")
Back to Journals » OncoTargets and Therapy » Volume 14
A c-Met Inhibitor Suppresses Osteosarcoma Progression via the ERK1/2 Pathway in Human Osteosarcoma Cells
Authors Chen W, Wu S, Huang Y, Zhang T, Dong H, Zheng X, Chen T, Gong X, Liu G, Zhao X
Received 13 May 2021
Accepted for publication 14 August 2021
Published 10 September 2021 Volume 2021:14 Pages 4791—4804
DOI https://doi.org/10.2147/OTT.S317122
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Alberto Bongiovanni
Weijie Chen,1– 3,* Su Wu,4,* Yang Huang,3,* Tingting Zhang,5 Hao Dong,6 Xing Zheng,3 Tao Chen,3 Xiaokang Gong,3 Gang Liu,1,2 Xing Zhao1
1Department of Orthopaedic Surgery, Sir Run Run Shaw Hospital, Zhejiang University School of Medicine, HangZhou, People’s Republic of China; 2Key Laboratory of Musculoskeletal System Degeneration and Regeneration Translational Research of Zhejiang Province, HangZhou, People’s Republic of China; 3Department of Orthopedics, Taizhou Municipal Hospital, Taizhou, People’s Republic of China; 4Department of Orthopedics, The Third People’s Hospital of JingDeZhen, JingDeZhen, People’s Republic of China; 5Taizhou Public Security Supervision Hospital, Taizhou Municipal Hospital, Taizhou, People’s Republic of China; 6Department of Gastrointestinal Surgery, Taizhou Municipal Hospital, Taizhou, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xing Zhao Email [email protected]
Gang Liu Email [email protected]
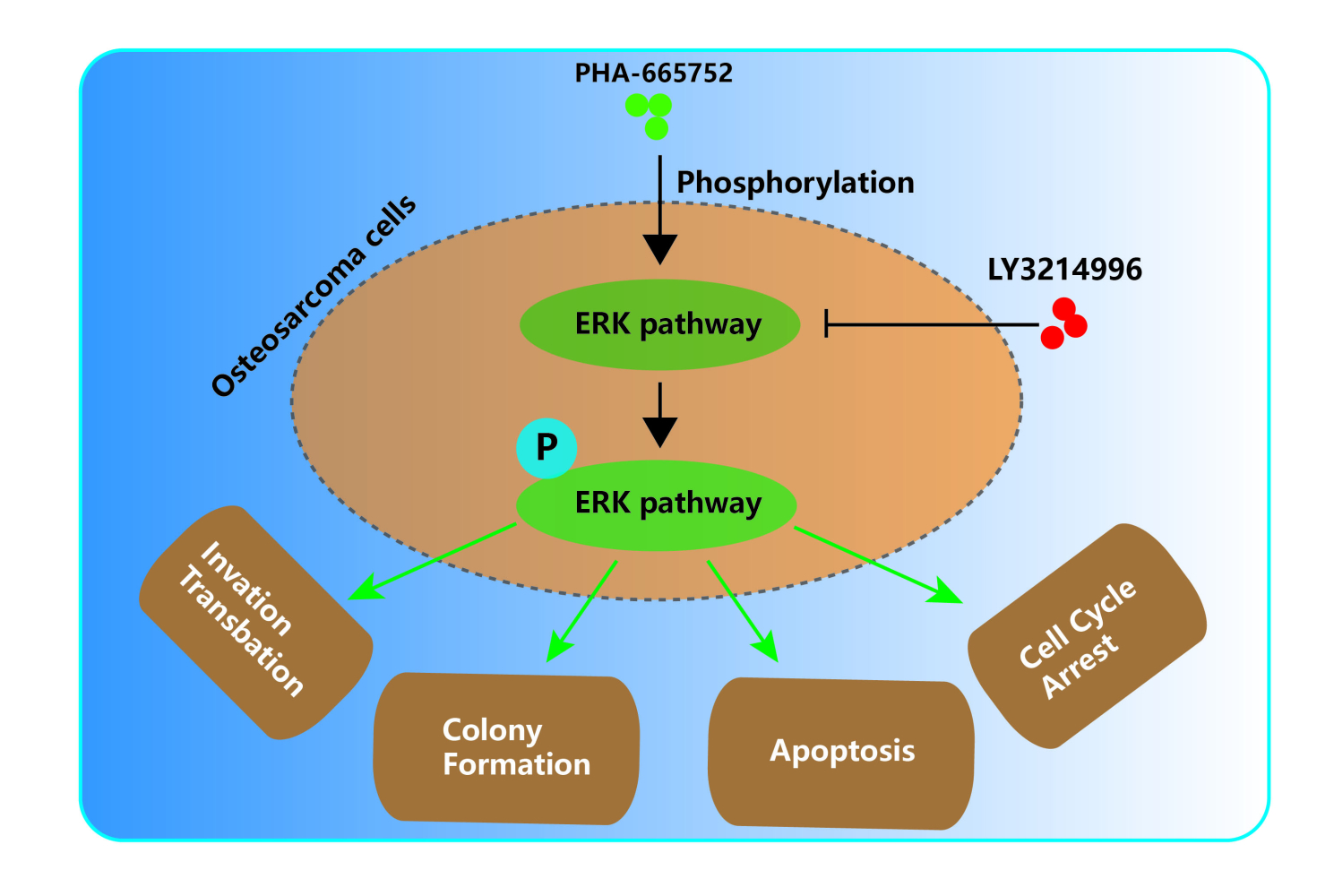
Introduction: Osteosarcoma is the most common primary malignancy of the bone among adolescents and children. Despite intensive chemotherapy and aggressive surgery, the 5-year survival rate of osteosarcoma still falls under 70%, mainly due to its tendency to metastasize and to develop drug resistance. Therefore, new treatments for osteosarcoma are urgently needed. HGF/c-Met signaling pathway, when dysregulated, is involved in the onset, progression and metastasis of various cancers, making the HGF/c-Met axis a promising therapeutic target.
Methods: In this study, we found Met to be a cancer-promoting gene in osteosarcoma as well, and aimed to investigate the role of a c-met inhibitor (PHA-665752) in osteosarcoma. For this purpose, two human osteosarcoma cell lines (143B and U2OS) were introduced in this study and treated with PHA-665752. CCK8 cell proliferation assay was performed to obtain the IC50 value of PHA-665752 for 143B and U2OS. After that, colony formation assay, transwell migration and invasion assay and wound-healing assay were performed. Furthermore, a tumor-transplanted mouse model was used for in vivo experiments.
Results: Our results showed that PHA-665752 could suppress osteosarcoma progression, promote apoptosis and inhibit proliferation of human osteosarcoma cells. Moreover, we found ERK1/2 pathway to be an important mediator underlying the osteosarcoma-suppressing function of PHA-665752. LY3214996, a highly selective inhibitor of the ERK1/2 pathway, was able to antagonize the effects of PHA-665752 in osteosarcoma. Finally, in vivo experiments indicated that PHA-665752 suppressed tumor growth in a tumor-transplanted mouse model.
Conclusion: Taken together, Met provided a druggable target for osteosarcoma and PHA-665752 is a promising candidate for anti-osteosarcoma treatments.
Keywords: osteosarcoma, Met, PHA-665752, ERK signaling, LY3214996
Graphical Abstract:
Introduction
Osteosarcoma (OS) originates from primitive bone-forming mesenchymal stem cells and represents the most common primary bone malignancy in children and adolescents.1,2 Before the 1970s, amputation surgery was the main treatment for osteosarcoma due to the lack of effective chemotherapy. However, surgeries pose a negative impact on the quality of life, and displayed a limited efficacy with more than 80% of the patients still dying of lung metastasis.3 In the past 30 years, with the advent of advanced medical methods such as neoadjuvant chemotherapy, the overall 5-year survival rate of osteosarcoma has increased from 20% to 70%.4 The four basic drugs for osteosarcoma chemotherapy are methotrexate, cisplatin, doxorubicin and ifosfamide. High-dose methotrexate (HDMtx) therapy, such as MAP (methotrexate, doxorubicin, and cisplatin) has become the standard treatment in North America and Europe.5 However, for those with metastasis or drug resistance, the efficacy of chemotherapy is limited with an overall 5-year survival rate less than 20%.6,7 Therefore, novel treatments are urgently needed, especially against the emergence of drug resistance and effective for distant metastasis.
Hepatocyte growth factor (HGF) and its high-affinity receptor, mesenchymal epithelial transition factor (c-Met) serves multiple functions in embryonic development,8 organogenesis9 and wound healing.10 Previous studies also found that the physiological function of HGF/c-Met axis revolves around cell motility and invasive growth, and displayed a higher activity in many cancers.11–14
PHA-665752 is a potent, selective and ATP competitive c-Met inhibitor. As early as in 2003, Christensen et al15 have found that PHA-665752 could inhibit the c-met-dependent phenotype in vitro and exhibit anti-tumor activity in vivo. Since then, Ma et al16 showed that PHA-665752 was able to cooperate with rapamycin to inhibit the growth of non-small cell lung cancer cells. Puri et al17 reported that PHA-665752 could inhibit the tumorigenicity and angiogenesis of lung cancer xenografts in mice to a degree reaching more than 85%. However, the therapeutic effect of PHA-665752 in osteosarcoma remains uninvestigated. Based on its significant inhibition on other malignancies, we speculated that PHA-665752 also has a therapeutic effect on osteosarcoma.
Herein, we identified a novel c-Met inhibitor (PHA-665752), which was able to suppress osteosarcoma progression via ERK signaling pathway. Our study provided a theoretical basis for the clinical use of PHA-665752 to treat osteosarcoma.
Methods
OS Mouse Model
All animal experiments were carried out according to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health and approved by the Ethics Committee of Sir Run Run Shaw Hospital. Eighteen female BALB/c-nu mice, aged 4 weeks, were purchased from Slac Laboratory (Shanghai). About 2× 143B cells were digested and mixed in 2 mL cold PBS. Then, a volume of 100μL cell suspension was injected subcutaneously. When the tumors in the dorsal area were macroscopic, mice were randomly divided into three groups (control group and two PHA-665752 groups) with six mice in each group. For the control group, a volume of 100μL 5% DMSO was injected into the peritoneum. While the PHA-665752 groups were received intraperitoneal injection of 100μL PHA-665752 (7.5mg/kg and 15mg/kg) diluted with 5% DMSO. The intraperitoneal injection was done every day, the body weight and tumor volume of mice were measured every 5 days. Thirty days later, the mice were sacrificed and the tumors were removed, weighted and fixed.
143B cells were digested and mixed in 2 mL cold PBS. Then, a volume of 100μL cell suspension was injected subcutaneously. When the tumors in the dorsal area were macroscopic, mice were randomly divided into three groups (control group and two PHA-665752 groups) with six mice in each group. For the control group, a volume of 100μL 5% DMSO was injected into the peritoneum. While the PHA-665752 groups were received intraperitoneal injection of 100μL PHA-665752 (7.5mg/kg and 15mg/kg) diluted with 5% DMSO. The intraperitoneal injection was done every day, the body weight and tumor volume of mice were measured every 5 days. Thirty days later, the mice were sacrificed and the tumors were removed, weighted and fixed.
Cell Culture
143B (ATCC: CRL-8303) and U2OS (ATCC: HTB-96TM) human osteosarcoma cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). The cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% FBS (Thermo Fisher Scientific, Waltham, MA, USA). The culture was maintained in an incubator set to 37°C with 5% CO2 and 100% humidity.
Transfection
Three different siRNAs of Met gene were designed and constructed by RiboBio (Guangzhou, China). The transfection of siRNAs was performed using Lipofectamine RNAiMAX (ThermoFisher), according to the manufacturer’s instructions. Relative sequences are shown in Table 1.
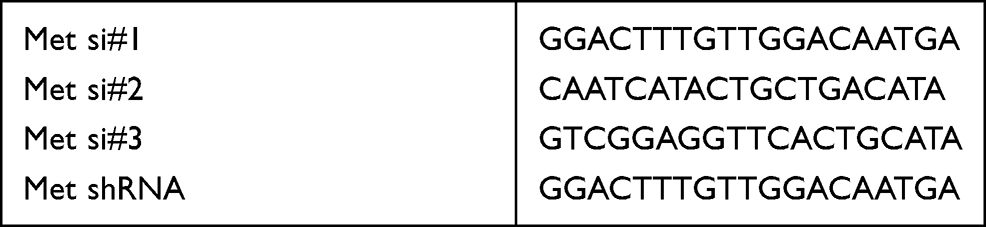 |
Table 1 Sequences of Si/shRNAs Used in This Study |
CCK8 Cell Proliferation Assay
Treated 143B and U2OS cells were seeded into a 96-well plate at a density of 2×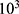 cells/well. The next day, cell proliferation was detected using CCK8 (Sigma-Aldrich), after 4 h of incubation, the absorbance of solution at 450 nm was measured using VersaMax microplate reader (Molecular Devices, CA, USA).
cells/well. The next day, cell proliferation was detected using CCK8 (Sigma-Aldrich), after 4 h of incubation, the absorbance of solution at 450 nm was measured using VersaMax microplate reader (Molecular Devices, CA, USA).
Colony-Formation Assay
Transfected or untreated cells were seeded into a 12-well plate at a density of 5×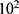 cells/well. In the drug treatment group, the medium was changed with fresh medium containing PHA-665752 (3–5 nM) or LY3214996 (5–40 nM) for about 14 days until the cells grew to visible colonies. The cells were washed with phosphate-buffered saline (PBS) and fixed with 4% paraformaldehyde for 20 min. Then, 0.5% crystal violet solution was used for staining, followed by image capture. Finally, the colonies were counted.
cells/well. In the drug treatment group, the medium was changed with fresh medium containing PHA-665752 (3–5 nM) or LY3214996 (5–40 nM) for about 14 days until the cells grew to visible colonies. The cells were washed with phosphate-buffered saline (PBS) and fixed with 4% paraformaldehyde for 20 min. Then, 0.5% crystal violet solution was used for staining, followed by image capture. Finally, the colonies were counted.
Transwell Migration and Invasion Assay
The migration and matrigel invasion assays were conducted using transwell chamber (for migration assay) or transwell precoated matrigel chamber (for invasion assay) according to the manufacturer’s protocol (BD Science, Bedford, MA, USA). The homogeneous single-cell suspensions (5 × 104 cells/well for migration, 1 × 105/well for invasion) were added to the upper chambers and incubated for 24 h. In the drug treatment group, the cells were treated with PHA-665752 or LY3214996. The migration and invasion rates were quantified by counting the migratory and invasive cells at least three random fields.
Wound-Healing Assay
Transfected or untreated cells were seeded into a six-well plate at a density of 2×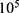 cells/well. In the drug treatment group, the cells were treated with PHA-665752 or LY3214996. For wound-healing assay, cells were scraped with the tip of 200 μL pipette tips. All images were acquired using 10 high-power fields at 0 and 24 h after injury. Remodeling was measured as diminishing distance across the induced injury, normalized to the 0 h control, and expressed as relative migration.
cells/well. In the drug treatment group, the cells were treated with PHA-665752 or LY3214996. For wound-healing assay, cells were scraped with the tip of 200 μL pipette tips. All images were acquired using 10 high-power fields at 0 and 24 h after injury. Remodeling was measured as diminishing distance across the induced injury, normalized to the 0 h control, and expressed as relative migration.
Western Blot
Treated 143B and U2OS cells or tissues were lysed with radioimmunoprecipitation assay buffer (RIPA, Beyotime, China) supplemented with 100 mM phenylmethanesulfonyl fluoride (PMSF) on ice. Total proteins were qualified using bicinchoninic acid (BCA) analysis (Beyotime, China). Then, proteins were separated by 8% SDS-PAGE and transferred to PVDF membranes (Bio-Rad), followed by blocking with 5% nonfat milk at room temperature for 1 h and incubating with primary antibody at 4°C overnight. The next day, the membranes were washed by TBST and incubated with a secondary antibody at room temperature for 1 h. Protein bands were visualized using FDbio-Femto ECL (Fudebio, Hangzhou, China) and a chemiluminescence system (Bio-Rad, USA). Anti-GAPDH, anti-N-cadherin and anti-vimentin antibodies were purchased from Proteintech (Chicago, USA); Anti-Cyclin D1, anti-CDK4, anti-ERK1/2 and anti-pERK1/2 (Thy202/Tyr204) antibodies were purchased from Abcam (Cambridge, UK).
Quantitative RT-PCR
Total RNA were extracted from 143B and U2OS cells using Ultrapure RNA Kit (CWBIO) according to the manufacturer’s protocol. Quantification of Met gene was performed using UltraSYBR one step RT-qPCR Kit (CWBIO) according to the manufacturer’s instructions. Each sample is repeated three times independently. The quantity of Met was normalized to GAPDH. Relative primers are shown in Table 2.
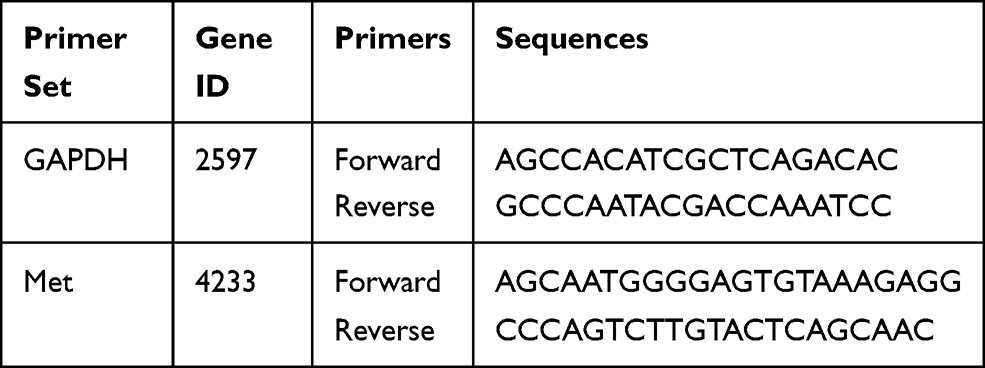 |
Table 2 Primer Sequences Used in This Study |
Cell Apoptosis Assay by Flow Cytometry
Transfected or untreated cells were seeded into a six-well plate at a density of 2×cells/well. In the drug treatment group, the cells were treated with PHA-665752 or LY3214996 for 24 h. An Annexin V-FITC/propidium iodide (PI) kit (BD Biosciences, San Diego, CA, USA) was used according to the manufacturer’s instructions. Cells were analyzed using a flow cytometer (BD FACSCANTO II, BD Biosciences, San Jose, CA, USA) and FlowJo software.
Cell Cycle Assay by Flow Cytometry
Transfected or untreated cells were seeded into a six-well plate at a density of 2×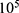 cells/well. In the drug treatment group, the cells were treated with PHA-665752 or LY3214996 for 24 h. The cells were harvested, washed with PBS and fixed with cold 75% ethyl alcohol at 4 °C overnight. The next day, cells were washed twice with PBS and incubated with RNase A for 30 min, followed by staining with 500 μL propidium iodide for 30 min at room temperature. Cell cycle analysis was performed on the Accuri C6 (BD Biosciences, Mountain View, CA, USA).
cells/well. In the drug treatment group, the cells were treated with PHA-665752 or LY3214996 for 24 h. The cells were harvested, washed with PBS and fixed with cold 75% ethyl alcohol at 4 °C overnight. The next day, cells were washed twice with PBS and incubated with RNase A for 30 min, followed by staining with 500 μL propidium iodide for 30 min at room temperature. Cell cycle analysis was performed on the Accuri C6 (BD Biosciences, Mountain View, CA, USA).
Histopathology and Immunohistochemistry
Tumor tissues were fixed in 4% paraformaldehyde, embedded in paraffin and sectioned at 5μm. The sections were incubated with primary antibodies at 4°C overnight. After washing thrice with PBST, the sections were incubated with a secondary antibody (Beyotime Institute of Biotechnology, Inc., Jiangsu, China) for 2 h at room temperature. The assessment was independently reviewed in parallel by two experienced pathologists.
TUNEL Staining
Tumor tissues were fixed in 4% paraformaldehyde, embedded in paraffin and sectioned at 5μm. An In Situ Cell Death Detection kit, POD (Sigma-Aldrich) was used for TUNEL staining according to the manufacturer’s instructions. All images were acquired using a fluorescence microscope (Eclipse E600; Nikon Corporation, Tokyo, Japan).
Statistical Analysis
Statistical analysis was performed using the SPSS version 18.0 software (IBM Corporation, USA). Data were analyzed with Student’s t-test, Fisher’s Exact test, and one-way ANOVA. The results are presented as the mean ± SD. Group differences were considered statistically different for p<0.05 between groups.
Results
Silencing Met Inhibits the Proliferation, Migration and Invasion, and Promotes Apoptosis in OS Cells
We examined the expression of Met in multiple OS cell lines (HOS, 143B, MG63, U2OS and SJSA-1) compared with hFOB1.19 cell line. Among the OS cell lines, 143B and U2OS cells exhibited the highest levels of Met (Supplementary Figure 1A). To explore the role of Met in OS cells (143B and U2OS), we first constructed a small hairpin RNA (shRNA) by selecting from three small-interfering RNAs (siRNAs). After transfecting the shRNA into 143B and U2OS cells, we found the expression level of Met considerably down-regulated. (Figure 1A). The results of the colony formation assay showed that the knockdown of Met expression considerably suppressed the colony-forming ability in OS cells (Figure 1B). As shown in Figure 1C, transwell migration and invasion assay demonstrated that the migration and invasion of 143B and U2OS cells were suppressed by the Met shRNA. These results were confirmed by wound-healing assay (Figure 1D). Moreover, Western blot analysis indicated that the expression of endothelial–mesenchymal transition (EMT) markers, including N-cadherin and vimentin were significantly downregulated upon Met knockdown (Figure 1E). We also performed cell apoptosis assay by flow cytometry, which showed that the downregulation of Met promoted cell apoptosis in both 143B and U2OS (Figure 1F). Taken together, these results demonstrated that Met was a tumor-promoting gene in OS cells. Silencing Met inhibited the proliferation, migration and invasion, and promoted apoptosis in OS cells.
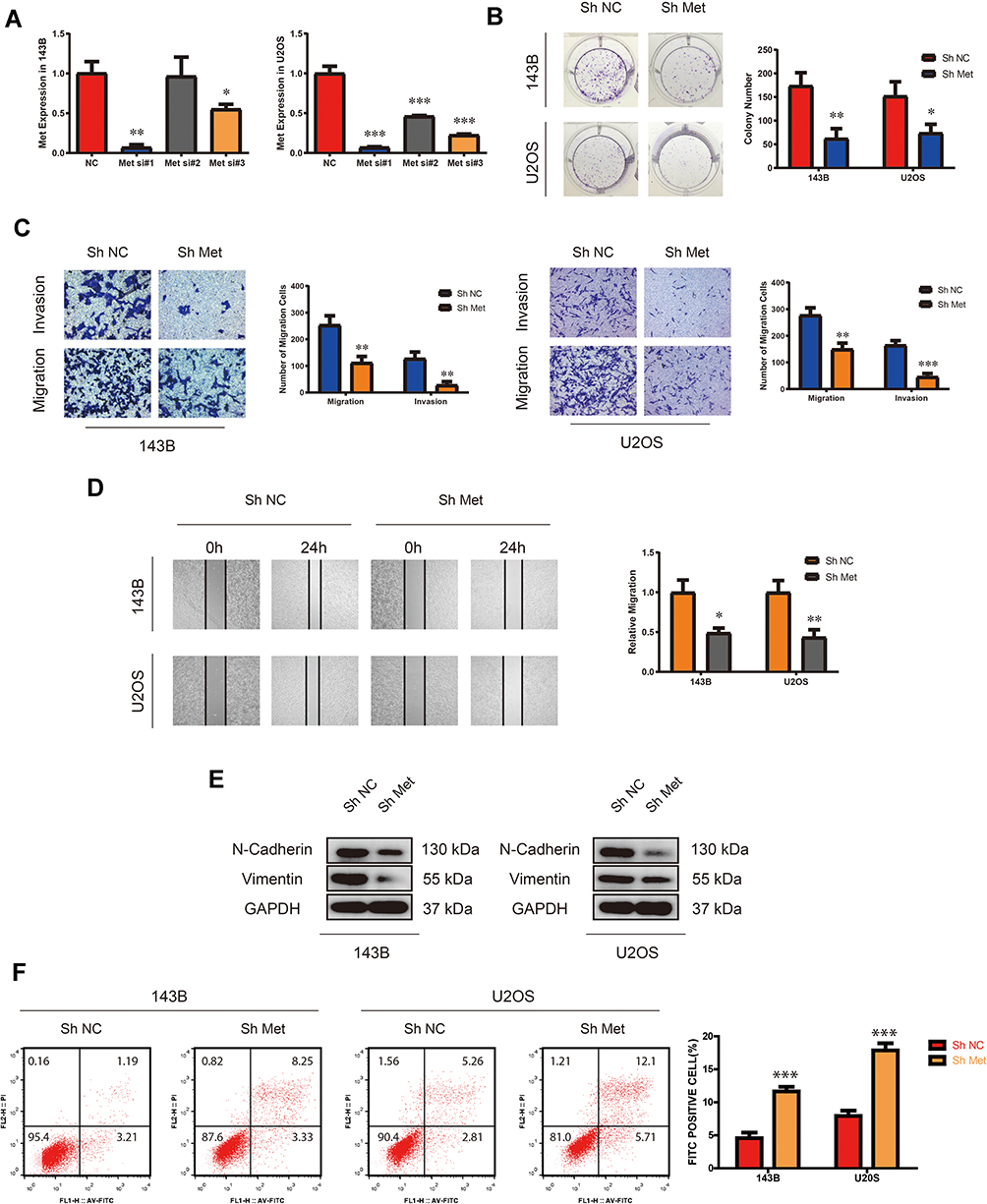 |
Figure 1 Silencing Met inhibits the proliferation, migration and invasion, while promotes apoptosis in OS cells. (A) The expression of Met after transfection of three different Met siRNAs in 143B and U2OS detected by qRT-PCR. (n=3) *p<0.05, **p<0.01, ***p<0.001. (B) Colony-formation assay of 143B and U2OS after stable transfection of Met Sh Met or N.C. (vector plasmids). (n=3) *p<0.05, **p<0.01. (C) Transwell migration and invasion assay of 143B and U2OS after stable transfection of Met Sh Met or N.C. (vector plasmids). (n=3) **p<0.01, ***p<0.001. (D) Wound-healing assay of 143B and U2OS after stable transfection of Met Sh Met or N.C. (vector plasmids). (n=3) *p<0.05, **p<0.01. (E) The expression of N-cadherin and vimentin in 143B and U2OS detected by Western blot analysis after stable transfection of Met Sh Met or N.C. (vector plasmids). (F) Cell apoptosis rate of 143B and U2OS after stable transfection of Met Sh Met or N.C. (vector plasmids). (n=3) ***p<0.001. |
PHA-665752 Inhibits Proliferation, Migration and Invasion of OS Cells
A novel selective c-Met inhibitor, PHA-665752, was acquired, and the molecular formula is shown in Figure 2A. To explore the function of PHA-665752 in OS cells, 143B and U2OS cells were treated with various concentration of PHA-665752 for 24 h (Figure 2B). The IC50 value was 3nM for 143B and U2OS. As shown in Figure 2C, treatment of PHA-665752 led to a compromised tumor formation. The migration and invasion of 143B and U2OS cells were suppressed by PHA-665752 in transwell migration and invasion assay (Figure 2D). Similar results were acquired by wound-healing assay (Figure 2E). Furthermore, N-cadherin and vimentin were significantly downregulated upon treatment of PHA-665752 (Figure 2F). Furthermore, to assess possible off-targets effects of c-Met inhibitor, we examined the function of PHA-665752 in Met-silenced OS cells, and the results showed that PHA-665752 could not play a role in Met-silenced OS cells (Supplementary Figure 1B–D), which indicated that c-Met was the specific target of PHA-665752. Taken together, these results proved the protective role of PHA-665752 in osteosarcoma by inhibiting cell proliferation, migration and invasion in OS cells in a dose-dependent manner.
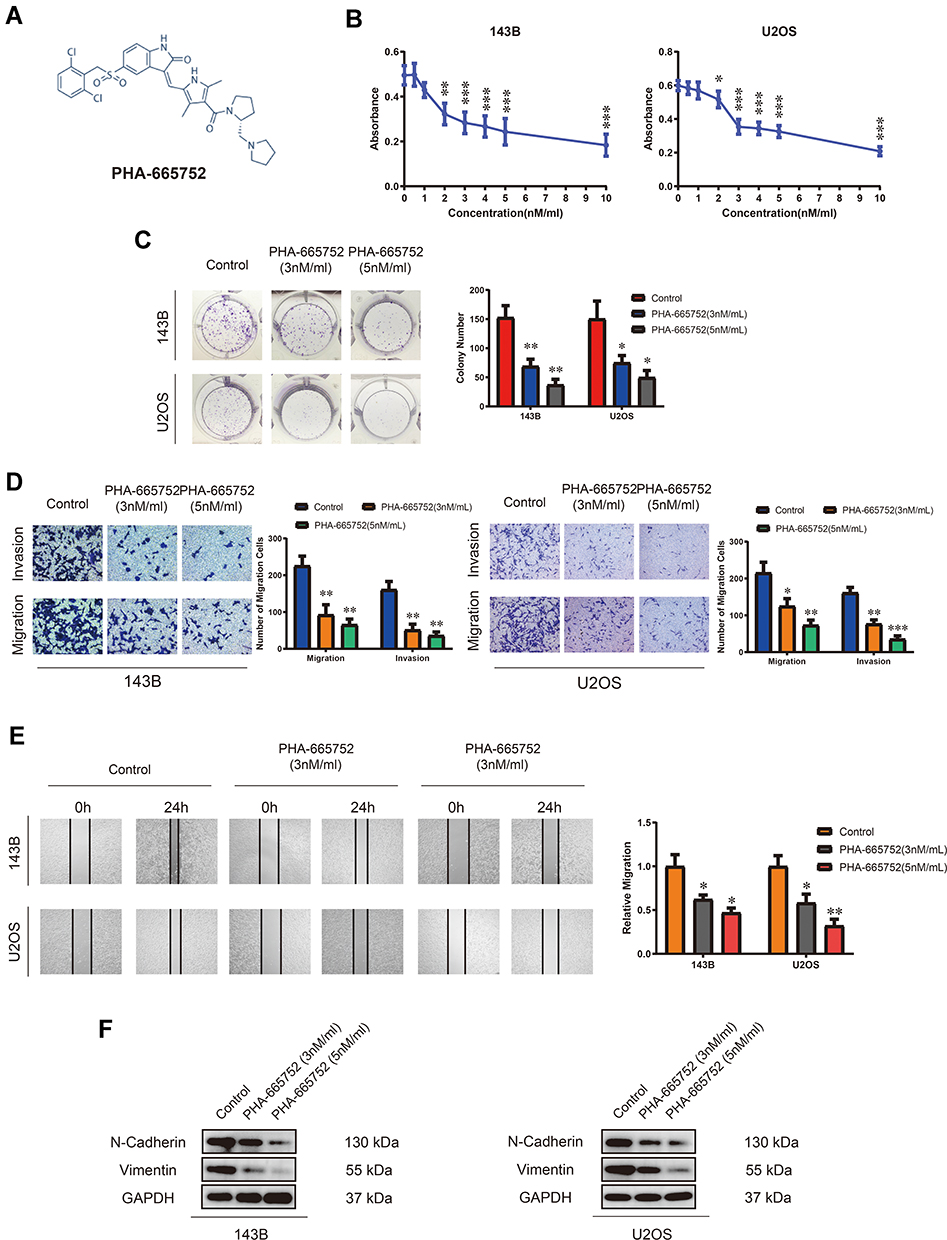 |
Figure 2 PHA-665752 inhibits proliferation, migration and invasion of OS cells. (A) The molecular formula of PHA-665752. (B) The anti-proliferative effect of PHA-665752 on 143B and U2OS was detected by CCK-8 assay. (n=5) Cells were treated with different concentration of PHA-665752. Control group contained 0.1% DMSO. *p<0.05, **p<0.01, ***p<0.001. (C) Colony-formation assay of 143B and U2OS with control or PHA-665752. (n=3) *p<0.05, **p<0.01. (D) Transwell migration and invasion assay of 143B and U2OS with control or PHA-665752. (n=3) *p<0.05, **p<0.01, ***p<0.001. (E) Wound-healing assay of 143B and U2OS with control or PHA-665752. (n=3) *p<0.05, **p<0.01. (F) The expression of N-cadherin and vimentin in 143B and U2OS with control or PHA-665752 detected by Western blot analysis. |
PHA-665752 Induces G0/G1 Phase Arrest and Promotes Cell Apoptosis in OS Cells
Considering the inhibiting function of PHA-665752, we treated OS cells with PHA-665752 for 24 h and performed cell cycle assay by flow cytometry afterwards. Results showed that PHA-665752 induced cell accumulation in the G1 phase, suggesting that PHA-665752 led to G0/G1 cell cycle arrest (Figure 3A). To elucidate the underlying mechanisms, expression levels of cycle-regulated proteins including CDK4 and Cyclin D1 were investigated by Western blot analysis. As shown in Figure 3B, treatment with PHA-665752 down-regulated CDK4 and Cyclin D1 expressions. Furthermore, TUNEL staining assay indicated that PHA-665752 promoted cell apoptosis in 143B and U2OS (Figure 3C). These results suggested that PHA-665752 induces G0/G1 phase arrest in OS cells by regulating the key regulators G0/G1 transition and promoted apoptosis of OS cells.
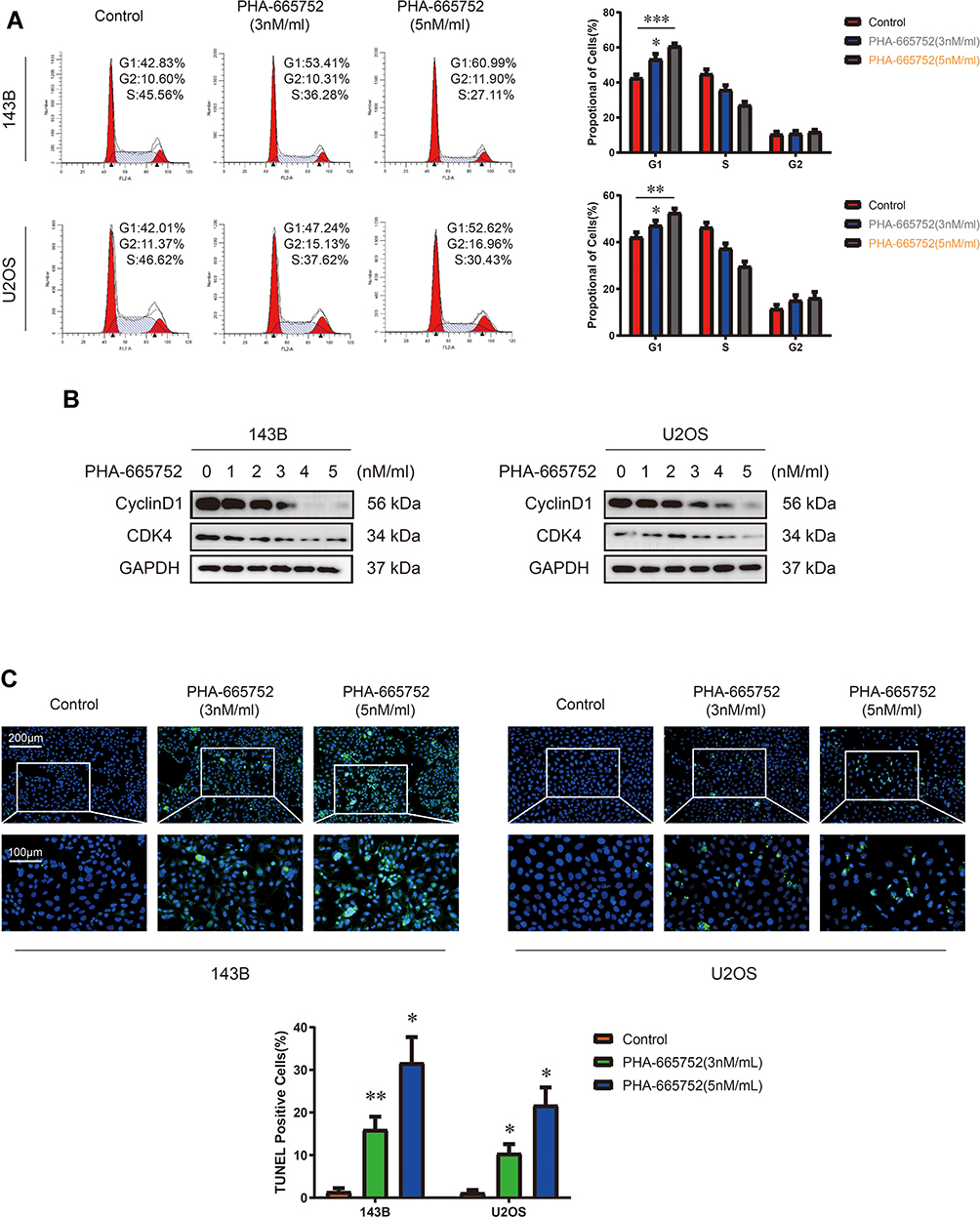 |
Figure 3 PHA-665752 induces G0/G1 phase arrest and promotes cell apoptosis in OS cells. (A) Cell cycle assay of 143B and U2OS with control or PHA-665752. (n=3) *p<0.05, **p<0.01, ***p<0.001. (B) The expression of Cyclin D1 and CDK4 in 143B and U2OS was detected by Western blot analysis. Cells were treated with different concentration of PHA-665752. (C) TUNEL staining assay of 143B and U2OS with control or PHA-665752. Scale bars, 100μm and 200μm. (n=3) *p<0.05, **p<0.01. |
The ERK1/2 Pathway Mediates the Role of PHA-665752 in Proliferation, Migration and Invasion in OS Cells
Accumulating evidences indicated that c-Met is canonically mediated by ERK1/2 pathway.18,19 Therefore, we speculated that PHA-665752 inhibits the proliferation, migration and invasion of OS cells through the ERK1/2 pathway. To verify this conjecture, we performed Western blot analysis and found that the expression of pERK1/2 in OS cells was considerably upregulated when treated with PHA-665752, while the expression of ERK1/2 had no change (Figure 4A). LY3214996 (Figure 4B), a highly selective inhibitor of the ERK1/2 pathway was then introduced in this study. We confirmed that LY3214996 downregulated pERK1/2 in OS cells as demonstrated by Western blot analysis, and the expression levels of N-cadherin and vimentin were downregulated under the treatment of PHA-665752, which was increased by adding LY3214996 (Figure 4C). 143B and U2OS were treated with 3nM/mL of PHA665752 and various concentrations of LY3214996 for 24 h. As shown in Figure 4D, the function of PHA-665752 was antagonized by LY3214996, which exhibited a ceiling effect when the concentration of LY3214996 exceeded 30 nM/mL. Therefore, subsequent experiments were performed with LY3214996 in a concentration of 30 nM/mL. Colony-formation assay showed that LY3214996 reversed the results of impaired tumor formation induced by PHA-665752 (Figure 4E). The effects of PHA-665752 on cell migration and invasion were also reversed by LY3214996 in transwell migration and invasion assay (Figure 4F). Similar results were acquired by wound-healing assay (Figure 4G). These results confirmed that the ERK1/2 pathway mediates the role of PHA-665752 in proliferation, migration and invasion in OS cells.
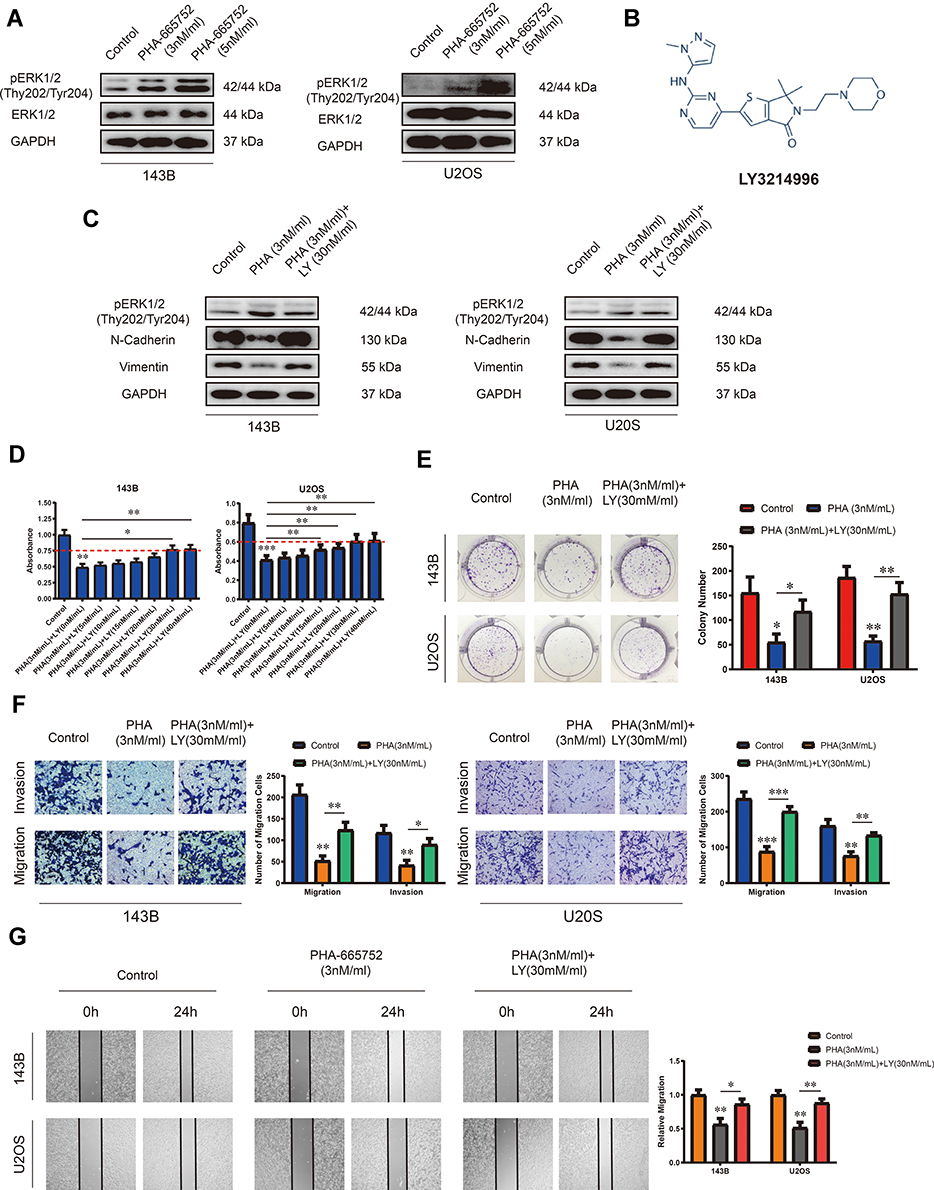 |
Figure 4 The ERK1/2 pathway mediates the role of PHA-665752 in proliferation, migration and invasion in OS cells. (A) The expression of ERK1/2 and pERK1/2 (Thy202/Tyr204) in 143B and U2OS was detected by Western blot analysis. Cells were treated with different concentration of PHA-665752. (B) The molecular formula of LY3214996. (C) The effects of PHA-665752 and LY3214996 on the expression of pERK1/2 (Thy202/Tyr204), N-cadherin and vimentin in OS cells as demonstrated by Western blot assay. (D) Proliferation of OS cells treated with PHA-665752 (3nM/mL) and various concentrations of LY3214996 was evaluated by CCK-8 assay. (n=5) *p<0.05, **p<0.01, ***p<0.001. (E) The effects of PHA-665752 and LY3214996 on the colony-formation ability in OS cells as demonstrated by colony-formation assay. (n=3) *p<0.05, **p<0.01. (F) Transwell migration and invasion assay demonstrated the effects of PHA-665752 and LY3214996 on the migration and invasion ability in OS cells. (n=3) *p<0.05, **p<0.01. Wound-healing assay of demonstrated the effects of PHA-665752 and LY3214996 on the migration ability in OS cells. (n=3) *p<0.05, **p<0.01. (G) The effects of PHA-665752 and LY3214996 on the migration ability in OS cells as demonstrated by wound-healing assay. (n=3) *p<0.05, **p<0.01. |
LY3214996 Antagonizes PHA-665752 Induced G0/G1 Phase Arrest and Apoptosis in OS Cells
Since PHA-665752 inhibited cell proliferation by inducing G0/G1 phase arrest and ERK1/2 pathway mediated the proliferation-promoting function of PHA-665752, we then performed cell cycle assay by flow cytometry after treating with PHA-665752 and LY3214996 for 24 h. As shown in Figure 5A, PHA-665752 led to G0/G1 cell cycle arrest in OS cells, and the arrest effect was canceled after LY3214996 was added. The Western blot analysis showed that the effects of PHA-665752 on CDK4 and Cyclin D1 were also reversed by LY3214996 (Figure 5B). Furthermore, TUNEL staining assay demonstrated that LY3214996 decreased cell apoptosis rate in OS cells (Figure 5C). Taken together, these results indicated that LY3214996 antagonized PHA-665752-induced G0/G1 phase arrest and apoptosis in OS cells.
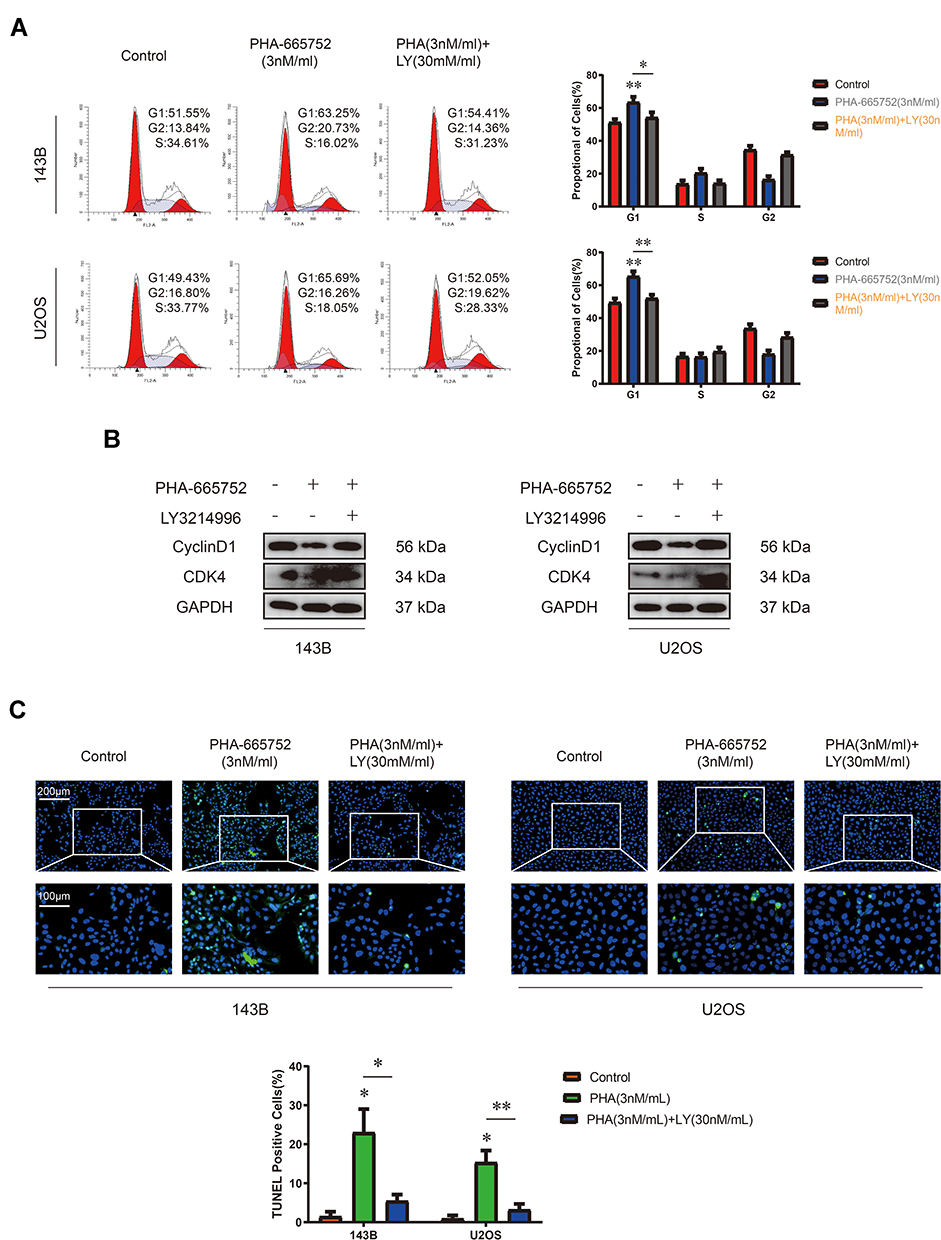 |
Figure 5 LY3214996 antagonizes PHA-665752 induced G0/G1 phase arrest and apoptosis in OS cells. (A) Cell cycle assay of 143B and U2OS was performed when treated with PHA-665752 and LY3214996. (n=3) *p<0.05, **p<0.01. (B) The expression of Cyclin D1 and CDK4 in 143B and U2OS among the above three groups was detected by Western blot analysis. (C) The effects of PHA-665752 and LY3214996 on cell apoptosis in 143B and U2OS as demonstrated by TUNEL staining assay. Scale bars, 100μm and 200μm. (n=3) *p<0.05, **p<0.01. |
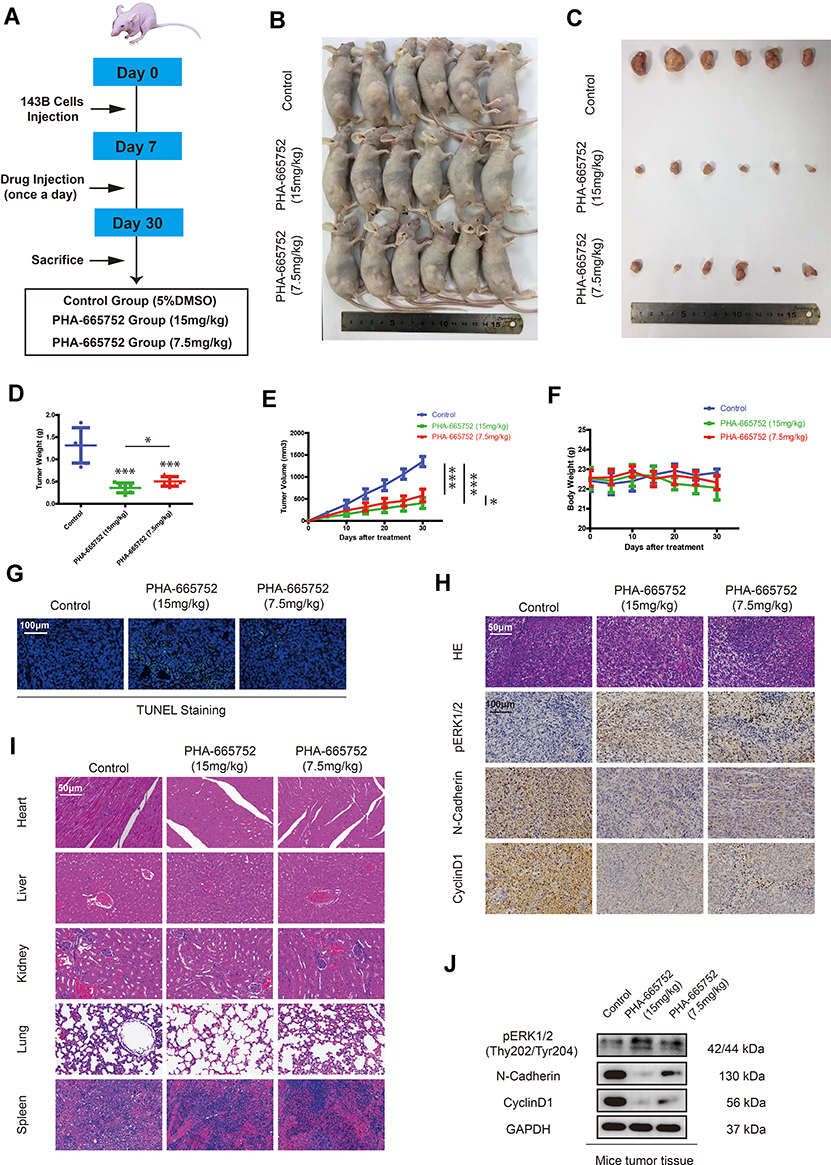
PHA-665752 Inhibits Growth of Osteosarcoma in vivo
A tumor-transplanted mouse model was introduced in this study to investigate the effect of PHA-665752 in vivo (Figure 6A). PHA-665752 at doses of 7.5mg/kg and 15mg/kg both resulted in considerably decreased tumor volume and weight after 24 days of drug administration, and 15mg/kg of PHA-665752 showed a more obvious effect (Figure 6B–E). However, there were no obvious differences of body weight among the three groups (Figure 6F). TUNEL staining assay showed significantly increased apoptosis cells in PHA-665752-treating groups with higher TUNEL-positive cell rate in 15mg/kg of PHA-665752 (Figure 6G). As shown in Figure 6H and J, PHA-665752 treatment led to an increase in the level of pERK1/2, while a decreased level of N-cadherin and Cyclin-D1 in a dose-dependent manner. The major organs, including heart, liver, kidney, lung and spleen, were collected to investigate the potential cytotoxic effects of PHA-665752 on normal tissues. Hematoxylin and eosin (H&E) staining of organs revealed no major organ-related toxicities of PHA-665752 (Figure 6I). These results showed that PHA-665752 exhibited potent antitumor activity with low toxicity in vivo.
Discussion
Current treatment of osteosarcoma mainly includes surgical intervention and systemic chemotherapy. This yields an overall 5-year survival rate of approximately 70% for patients with localized osteosarcoma. However, the survival rate plunges to less than 20% for patients with metastatic or recurrent disease.7,20 A major, as yet unsolved, problem is the dismal prognosis for patients with unresectable or relapsed osteosarcomas. Novel approaches and molecular targets are needed to improve prognosis.4
Aberrant activation of the HGF/c-Met signaling pathway was reported to be associated with several tumors, including cervical cancer,21 non-small-cell lung cancer22 and gastrointestinal cancer.23 However, as the most common primary bone malignancy, the role of HGF/c-met signaling pathway in osteosarcoma was less studied. With the in-depth study of c-Met gene, researches have confirmed that c-Met oncogene plays a crucial role in OS initiation and progression, and some of its inhibitors were proved to be effective for the treatment of osteosarcoma. Fioramonti et al24 Reported that a c-Met inhibitor cabozantinib affects osteosarcoma growth through a direct effect on tumor cells and modifications in bone microenvironment. Sampson et al25 showed met inhibitor PF-2341066 inhibits osteosarcoma growth and osteolysis/matrix production in a xenograft model. In our present study, we used loss-of-function approaches in vitro and demonstrate that Met gene was an oncogene in osteosarcoma, mainly by promoting cell proliferation, migration and invasion, and inhibiting cell apoptosis in 143B and U2OS. Therefore, HGF/c-Met signaling pathway serves as a promising candidate for osteosarcoma treatment.
PHA-665752, a novel c-Met inhibitor, was introduced in this study, with an IC50 value of 3nM for 143B and U2OS. The role of PHA-665752 was investigated in this study. We found that PHA-665752 could inhibit cell proliferation, migration and invasion in OS cells in a dose-dependent manner. Previous researches have only reported the role of PHA-665752 in OS cells migration and invasion.26,27 In this study, we found PHA-665752 induced G0/G1 phase arrest and promoted cell apoptosis through the key regulators of G0/G1 transition. Moreover, in a tumor-transplanted mouse model, we exhibited the potent antitumor activity of PHA-665752 as well as its low toxicity in vivo for the first time.
Previous studies demonstrated that c-Met were canonically mediated by ERK1/2 pathway and PI3K-AKT pathway.26,28,29 The ERK signaling is activated by numerous extracellular signals and was reported to participate in various biological cellular functions, including proliferation, migration and differentiation.30,31 Most cancer-associated lesions leading to an activation of ERK signaling occur early in the pathway.32 Our results further indicated that ERK signaling pathway was activated by PHA-665752, while LY3214996, a highly selective inhibitor of the ERK1/2 pathway, significantly downregulated the expression of pERK1/2 and antagonized the effects of PHA-665752 in osteosarcoma. We demonstrated that PHA-665752 suppressed osteosarcoma progression via the ERK1/2 pathway.
However, inhibiting the ERK1/2 pathway failed to completely reverse the effects of PHA-665752, suggesting the presence of other downstream pathways of PHA-665752. Moreover, we proved that ERK1/2 pathway was an important mediator of PHA665752 in osteosarcoma, though the specific target of the PHA-665752 -ERK1/2 interaction was unknown, which merit additional studies and further clarification.
In conclusion, we found a c-Met inhibitor, PHA-665752, effective in suppressing osteosarcoma progression via the ERK1/2 pathway. Meanwhile, it posed little toxicity in vivo. Our study provided a theoretical basis for the clinical use of PHA-665752 to treat osteosarcoma.
Conclusions
This study demonstrates that met provided a druggable target for osteosarcoma, and PHA-665752 is a promising candidate for anti-osteosarcoma treatments, which suppress osteosarcoma progression via the ERK1/2 pathway in human osteosarcoma cells.
Ethics Approval and Consent to Participate
The study design was approved by the Ethics Committee of Sir Run Run Shaw Hospital.
Acknowledgments
The authors gratefully acknowledge all the people who helped us to do this project.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Funding
This study was sponsored by Innovative Talents Support Plan Program of Zhejiang Province (2020383422). No benefits in any form have been or will be received from a commercial party related directly or indirectly to the subject of this manuscript.
Disclosure
The authors have declared that no competing interests exist.
References
1. Lindsey BA, Markel JE, Kleinerman ES. Osteosarcoma overview. Rheumatol Ther. 2017;4:25–43. doi:10.1007/s40744-016-0050-2
2. Gorlick R, Anderson P, Andrulis I, et al. Biology of childhood osteogenic sarcoma and potential targets for therapeutic development: meeting summary. Clin Cancer Res. 2003;9:5442–5453.
3. Bishop MW, Janeway KA, Gorlick R. Future directions in the treatment of osteosarcoma. Curr Opin Pediatr. 2016;28(1):26–33. doi:10.1097/MOP.0000000000000298
4. Ritter J, Bielack SS. Osteosarcoma. Ann Oncol. 2010;21(Suppl 7):vii320–vii325. doi:10.1093/annonc/mdq276
5. Rastogi S, Aggarwal A, Tiwari A, Sharma V. Chemotherapy in nonmetastatic osteosarcoma: recent advances and implications for developing countries. J Glob Oncol. 2018;4:1–5.
6. Burns J, Wilding CP, Jones RL, Huang PH. Proteomic research in sarcomas - current status and future opportunities. Semin Cancer Biol. 2020;61:56–70.
7. Harrison DJ, Geller DS, Gill JD, Lewis VO, Gorlick R. Current and future therapeutic approaches for osteosarcoma. Expert Rev Anticancer Ther. 2018;18(1):39–50. doi:10.1080/14737140.2018.1413939
8. Latimer AJ, Jessen JR. Hgf/c-met expression and functional analysis during zebrafish embryogenesis. Dev Dyn. 2008;237(12):3904–3915. doi:10.1002/dvdy.21794
9. Trusolino L, Bertotti A, Comoglio PM. MET signalling: principles and functions in development, organ regeneration and cancer. Nat Rev Mol Cell Biol. 2010;11(12):834–848. doi:10.1038/nrm3012
10. Chalenko YM, Sysolyatina EV, Sobyanin KA, et al. Topical treatment with the bacterium-derived c-Met agonist InlB321/15 accelerates healing in the abrasion wound mouse model. Arch Dermatol Res. 2018;310(10):849–856. doi:10.1007/s00403-018-1870-4
11. Refaat T, Donnelly ED, Sachdev S, et al. c-Met overexpression in cervical cancer, a prognostic factor and a potential molecular therapeutic target. Am J Clin Oncol. 2017;40(6):590–597. doi:10.1097/COC.0000000000000203
12. Szturz P, Budikova M, Vermorken JB, et al. Prognostic value of c-MET in head and neck cancer: a systematic review and meta-analysis of aggregate data. Oral Oncol. 2017;74:68–76. doi:10.1016/j.oraloncology.2017.09.009
13. Xu CW, Wang WX, Wu MJ, et al. Comparison of the c-MET gene amplification between primary tumor and metastatic lymph nodes in non-small cell lung cancer. Thorac Cancer. 2017;8:417–422. doi:10.1111/1759-7714.12455
14. Hass R, Jennek S, Yang Y, Friedrich K. c-Met expression and activity in urogenital cancers – novel aspects of signal transduction and medical implications. Cell Commun Signal. 2017;15(1):10. doi:10.1186/s12964-017-0165-2
15. Christensen JG, Schreck R, Burrows J, et al. A selective small molecule inhibitor of c-Met kinase inhibits c-Met-dependent phenotypes in vitro and exhibits cytoreductive antitumor activity in vivo. Cancer Res. 2003;63:7345–7355.
16. Ma PC, Schaefer E, Christensen JG, Salgia R. A selective small molecule c-MET inhibitor, PHA665752, cooperates with rapamycin. Clin Cancer Res. 2005;11(6):2312–2319. doi:10.1158/1078-0432.CCR-04-1708
17. Puri N, Khramtsov A, Ahmed S, et al. A selective small molecule inhibitor of c-Met, PHA665752, inhibits tumorigenicity and angiogenesis in mouse lung cancer xenografts. Cancer Res. 2007;67(8):3529–3534. doi:10.1158/0008-5472.CAN-06-4416
18. Yin B, Liu Z, Wang Y, et al. RON and c-Met facilitate metastasis through the ERK signaling pathway in prostate cancer cells. Oncol Rep. 2017;37(6):3209–3218. doi:10.3892/or.2017.5585
19. Zhang S, Song Q, Wang X, et al. Virtual screening guided design, synthesis and bioactivity study of Benzisoselenazolones (BISAs) on inhibition of c-Met and its downstream signalling pathways. Int J Mol Sci. 2019;20:2489.
20. Gill J, Ahluwalia MK, Geller D, Gorlick R. New targets and approaches in osteosarcoma. Pharmacol Ther. 2013;137(1):89–99. doi:10.1016/j.pharmthera.2012.09.003
21. Boromand N, Hasanzadeh M, ShahidSales S, et al. Clinical and prognostic value of the C-Met/HGF signaling pathway in cervical cancer. J Cell Physiol. 2018;233(6):4490–4496. doi:10.1002/jcp.26232
22. Awad MM, Oxnard GR, Jackman DM, et al. MET exon 14 mutations in non–small-cell lung cancer are associated with advanced age and stage-dependent MET genomic amplification and c-Met overexpression. J Clin Oncol. 2016;34(7):721–730. doi:10.1200/JCO.2015.63.4600
23. Bahrami A, Shahidsales S, Khazaei M, et al. C-Met as a potential target for the treatment of gastrointestinal cancer: current status and future perspectives. J Cell Physiol. 2017;232(10):2657–2673. doi:10.1002/jcp.25794
24. Fioramonti M, Fausti V, Pantano F, et al. Cabozantinib affects osteosarcoma growth through a direct effect on tumor cells and modifications in bone microenvironment. Sci Rep. 2018;8(1):4177. doi:10.1038/s41598-018-22469-5
25. Sampson ER, Martin BA, Morris AE, et al. The orally bioavailable met inhibitor PF-2341066 inhibits osteosarcoma growth and osteolysis/matrix production in a xenograft model. J Bone Miner Res. 2011;26(6):1283–1294. doi:10.1002/jbmr.336
26. Wang K, Zhuang Y, Liu C, Li Y. Inhibition of c-Met activation sensitizes osteosarcoma cells to cisplatin via suppression of the PI3K–Akt signaling. Arch Biochem Biophys. 2012;526(1):38–43. doi:10.1016/j.abb.2012.07.003
27. De Maria R, Miretti S, Iussich S, et al. met oncogene activation qualifies spontaneous canine osteosarcoma as a suitable pre-clinical model of human osteosarcoma. J Pathol. 2009;218:399–408. doi:10.1002/path.2549
28. Qian L, Su H, Wang G, Li B, Shen G, Gao Q. Anti-tumor activity of bufalin by inhibiting c-MET mediated MEK/ERK and PI3K/AKT signaling pathways in gallbladder cancer. J Cancer. 2020;11(11):3114–3123. doi:10.7150/jca.38393
29. El Bezawy R, De Cesare M, Pennati M, et al. Antitumor activity of miR-34a in peritoneal mesothelioma relies on c-MET and AXL inhibition: persistent activation of ERK and AKT signaling as a possible cytoprotective mechanism. J Hematol Oncol. 2017;10(1):19. doi:10.1186/s13045-016-0387-6
30. Bonjardim CA. Viral exploitation of the MEK/ERK pathway - a tale of vaccinia virus and other viruses. Virology. 2017;507:267–275. doi:10.1016/j.virol.2016.12.011
31. Kyriakis JM, Avruch J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10-year update. Physiol Rev. 2012;92(2):689–737. doi:10.1152/physrev.00028.2011
32. Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007;26(22):3279–3290. doi:10.1038/sj.onc.1210421
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.