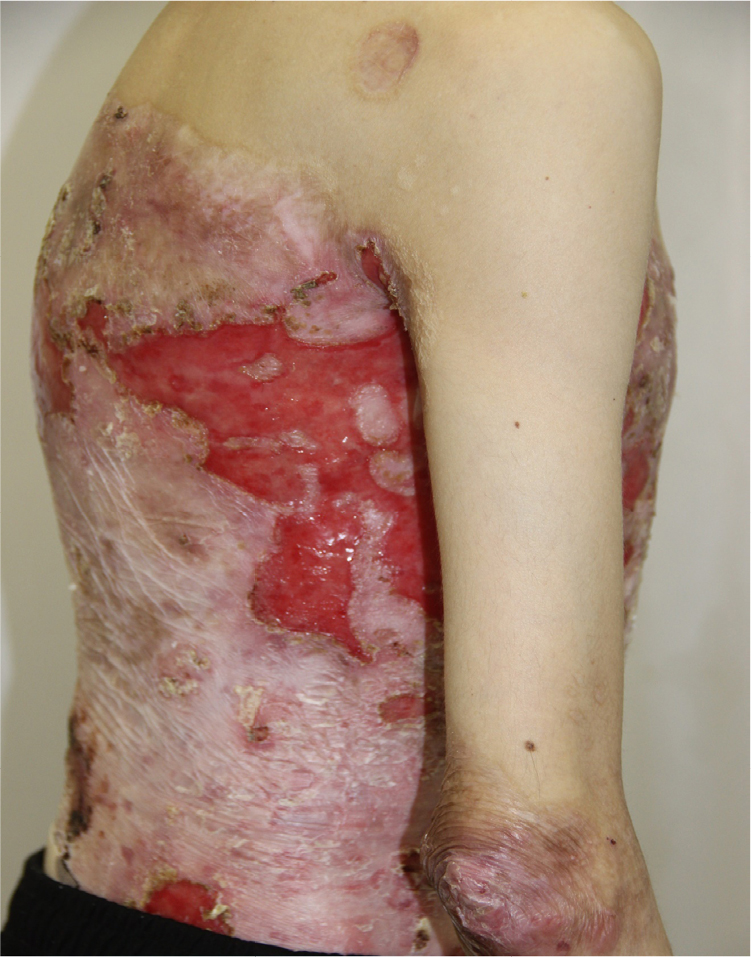 | Figure 6 Appearance of lesions on the patient’s lateral abdomen on presentation. |
In the ankle and dorsum of the feet, there were minor red-colored erosions of irregular shape, with clear boundaries and multiple brownish-yellow laminated crusts on the surface. Areas of skin atrophy and scarring were noted on the periphery of the lesions.
In addition, there were contractures and pseudosyndactyly in the first and second digits of the left foot, the first, second, and third digits of the right foot, and dislocation of the fifth digit of the left foot. Nail plates on the fingers and toes were absent. During the inspection of the oral cavity, absence of eight teeth, as well as limitation of language skills, was noted.
Investigations
- Positive biochemical analysis results: potassium 5.7 mmol/L (normal: 3.5–5.5 mmol/L), chloride 112 mmol/L (normal: 99–109 mmol/L), phosphorus 1.64 mmol/L (normal: 0.81–1.45 mmol/L), iron 5.5 mmol/L (normal: 9.0–30.4 mmol/L), transferrin 1.81 g/L (normal: 2.0–3.6 g/L), % saturation of iron 12.03% (normal: 20–55%), amylase 18.2 μ/L (normal: 30–118 μ/L), and IgA 3.59 g/L, IgM 1.27 g/L, and IgG 27.42 g/L (normal: 28–365 ng/mL);
- Immunological study results: total IgE (ELISA) 371.76 IU/mL (normal: 0–100 IU/mL);
- Immunological study results: antistreptolysin 0–232 IU/mL (normal: 0–125 IU/mL), C-reactive protein 11.40 mg/dL (normal: 0–0.8 mg/dL), and rheumatoid factor 25.30 IU/mL (normal: 0–20 IU/mL);
- Ultrasonography of the abdominal cavity: echographic signs of a slight increase in the right lobe of the liver and inflection of the gallbladder;
- Osteodensitometry results: bone density 0.503 g/cm2, which is found in 41% of the healthy people in this age group;
- Age determination by roentgenogram of the hand bones: bone age was 17 years, which is a younger number than chronological age of 23 years (flexion contracture of fingers, severe osteopenia); and
- Biopsy examination result: subepidermal blisters with variable inflammation; superficial dermis was fibrotic (collagen type IV positive, granular staining within basal and lower suprabasal keratinocytes; absent or markedly reduced staining along the dermoepidermal junction) (Figures 7 and 8).
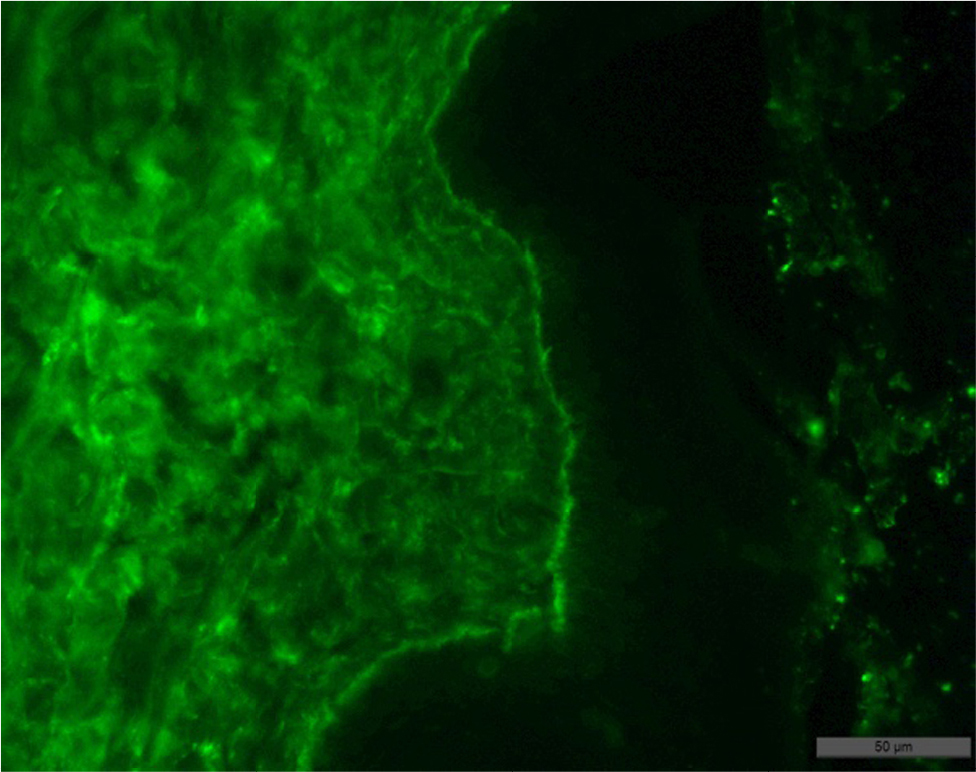 | Figure 7 Result of the patient’s skin biopsy showing fixation of IgG in the basement membrane of the epidermis, on the roof the subepidermal blisters, with minor migration in the intercellular space of the epidermis and moderate in the papillary layer of the dermis. |
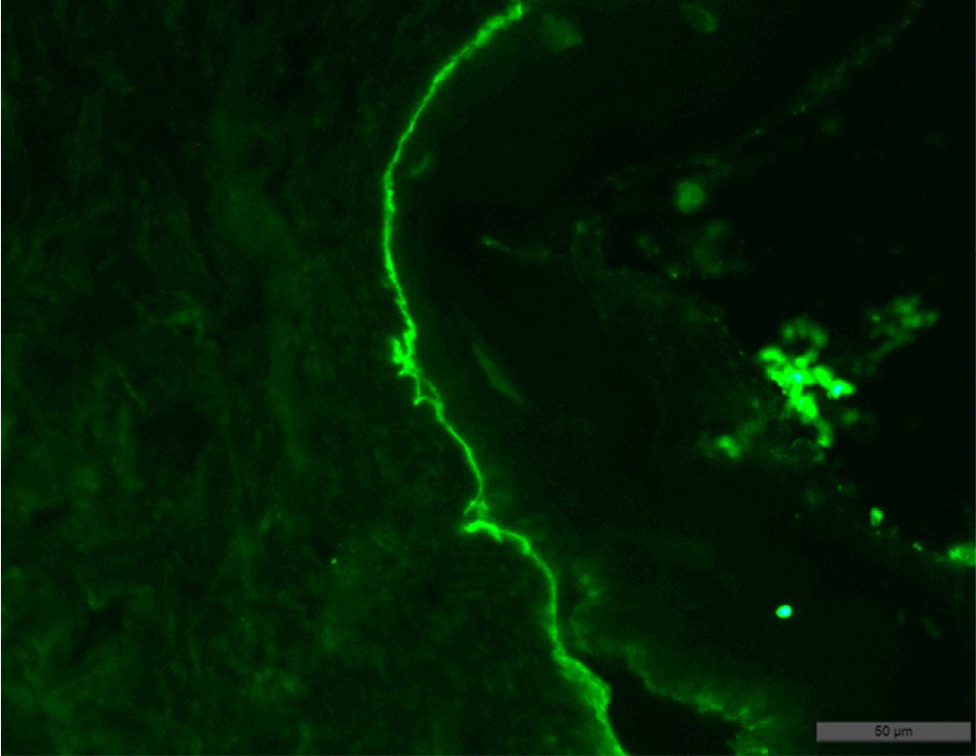 | Figure 8 Result of the patient’s skin biopsy showing fixation in the basal membrane of the epidermis, and slightly in the dermis, with permeation of some keratinocytes of the C3 component of complement. |
Progress throughout admission
During admission, the patient received the following treatments:
- Actovegin 5 mL (200 mg:1 mL) intravenous infusion daily;
- Rifampicin 400 mg one tablet two times/day for a total of 14 days;
- Venofer 10 mL infusion three times/week after dilution of the drug with 0.9% NaCl solution in a ratio of 1:20 intravenously;
- Albumin 100 mL 5% intravenous infusion with the aim of maintaining the plasma albumin level of 3–4 mg/dL;
- Vitamin B12 1 mL+0.9% NaCl 10 mL/jet once a week for 3 weeks;
- Folic acid one tablet three times a day for 6 days;
- Chlorpheniramine one tablet three times a day PRN;
- Metoclopramide 10 mg three times a day PRN.
The overall management included antibiotic rifampicin to fight the existing infections and prevent secondary skin infections from developing, venofer iron sucrose to replenish the iron stores and treat anemia, albumin to treat hypoalbuminemia, and antihypoxic effect with actovegin which normalizes pH, promotes blood circulation, intensifies energy-intensive processes of regeneration and repair, and improves tropism of tissues. In addition, the patient was supplemented with vitamin B12 and folic acid due to their deficiencies and was given antihistamine and antipruritic as per the need to control pruritus and nausea as a side effect of treatment.
Local treatment included dioxomethyltetrahydropyrimidine and collagen sponge (Meturacol), spray dexpanthenol, algipor, octenidine dihydrochloride and phenoxyethanol antiseptic spray (Octenisept), betamethasone + gentamicin + clotrimazole ointment (Triderm), and hydrocortisone + natamycin + neomycin ointment (Pimafucort). Wound care was done daily using nonadhesive silicone dressings, which absorb exudate without causing detachment of the epidermis and shedding, and coarse atraumatic dressings saturated with anhydrous ointment. Before bandaging, erosive surfaces were treated with the abovementioned antiseptic preparations, aerosols, antibiotic ointments, glucocorticosteroids, agents stimulating tissue regeneration, azulene cream, and Unna zinc paste.
To stimulate epithelialization of erosions in the complex treatment of the patient, we included low-level laser therapy in order to provide trophic and regenerative action.
- Given this complex treatment regimen, the patient’s skin in the trunk and lower extremities showed improvement with a decrease in the area of erosions, accompanied by a decrease in pain and itching of the lesions. The patient was discharged with the following recommendations in addition to close follow-up with the dermatologist (Figures 9 and 10): vitamin A and E; Aevit one capsule two times/day for 1 month;
- Ascorbic acid (vitamin C);
- Ascorutin one capsule two times a day for 1 month;
- Actovegin 200 mg one tablet three times a day for 6 weeks;
- Methyluracil 0.5 g one tablet three times a day for 1 month;
- Likopid one tablet sublingually for 10 days;
- Folic acid one tablet three times a day for 2 months;
- Human immunoglobulin 3 mL intramuscularly two times per week;
- Aktinolizat immunostimulant 3 mL intramuscularly two times a week;
- Local: collagen sponge: metrical and algipore.
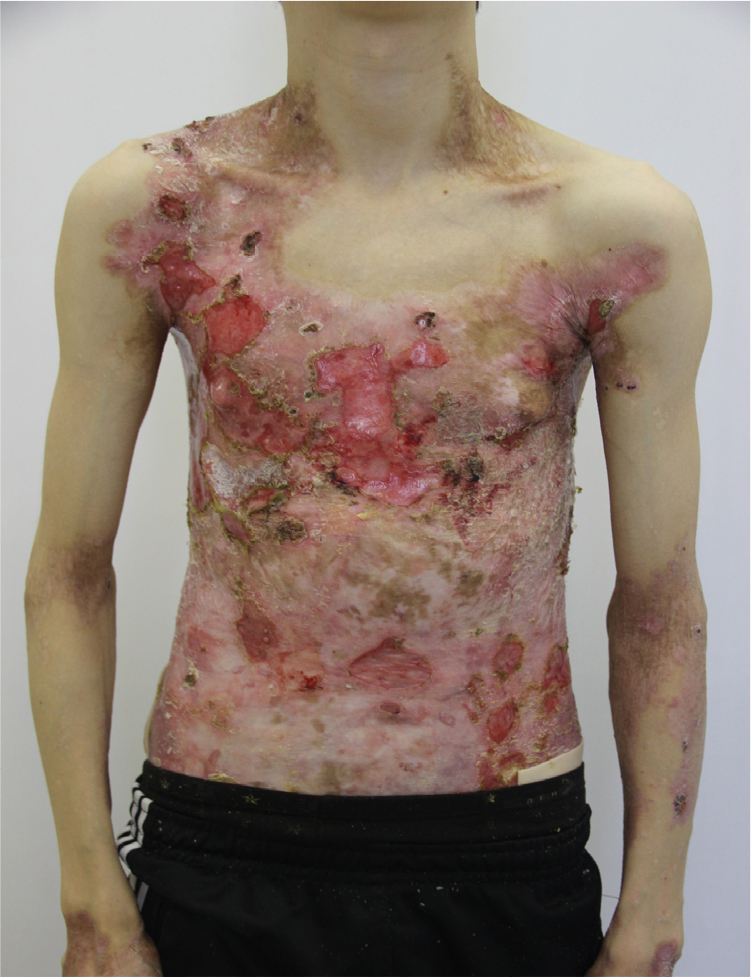 | Figure 9 Appearance of lesions on the patients chest and abdomen on day 1 after discharge. |
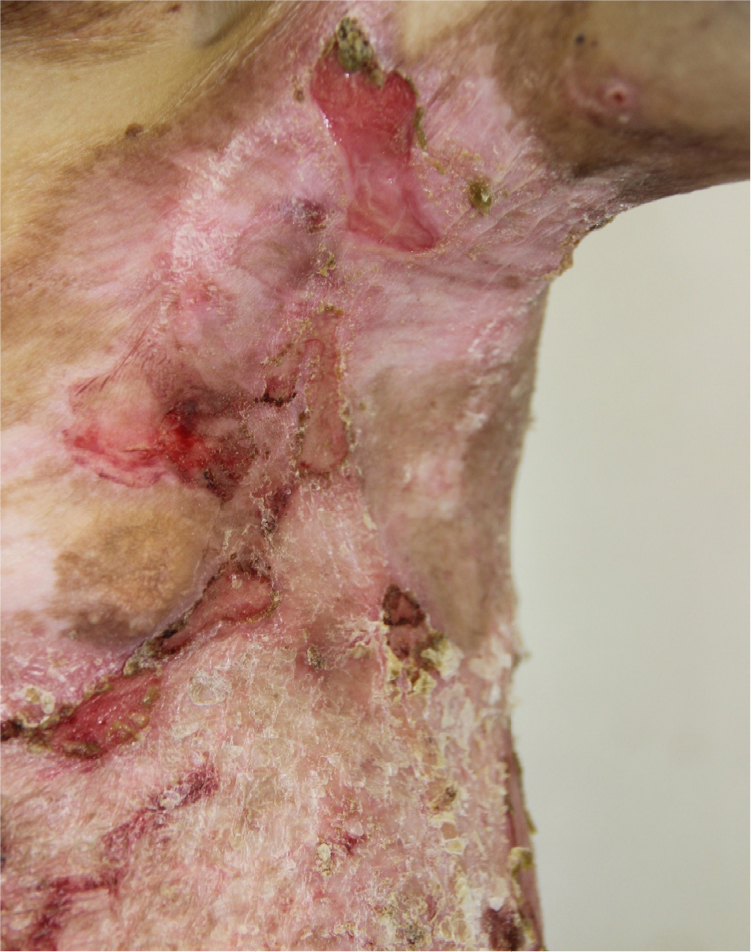 | Figure 10 Appearance of lesions on the patients axilla on day 1 after discharge. |
Discussion
EB is a chronic, recurrent, disfiguring, and painful inherited connective tissue disease. The severe type can be challenging and difficult to manage; hence, it can dramatically affect the patient’s quality of life. Patients with EB need special ongoing care, frequent dressings, and a multidisciplinary approach, including a dermatologist, pediatrician and/or neonatologist or internist, anesthetist, pathologist, medical geneticist, pain specialist, specialized nurses, and psychiatrist or psychologist. In addition, an ophthalmologist, gastroenterologist, dentist, otolaryngologist, and endocrinologist may be part of the team.4 Lifelong follow-up is required.
Diagnosis can be clinical, if there is a well-known informative family history. Otherwise, a skin biopsy examination is necessary, including immunofluorescent antigen mapping, transmission electron microscopy, and genetic analysis (molecular or DNA diagnosis).5
In order to establish the subtype of EB, transmission electron microscopy is capable of detecting ultrastructural changes in the skin of patients. Immunofluorescent staining of the skin is utilized to determine the zone of dermoepidermal junction and to establish presence, reduction, or absence of structural skin proteins.6 While the clinical picture is highly variable, diagnosis relies on direct IF microscopy of a perilesional skin biopsy. Histopathology by microscopy of a lesional skin biopsy allows distinguishing between intraepidermal and subepidermal blistering characteristic for pemphigoid disorders such as EB. However, histopathology alone cannot differentiate between the different pemphigoid disorders. Direct immunogold electron microscopy allows visualization of the deposits of autoantibodies in the sublamina densa, which clearly differentiates EBA from other pemphigoid diseases located either in the lamina lucida or lamina densa.
In direct IF microscopy of EB, like in all pemphigoid diseases, linear binding of IgG and/or IgA and/or C3 can be seen along the dermal–epidermal junction. IgG/IgA linear deposits at the basement membrane zone (BMZ) show a subtle but well remarkable pattern described as u-serrated. Indirect IF microscopy can be applied for the characterization of circulating antibodies and detection of antitype VII collagen antibodies.7
Other two investigations that could be done but are not widely available are Western blotting which is based on cell-derived or recombinant forms of type VII collagen that had been subjected to SDS-PAGE. Antitype VII collagen antibodies bound to the nitrocellulose-attached antigen can then be visualized. The other one is ELISA that allows the simultaneous testing of sera for IgG autoantibodies against the most relevant target antigens of autoimmune blistering diseases including type VII collagen.8
Currently, according to the recommendations of the Third International Consensus Meeting on Diagnosis and Classification of EB (2008) and the revision in 2014, there are four main groups and six subgroups of congenital EB. The classification is based on ultrastructural analysis of damage examined using electron microscopy.9
DEB is a group of genetic diseases, the development of which is caused by a mutation in COL7A1, a gene encoding collagen type VII, which is a major component of anchoring fibrils.10 DEB is characterized by deep skin lesions, frequent involvement of the mucous membranes with the formation of scarring, stricture formation, and milia. There are two subgroups of DEB depending on the type of inheritance: autosomal dominant or autosomal recessive11 (Table 1).
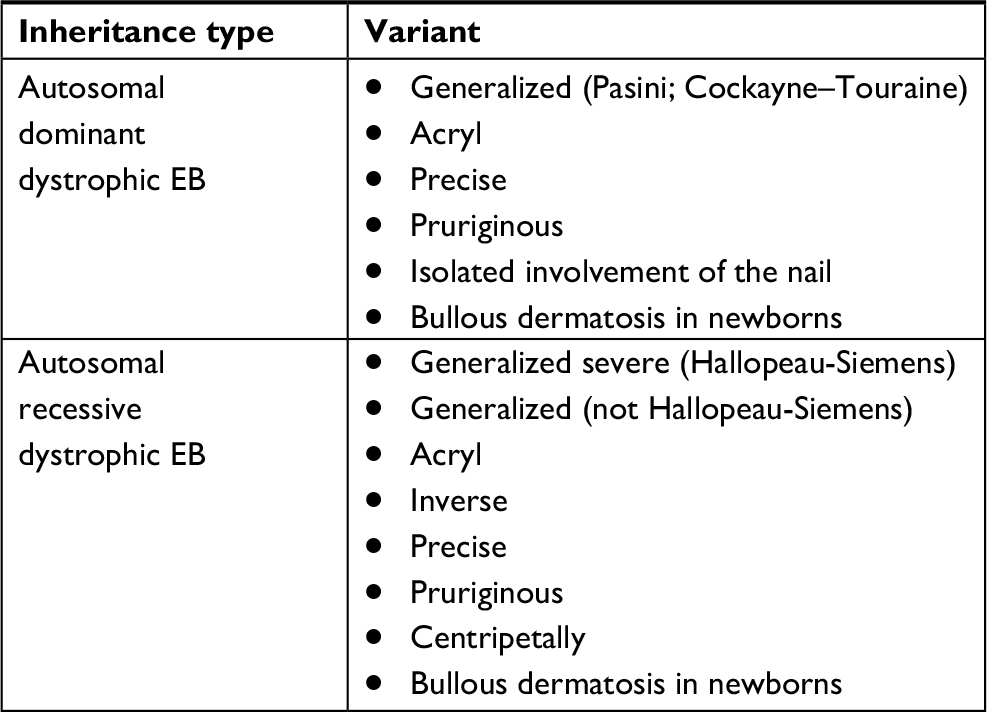 | Table 1 Classification of EB Abbreviation: EB, epidermolysis bullosa. |
The severity and poor prognosis are related to the fact that such patients exhibit persistent erosive-ulcerous defects of the skin and mucous membranes. These vesicles are accompanied by pain and itching, followed by scarring, which may lead to dysfunction of affected organs, defect of the cornea, anemia of multifactorial origin, profound growth retardation, esophageal strictures, pseudosyndactyly, ankyloglossia, and microstomia. Complications also include chronic renal failure and potentially fatal dilation cardiomyopathy. Malignant tumors, such as squamous cell carcinoma of the skin, often develop, which may lead to death despite radical removal of the tumor.12
Table 2 includes a summary of other complications associated with EB and their management.
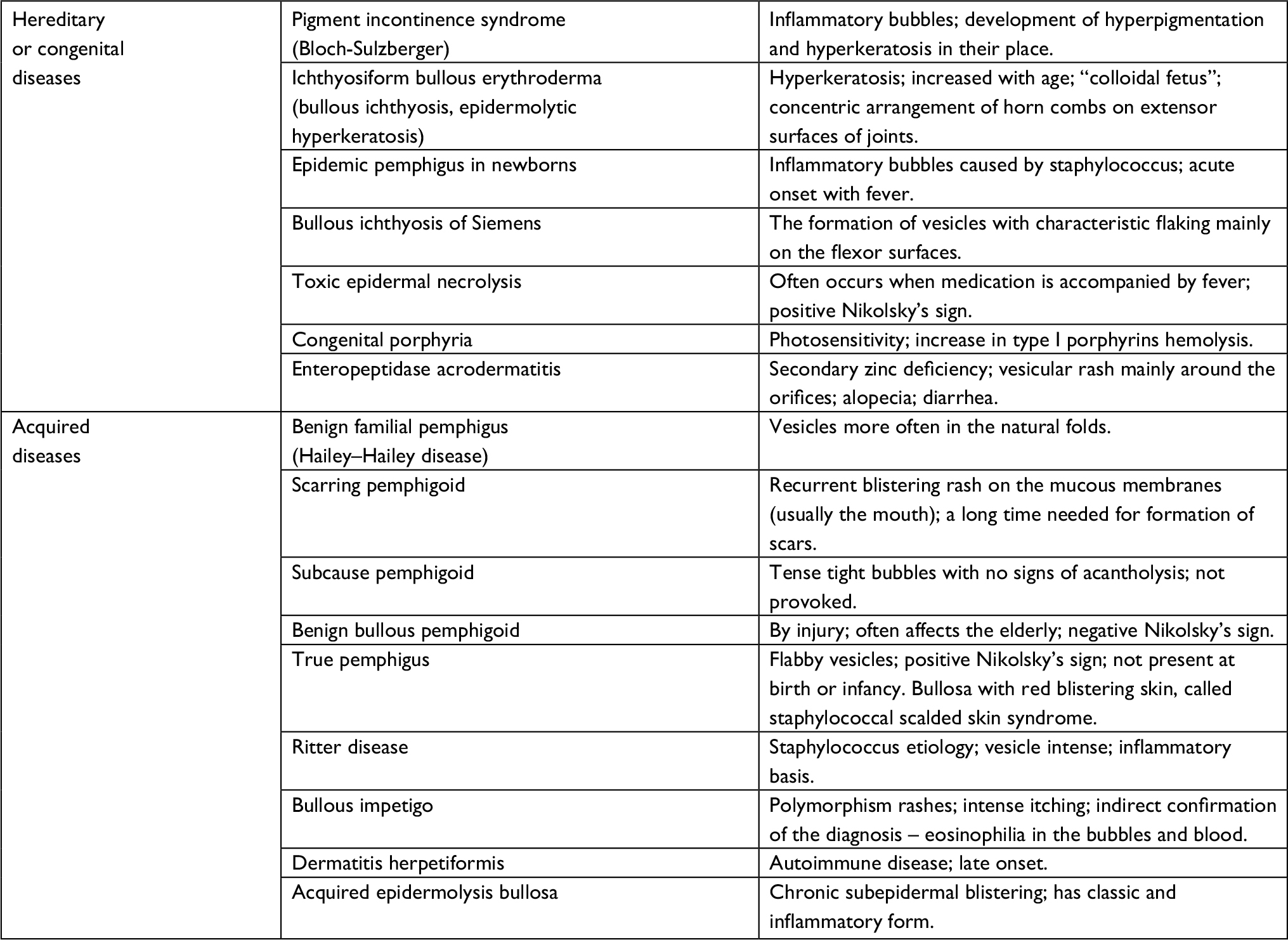 | Table 2 Differential diagnosis of EB Abbreviation: EB, epidermolysis bullosa. |
When considering the differential diagnoses of EB, it is necessary to take into account the age of the patient13 (Table 3).
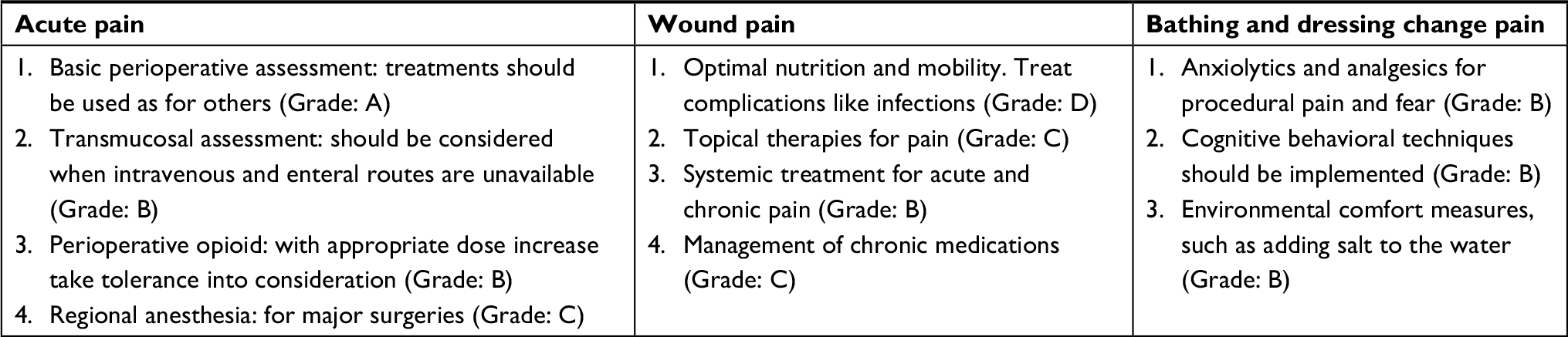 | Table 3 Summary of important recommendations in the guidelines for EB pain assessment Abbreviation: EB, epidermolysis bullosa. |
There is no specific therapy for most forms of EB. Treatment is largely supportive and includes wound care, control of infection, nutritional support, and prevention and treatment of complications. Nutritional support includes monitoring albumin levels (<3 g/dL), body mass index, growth curves, and caloric intake. Pain management and more importantly prevention are required by using protective atraumatic dressings, releasing tense blisters, avoiding anything adhesive to the skin such as dressings or products, and padding trauma prone areas. Anemia should be addressed, and if hemoglobin is <100 g/L, then iron supplementation should be initiated. Pruritus could be very distressing for the patients and hence has to be evaluated by the Pruritus Visual Analog Scale and treated accordingly with antihistamines. In addition, both the patient and their family should undergo a psychological evaluation since this chronic condition can be distressing. Antibiotic treatment may be indicated depending on the infectious status of wound.
The first evidence-based care guidelines for pain control in patients with EB were published in 2014. The guidelines explain the persistence and severity of pain that may require constant analgesia. Analgesia may be initiated with a mild painkiller, such as acetaminophen, and then gradually increased in potency; opioids can be administered based on the patient’s need. The important recommendations in the guidelines are summarized below14 (Table 4).
 | Table 4 Complications associated with EB and their management Abbreviation: EB, epidermolysis bullosa. |
Some clinical trials using protein therapy and viral gene therapies are being conducted for the treatment of EB. Gene therapies were conducted by injection of genetically engineered human fibroblasts overexpressing C7 into the systemic circulation of a murine model was reported to deliver C7 at wound sites where they were incorporated into the skin’s BMZ, forming AF. Also, bone marrow and cord blood transplantation of HLA-matched donors led to clinical improvement in patients with severe disease. Protein therapy consists of delivering recombinant human type VII collagen C7 protein, which resulted in correction of the subepidermal blistering and restoration of type VII collagen expression.15,16
Cutaneous complications associated with EB include chronic blisters, wounds/ulcers, pruritus, palmoplantar keratoderma, EB nevi, scarring, contractures, and squamous cell carcinoma.17
Conclusion
The described case is of considerable clinical interest due to the rarity and severity of EB. The treatment of patients with EB presents great difficulties. Exact causes of EB remain unclear and, therefore, palliative therapy comprising a multidisciplinary approach involving dermatologist, nutritionist, and general physician is the only way to effectively approach a patient with EB. In conclusion, severe variants are very rare, and there is need for large randomized trials to be conducted regarding management approach. For that reason, recommendations for treatment come largely from expert consensus, literature review, and observations.
Disclosure
The authors report no conflicts of interest in this work.
References
Laimer M, Bauer J, Murrell D [webpage on the Internet]. Epidemiology, Pathogenesis, Classification, and Clinical Features of Epidermolysis Bullosa. UptoDate®; 2018. Available from: https://www.uptodate.com/contents/epidemiology-pathogenesis-classification-and-clinical-features-of-epidermolysis-bullosa. Accessed August 27, 2018. | ||
Watkins J. Diagnosis, treatment and management of epidermolysis bullosa. Br J Nurs. 2016;25(8):428–431. | ||
Tabor A, Pergolizzi JV, Marti G, Harmon J, Cohen B, Lequang JA. Raising Awareness Among Healthcare Providers about Epidermolysis Bullosa and Advancing Toward a Cure. J Clin Aesthet Dermatol. 2017;10(5):36–38. | ||
El Hachem M, Zambruno G, Bourdon-Lanoy E, et al. Multicentre consensus recommendations for skin care in inherited epidermolysis bullosa. Orphanet J Rare Dis. 2014;9:76. | ||
Dermnet [webpage on the Internet]. Epidermolysis Bullosa. Available from: https://www.dermnetnz.org/topics/epidermolysis-bullosa. Accessed September 3, 2017. | ||
Cepeda-Valdés R, Pohla-Gubo G, Borbolla-Escoboza JR, et al. Immunofluorescence mapping for diagnosis of congenital epidermolysis bullosa. Actas Dermosifiliogr. 2010;101(8):673–682. Spanish. | ||
Nieboer C, Boorsma DM, Woerdeman MJ, Kalsbeek GL. Epidermolysis bullosa acquisita. Immunofluorescence, electron microscopic and immunoelectron microscopic studies in four patients. Br J Dermatol. 1980;102(4):383–392. | ||
Kim JH, Kim YH, Kim S, et al. Serum levels of anti-type VII collagen antibodies detected by enzyme-linked immunosorbent assay in patients with epidermolysis bullosa acquisita are correlated with the severity of skin lesions. J Eur Acad Dermatol Venereol. 2013;27(2):e224–e230. | ||
Fine JD, Eady RA, Bauer EA, et al. The classification of inherited epidermolysis bullosa (EB): Report of the Third International Consensus Meeting on Diagnosis and Classification of EB. J Am Acad Dermatol. 2008;58(6):931–950. | ||
Megahed M. Histopathology of Blistering Diseases. Heidelberg: Springer-Verlag Berlin Heidelberg; 2004:238–255. | ||
Kane K, Lio P, Stratigos A, Johnson R. Children’s dermatology: color atlas and handbook. Beanom. 2011:94–104. | ||
Park J, Chae H, Kim M, et al. A novel COL7A1 mutation in a Korean patient with Hallopeau-Siemens recessive dystrophic epidermolysis bullosa. Genet Mol Res. 2013;12(1):678–682. | ||
Fine JD. Inherited epidermolysis bullosa: past, present, and future. Ann N Y Acad Sci. 2010;1194:213–222. | ||
Goldschneider KR, Good J, Harrop E, et al; Dystrophic Epidermolysis Bullosa Research Association International (DEBRA International). Pain care for patients with epidermolysis bullosa: best care practice guidelines. BMC Med. 2014;12:178. | ||
Woodley DT, Wang X, Amir M, et al. Intravenously injected recombinant human type VII collagen homes to skin wounds and restores skin integrity of dystrophic epidermolysis bullosa. J Invest Dermatol. 2013;133(7):1910–1913. | ||
Hovnanian A. Systemic protein therapy for recessive dystrophic epidermolysis bullosa: how far are we from clinical translation? J Invest Dermatol. 2013;133(7):1719–1721. | ||
USNLo M [webpage on the Internet]. Dystrophic Epidermolysis Bullosa. Genetics Home Reference; 2016. Available from: https://ghr.nlm.nih.gov/condition/dystrophic-epidermolysis-bullosa. Accessed September 13, 2017. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.