")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 12
5-Aza-2'-deoxycytidine protects against emphysema in mice via suppressing p16Ink4a expression in lung tissue
Authors He ZH, Chen Y, Chen P, He SD, Zeng HH, Ye JR, Liu D, Cao J
Received 26 December 2016
Accepted for publication 24 July 2017
Published 30 October 2017 Volume 2017:12 Pages 3149—3158
DOI https://doi.org/10.2147/COPD.S131090
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Richard Russell
Zhi-Hui He,1 Yan Chen,2 Ping Chen,2 Sheng-Dong He,2 Hui-Hui Zeng,2 Ji-Ru Ye,2 Da Liu,2 Jun Cao3
1Intensive Care Unit, 2Department of Respiratory Medicine, Second Xiangya Hospital, Central South University, Changsha, 3Department of Respiratory Medicine, Hunan Provincial People’s Hospital, Changsha, China
Background: There is a growing realization that COPD, or at least emphysema, involves several processes presenting in aging and cellular senescence. Endothelial progenitor cells (EPCs) contribute to neovascularization and play an important role in the development of COPD. The gene for p16Ink4a is a major dominant senescence one. The aim of the present study was to observe changes in lung function, histomorphology of lung tissue, and expression of p16Ink4a in lung tissue and bone marrow-derived EPCs in emphysematous mice induced by cigarette-smoke extract (CSE), and further to search for a potential candidate agent protecting against emphysema induced by CSE.
Materials and methods: An animal emphysema model was induced by intraperitoneal injection of CSE. 5-Aza-2'-deoxycytidine (5-Aza-CdR) was administered to the emphysematous mice. Lung function and histomorphology of lung tissue were measured. The p16Ink4a protein and mRNA in EPCs and lung tissues were detected using Western blotting and quantitative reverse-transcription polymerase chain reaction, respectively.
Results: CSE induced emphysema with increased p16Ink4a expression in lung tissue and bone marrow-derived EPCs. 5-Aza-CdR partly protected against emphysema, especially in the lung-morphology profile, and partly protest against the overexpression of p16Ink4a in EPCs and lung tissue induced by CSE.
Conclusion: 5-Aza-CdR partly protected against emphysema in mice via suppressing p16Ink4a expression in EPCs and lung tissue.
Keywords: 5-Aza-2'-deoxycytidine, cigarette smoke, emphysema, endothelial progenitor cells, p16Ink4a
Introduction
Increasing research has indicated that COPD, or at least emphysema, represents premature aging or premature senescence of lung parenchymal cells, which are induced in part by oxidative damage from cigarette-smoke (CS) components, resulting in accelerated lung aging/accelerated lung senescence.1–3 Moreover, the key pathogenetic processes involved in COPD are considered to involve those senescent cells, notably progenitor cells, decreasing regenerative properties.4 Bone marrow-derived endothelial progenitor cells (EPCs), one of the major components of parenchymal cells, provide an alternative source of endothelial cells (ECs) and play a fundamental role in the maintenance of endothelial integrity and function, postnatal vasculogenesis, vascular repair, and tissue regeneration through pivotal bioactivity, differentiating into ECs and secretion of vasoactive substances that promote angiogenesis and maintain vascular homeostasis.5–7 The normal function of EPCs is required for tissue repair and airway remodeling in lungs.8–10 Our previous study showed decreased and dysfunctional circulating EPCs in patients with COPD.11
p16Ink4a was initially discovered as a tumor-suppressor factor composed of 148 amino-acid residues with molecular weight 16 kD.12 With increased tumor investigations, it was found that relationships between p16Ink4a and tumor cells were not all the same. p16Ink4a is a cyclin-dependent kinase inhibitor that controls cell-cycle progression,13 and could be regarded as a major dominant senescence gene.14 ECs have higher expression rates of p16Ink4a, inducing cell senescence in COPD patients.15 p16Ink4a expression in EPCs and emphysematous lung tissue has been little studied.
The advent of genome-wide epigenetic studies allowed for more comprehensive study of the epigenome in many diseases. Hypermethylation of genes associated with CS has been reported.16,17 5-Aza-2′-deoxycytidine (5-Aza-CdR), an S-phase-specific inhibitor of DNA methyltransferase, is the most widely used inhibitor of DNA methylation and triggers demethylation, leading to a consecutive reactivation of epigenetically silenced genes in vitro and in vivo.18 In this study, in an attempt to elucidate pathophysiological mechanisms of emphysema with regard to gene hypermethylation, we detected p16Ink4a expression in bone marrow-derived EPCs and lung tissue of mice with emphysema induced by CS extract (CSE) and compared the results with those in mice with emphysema treated with 5-Aza-CdR. Lung function, histomorphology, and apoptosis in lung tissue were the indicators for evaluating the severity of emphysema in mice.
Materials and methods
Animals
A total of 24 C57BL/6J male mice aged 4–6 weeks were randomly enrolled in this study. All animals were purchased from the Shanghai Laboratory Animal Center of the Chinese Academy of Sciences and fed in a cleaning unit at 23°C–25°C and 50%–60% humidity, with a 12-hours light–dark cycle. The study was approved by the institutional review board of Central South University and conformed to the guiding principles for research involving animals and human beings.
Preparation of CSE
CSE was prepared according to a previous publication,19 with some modifications. Briefly, one unfiltered Furong cigarette (tar 13 mg, nicotine 1 mg, carbon monoxide 14 mg/cigarette; China Tobacco Hunan Industrial, Changsha, China) was burned and the smoke passed through 4 mL PBS via a vacuum pump at a constant pressure of −0.1 kPa. This product was further filtered through a filter with 0.22 μM pores (Thermo Fisher Scientific, Waltham, MA, USA) to remove particles and bacteria and used for intraperitoneal injection. The solution was prepared freshly for each injection.
Preparation of 5-Aza-2′-deoxycytidine
5-Aza-CdR powder (5 g; Sigma-Aldrich, St Louis, MO, USA) was dissolved in 2 mL PBS and further diluted to 25 mg/mL, subpackaged, and stored under −80°C until experiments.
Animal modeling
The mouse emphysema model was established as previously described.20 C57BL/6J mice were divided into three groups: controls, CSE, and CSE + 5-Aza-CdR (n=8 per group). The total experimental period was 4 weeks, with intraperitoneal injection of PBS, CSE, or 5-Aza-CdR (Table 1). According to animal weight, intraperitoneal injection doses of PBS, CSE, 5-Aza-CdR were 0.3 mL/20 g, 0.3 mL/20 g, and 2.5 mg/kg (0.3 mL/20 g constant volume), respectively. At day 28, mice were killed for measurement of lung function, detection of histomorphology of lung tissue, and separation of bone marrow-derived EPCs.
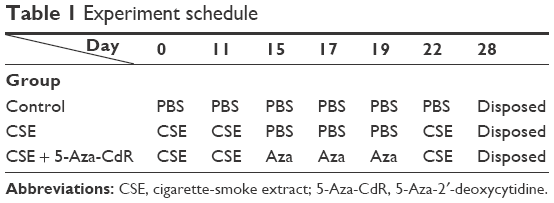 | Table 1 Experiment schedule |
Isolation, culture, and identification of EPCs
Ficoll density-gradient centrifugation (Histopaque-1083; Sigma-Aldrich) was used to isolate mononuclear cells (MNCs) from bone marrow of C57BL/6J mice according to a previously published method.21,22 Isolated MNCs were cultured with EGM-2 growth medium in the presence of 5% FBS (SingleQuots; Lonza, Basel, Switzerland) under an atmosphere of 95% humidity, 5% CO2, and 37°C for EPC culture. Cells were inoculated into culture flasks at a density of 3–5×106/mL. Then, culture fluid was replaced totally by fresh culture medium on day 4 of the culture to remove unattached cells. Half replacement with the fresh medium was performed every 3 days. Cell harvesting was performed on day 7 of the culture. To identify EPCs, firstly photos were taken during the culture using phase-contrast microscopy (Olympus, Tokyo, Japan) to confirm the morphology of EPCs. Secondly, cells positively stained with both DiI-labeled acetylated low-density lipoprotein (acLDL) and fluorescein isothiocyanate (FITC)-labeled Ulex europaeus agglutinin (UEA)-1 were identified as EPCs.23,24 Briefly, cells were incubated with 7.5 μg/mL Dil-acLDL (Thermo Fisher Scientific) at 37°C for 4 hours and fixed with 4% paraformaldehyde for 10 minutes. After being washed, cells were treated with 10 μg/mL FITC-UEA1 (Sigma-Aldrich) for 30 minutes. Finally, cells were treated with 1 μg/mL DAPI for 5 minutes before identification through laser-scanning confocal microscopy (Olympus). Fifteen random-view fields were involved to calculate the positive rate of amphophilic cells.
Lung-function measurement
Lung-function measurement was performed using small-animal spirometry (PLY3211 system; Buxco Electronics, Wilmington, NC, USA) as previously described with a minor modification.20 Briefly, the mouse was anesthetized by intraperitoneal injection of 10% chloral hydrate (3 mL/kg body weight) and tracheostomized. The trachea was cannulated and the cannula connected to the computer-controlled small-animal spirometer. Airway resistance (Raw), lung dynamic compliance (Cdyn), peak expiratory flow (PEF), and inspiratory time/expiratory time (Ti/Te) were measured according to the manufacturer’s instructions.
Histomorphological detection
After lung-function measurement, animals were killed by overdose of anesthetic. The lower-left lobes of lungs were inflated with 4% paraformaldehyde at a pressure of 25 cm H2O, then fixed with 4% paraformaldehyde for 24 hours.3 Fixed lungs were embedded in paraffin (Sigma-Aldrich) and sliced into 4 μm sections. The slices were stained with H&E (Sigma-Aldrich). Pulmonary emphysema was quantified based on the measurement of the mean linear intercept (MLI) and destructive index (DI) in micrometers. The MLI was measured by dividing the length of a line drawn across the lung section by a total number of intercepts counted within this line at 100× magnification. A total of 36 lines per mouse lung were drawn and measured. The DI was calculated by dividing the defined destructive alveoli by the total number of alveoli counted. Destructive alveolus was defined if at least one of the following alveoli was observed: alveolar wall defects, intraluminal parenchymal rags in alveolar ducts, obviously abnormal morphology, and typically emphysematous changes. Analysis was performed using a microscopic point-count technique at 200× magnification.19 Ten randomly selected fields per slice were photographed in a blinded manner. Airways and vascular structures were eliminated from the analysis.
Apoptosis assay
Terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) was performed to label the DNA-damaged cells in the lungs of experimental mice using an in situ cell-death-detection kit (Hoffman-La Roche, Basel, Switzerland) following the manufacturer’s instructions. The apoptotic index (AI) was calculated as the percentage of TUNEL-positive nuclei in a total of more than 3,000 nuclei randomly counted for each lung at 400× magnification.
Western blotting
Briefly, EPCs were washed three times with ice-cold PBS, then lysed in radioimmunoprecipitation-assay lysate (Applygen Technologies Beijing, China) for 30 minutes on ice. Lung tissues were homogenized manually in a glass homogenizer and lysed in radioimmunoprecipitation-assay lysate for 30 minutes on ice. Solutions of EPCs or lung tissue were centrifuged at 4°C, 12,000 g for 5 minutes. A BCA protein-quantification kit (Wellbio, Changsha, China) was used for protein measurement. Protein (30–60 μg) was mixed 1:1 with 2× sodium dodecyl sulfate (SDS) loading buffer (20% glycerol, 4% SDS, 3.12% dithiothreitol DDT, 0.2% bromophenol blue, and 0.1 mol/L Tris HCl, pH 6.8; all Sigma-Aldrich) and incubated at 100°C for 4 minutes. Equal amounts of protein for each sample were separated by 10%–12% SDS–polyacrylamide gel run at 120 V for 90 minutes and blotted onto a polyvinylidene difluoride microporous membrane (EMD Millipore, Billerica, MA, USA). Membranes were incubated with a 1:200 dilution of primary antibody (mouse monoclonal antibody; Santa Cruz Biotechnology, Dallas, TX, USA) overnight, then washed for three times with Tris-buffered-saline with Tween (TBS-T) and revealed using secondary antimouse antibody with horseradish peroxidase conjugate (1:3,000, 1 hour), followed by washing with TBS-T again. Immunoreactive bands were developed using enhanced chemiluminescence substrate (Thermo Fisher Scientific).
RNA extraction and quantitative RT-PCR
p16Ink4a mRNA expression in bone marrow-derived EPCs and lung tissue was detected by quantitative reverse-transcription polymerase chain reaction (RT-PCR). Total RNA was extracted from cells or tissues using Trizol reagent (Thermo Fisher Scientific). First-strand cDNA was synthesized using a RevertAid first-strand cDNA-synthesis kit (Thermo Fisher Scientific) according to the manufacturer’s instructions, and used as the template for quantitative RT-PCR analysis. DNase-treated samples were subjected to RT-PCR using SYBR Green quantitative PCR master mix (Thermo Fisher Scientific) on a CFX96 real-time system (Bio-Rad Laboratories, Hercules, CA, USA), with β-actin used as an internal control. The PCR-amplification conditions were 10 minutes at 95°C followed by 40 cycles of denaturation at 95°C for 15 seconds and annealing and extension for 1 minute at 60°C. Data were analyzed using comparative Ct.25 The relative expression level of p16Ink4a was calculated by determining the ratio of p16Ink4a to that of the internal control. Melting-curve analysis (65°C–95°C) was used to determine melting temperatures of specific amplification products and primer dimmers. Each experiment was repeated twice in triplicate. Primer sequences were: β-actin, 5′-CATCCTGCGTCTGGACCTGG-3′ (forward), 5′-TAATGTCACGCACGATTTCC-3′ (reverse); p16Ink4a, 5′-CCGCCTCAGCCCGCCTTTTT-3′ (forward), 5′-CCGCCGCCTTCGCTCAGTTT-3′ (reverse).
Statistical analysis
Analyses were performed using SPSS for Windows 16.0 (SPSS, Chicago, IL, USA). All data are expressed as means ± SD. Analyses of differences among groups were performed using one-way analysis of variance, followed by post hoc analysis as appropriate. Values of P<0.05 were considered statistically significant.
Results
Culture and identification of EPCs
On day 1 of the culture, MNCs isolated from the murine bone marrow formed circularly, sizes of cells were almost uniform, and cells were suspended in culture media (Figure 1A). On day 4 of the culture, the cells were attached to one another and getting larger, and shapes became oval, spindle, or polygonal. The cells at this stage tended to gather to form ball-like structures (Figure 1B). On culture day 7, cells shaped into fusiform or polygon patterns and contacted one another to attempt to form capillary structures (Figure 1C). Cell shapes at this stage displayed well in the culture medium. In addition, laser-scanning confocal microscopy illustrated that cells on culture day 7 displayed red cytoplasm when stained with Dil-acLDL (Figure 2A), green cytomembrane when combined with FITC-UEA1 (Figure 2B), and orange confocal when double-positively stained with Dil-acLDL and FITC-UEA1 (Figure 2C). The positive rate of amphophilic cells was 95.25%±3.61% on culture day 7.
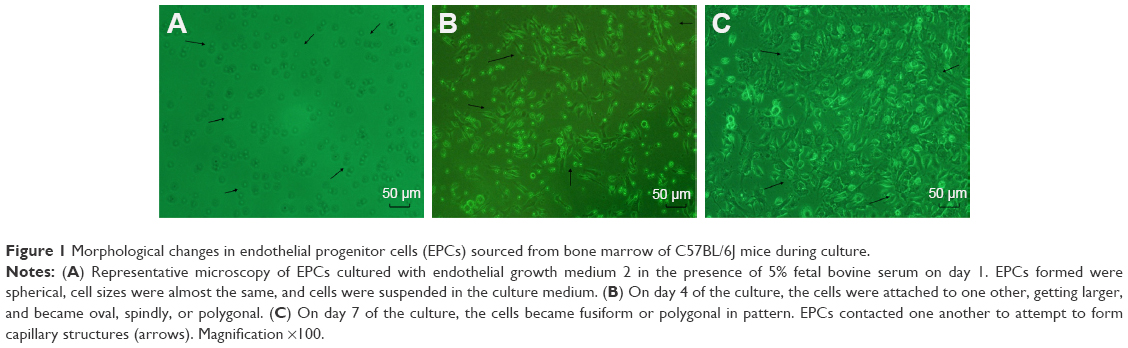 | Figure 1 Morphological changes in endothelial progenitor cells (EPCs) sourced from bone marrow of C57BL/6J mice during culture. |
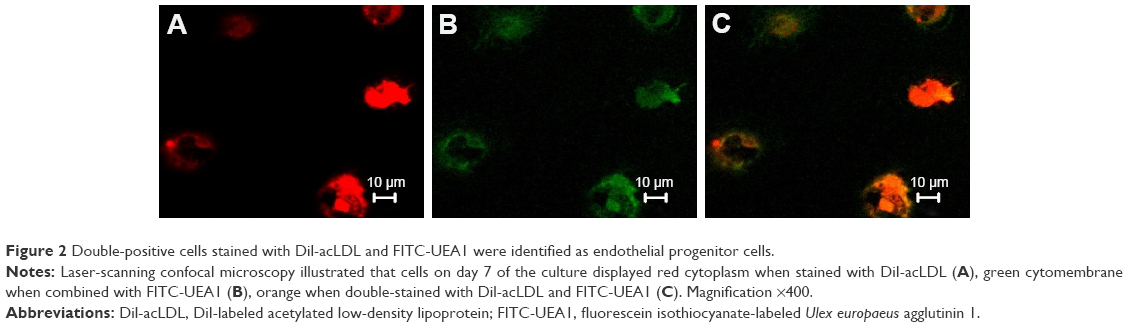 | Figure 2 Double-positive cells stained with Dil-acLDL and FITC-UEA1 were identified as endothelial progenitor cells. |
Lung-function test
As shown in Figure 3, the maximal expiratory flow-volume curve of the CSE group (Figure 3B) and CSE + 5-Aza-CdR group (Figure 3C) showed abrupt ascents, and descending limbs showed a prolonged expiratory phase compared with that in controls (Figure 3A). Cdyn (mL/cmH2O) was significantly lower in the CSE group (0.57±0.15, P<0.01) and CSE + 5-Aza-CdR group (0.67±0.19, P<0.05) than controls (1.03±0.29). Raw (cmH2O/mL/min) was significantly higher in the CSE group (2.49±0.52, P<0.01) and CSE + 5-Aza-CdR group (1.91±0.47, P<0.01) than controls (0.58±0.14). PEF (mL/second) was significantly lower in the CSE group (2.91±0.5, P<0.01) and CSE + 5-Aza-CdR group (3.24±0.62, P<0.05) than controls (4.4±0.74). The Ti/Te was significantly lower in the CSE group (0.63±0.17) than controls (0.89±0.17, P<0.05). There was no significant difference in Ti/Te between the CSE + 5-Aza-CdR group (0.7±0.15) and controls (P>0.05). There was no significant difference between the CSE group and CSE + 5-Aza-CdR group in terms of the parameters described (P>0.05, Figure 4).
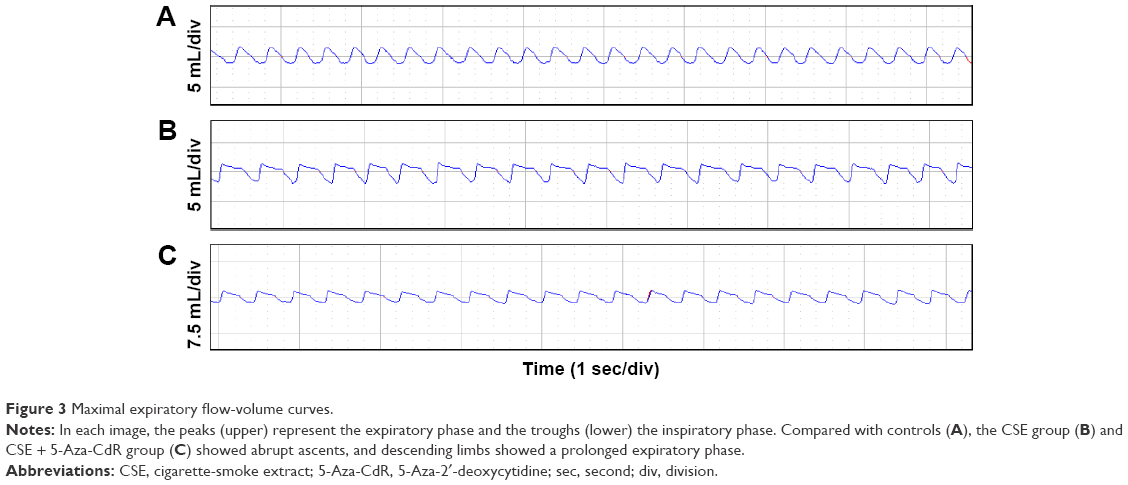 | Figure 3 Maximal expiratory flow-volume curves. |
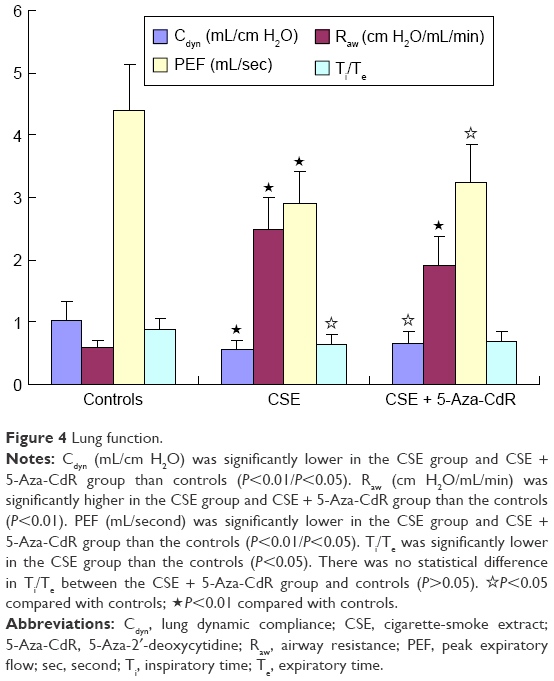 | Figure 4 Lung function. |
Histomorphological changes in lung tissue
As shown in Figure 5, lung tissue of the CSE group exhibited enlarged alveolar space, thinner alveolar septum, and destroyed alveolar wall. Lung tissue of the CSE + 5-Aza-CdR group also exhibited enlarged alveolar space, but less than the CSE group. The changes described were manifested in the MLI and DI (Figure 6). The MLI of the CSE group (67.63±9.87 μm) was significantly increased when compared with controls (29.2±4.64 μm, P<0.01). Interestingly, the MLI of the CSE + 5-Aza-CdR group (52.7±6.34 μm) was significantly smaller than the CSE group (P<0.01), though larger than controls (P<0.01). Similarly, the DI of the CSE group (42.41%±5.86%) was significantly increased when compared with controls (6.38%±1.57%, P<0.01). The DI of the CSE + 5-Aza-CdR group (33.26%±5.03%) was significantly less than the CSE group (P<0.05), though more than controls (P<0.01).
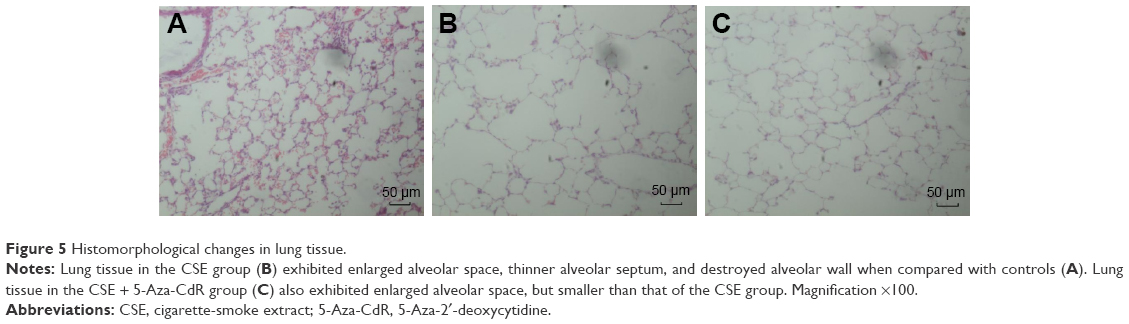 | Figure 5 Histomorphological changes in lung tissue. |
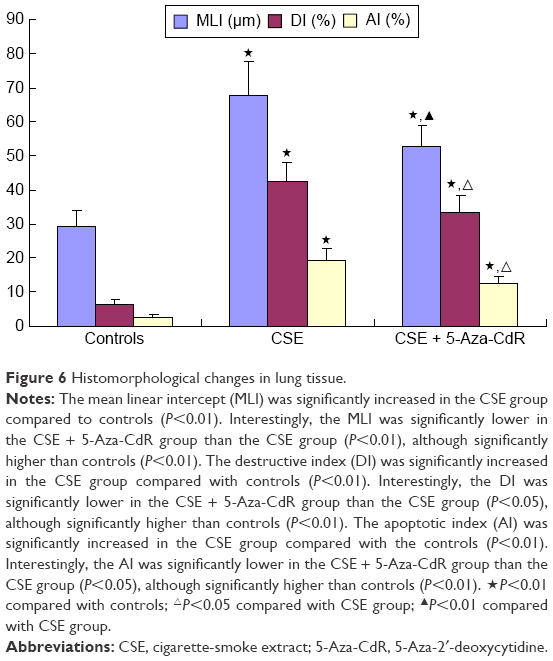 | Figure 6 Histomorphological changes in lung tissue. |
Apoptosis in lung tissue
As shown in Figure 7, numbers of apoptotic cells in alveolar septa in the CSE and CSE + 5-Aza-CdR groups were significantly increased comparing with controls. Quantitatively, the AI of the CSE group (19.5%±3.16%) was significantly increased when compared with controls (2.75%±0.46%, P<0.01). Interestingly, the AI of the CSE + 5-Aza-CdR group (12.75%±1.67%) was significantly lower than the CSE group (P<0.05), though higher than controls (P<0.01, Figure 6).
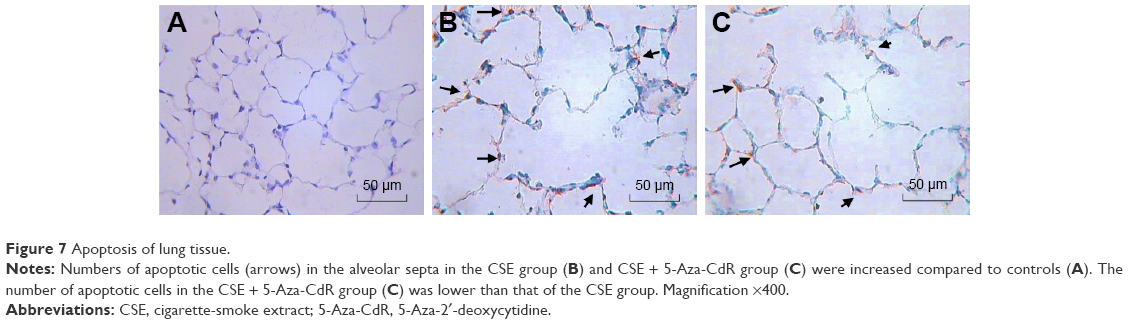 | Figure 7 Apoptosis of lung tissue. |
Expression of p16Ink4a protein in lung tissue and EPCs
As shown in Figure 8A and C, p16Ink4a/β-actin in lung tissue was significantly increased in the CSE group (0.59±0.05, P<0.01) and CSE + 5-Aza-CdR group (0.46±0.03, P<0.01) compared with controls (0.32±0.02). Interestingly, p16Ink4a/β-actin in lung tissue was significantly lower in the CSE + 5-Aza-CdR group than the CSE group (P<0.01).
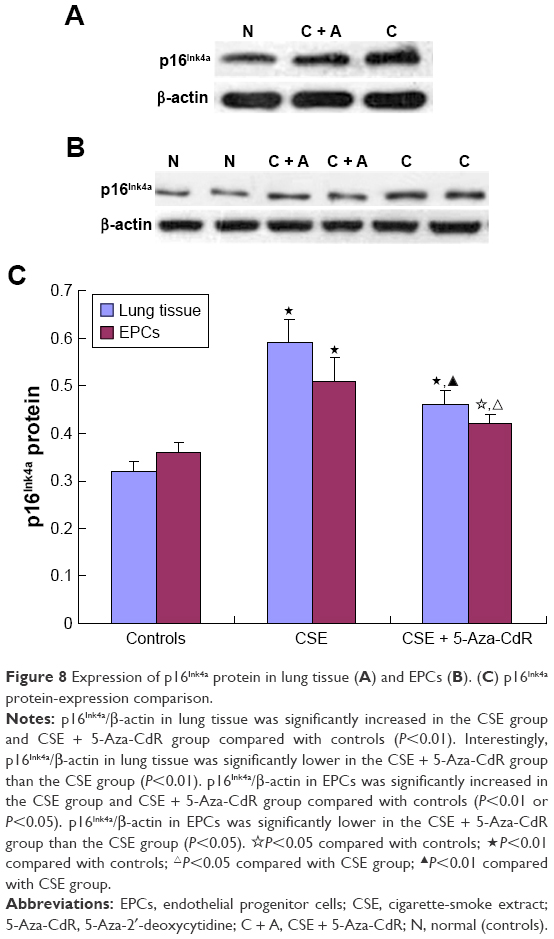 | Figure 8 Expression of p16Ink4a protein in lung tissue (A) and EPCs (B). (C) p16Ink4a protein-expression comparison. |
As shown in Figure 8B and C, p16Ink4a/β-actin in EPCs was significantly increased in the CSE group (0.51±0.05, P<0.01) and CSE + 5-Aza-CdR group (0.42±0.02, P<0.05) compared with controls (0.36±0.02). Interestingly, p16Ink4a/β-actin in EPCs was significantly lower in the CSE + 5-Aza-CdR group than the CSE group (P<0.05).
Expression of p16Ink4a mRNA in lung tissue and bone marrow-derived EPCs
As shown in Figure 9, p16Ink4a mRNA in lung tissue was significantly increased in the CSE group (5.24±0.67, P<0.01) and CSE + 5-Aza-CdR group (3.82±0.44, P<0.01) compared with controls (1±0.12). Interestingly, p16Ink4a mRNA in lung tissue was significantly lower in the CSE + 5-Aza-CdR group than the CSE group (P<0.05). p16Ink4a mRNA in EPCs was significantly increased in the CSE group (4.4±0.6, P<0.01) and CSE + 5-Aza-CdR group (1.99±0.25, P<0.05) compared with controls (1.01±0.13). p16Ink4a mRNA in EPCs was significantly lower of CSE + 5-Aza-CdR group than the CSE group (P<0.01).
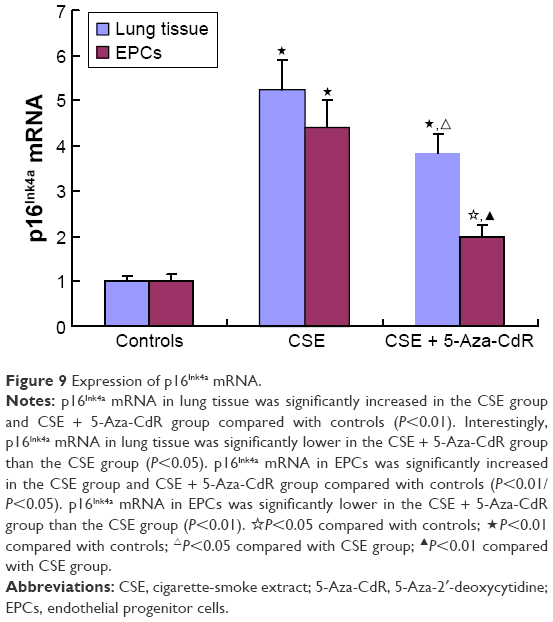 | Figure 9 Expression of p16Ink4a mRNA. |
Discussion
The present study showed that the expression of p16Ink4a in lung tissue and bone marrow-derived EPCs was increased in mice with CSE-induced emphysema, which suggested that CSE might induce p16Ink4a expression, resulting in EPC senescence that contributes to emphysema with overexpression of p16Ink4a in lung tissue of mice with emphysema. Most importantly, the present study demonstrated for the first time that 5-Aza-CdR can partly protect against emphysema in the mouse model induced by CSE, especially in the profile of lung morphology via suppressing expression of p16Ink4a in EPCs and lung tissue.
Cigarette smoking is by far the most critical risk factor for emphysema and COPD. CS induces significant increases in reactive oxygen species generation.26 CSE contains most of the compounds inhaled by cigarette smokers, and is usually used as a surrogate for CS.27 CSE directly induces inflammatory cytokines28,29 and superoxide generation,30 resulting in increased p16Ink4a expression that induces fibroblast senescence.31
There was evidence showing that expression of p16Ink4a in aged cells may be ten times more than in young cells. Inserting p16Ink4a cDNA into normal fibroblasts slowed cell growth, aggravated nonenzymatic glycosylation, increased senescence-associated β-galactosidase positivity, and shortened telomeres. On the other hand, significant delay of several senescent features was observed in fibroblasts, and the life span of fibroblasts was significantly extended by inserting antisense p16,Ink4a but the onset of replicative senescence could not be totally prevented.14 Therefore, p16Ink4a could be regarded as a major dominant senescence gene. p16Ink4a levels are increased in pulmonary vascular ECs in patients with COPD.32 A recent study showed that cord-blood EPCs in premature neonates exhibited overexpression of p16Ink4a, contributing to accelerated senescence of EPCs.33
The present study showed overexpression of p16Ink4a in both bone marrow-derived EPCs and lung tissue of emphysematous mice induced by CSE. There is a growing realization that COPD, or at least emphysema, involves several processes present in aging and cellular senescence. Aging and cellular senescence underlie loss-of-function diseases.34–36 Aging-associated inflammation/structural change is the result of failure of reactive oxygen species elimination, failure of repair of damaged DNA, and telomere shortening.1 Meanwhile, biological aging may occur before age-related aging, and is considered related to chronic inflammation, as evidenced in IL6, IL1β, and TNFα levels.37 Our previous study showed that IL6 in sputum from COPD patients was increased.38 On the other hand, cellular senescence is believed to induce inflammation by producing various inflammatory cytokines in tissue.36 Premature pulmonary vascular EC senescence is a major process perpetuating lung inflammation in COPD.32 CSE-induced apoptosis of pulmonary ECs exerts a direct effect on pulmonary vascular structure.39 EPCs are the precursors of ECs. Accumulating evidence indicates that EPCs derived from bone marrow contribute to “reendothelialization” of injured vessels, as well as neovascularization of ischemic lesions in either a direct or an indirect way under physiological and pathological conditions.40,41 EPCs have been demonstrated to be required for tissue repair and airway remodeling in lungs.8–10 EPC function is decreased after chronic stimulation by CSE.42 Peripheral infusion of rat bone marrow-derived EPCs is helpful in vascular repair and damage-healing processes by homing in on impaired locations.43 Autologous transplantation of circulating EPCs effectively attenuates acute lung injury by direct endothelial repair and indirect immunomodulation.44 Our study suggests that CSE can induce p16Ink4a expression, resulting in EPC senescence and contributing to emphysema and direct p16Ink4a expression in lung tissue.
Aging lungs exhibit both structural and functional alterations.1 The leading clinical symptom of COPD or emphysema is chronic airflow limitation, which means decreased lung function. In the present study, airflow limitation was detected in CSE-induced emphysematous mice and manifested by decreases in Cdyn, Raw, PEF, and Ti/Te. Lung tissue in emphysema mice showed enlarged alveolar space, thinner alveolar septum, and destroyed alveolar wall, manifested in increased MLI and DI. Alveolar septal cell apoptosis plays an important role in the development of emphysema.45,46 Oxidative stress also triggers apoptosis.47 In the present study, the AI of lung tissue, which reflects the apoptosis status of lung parenchyma, from emphysematous mice was increased compared with control mice.
In COPD, oxidative stress induced by cigarette smoking further damages the lung, leading to acquired genetic changes, including DNA methylation, due to inefficient DNA-repair machinery.48 DNA methylation is catalyzed by the DNA methyltransferase (DNMT) family,49,50 and plays an important role in maintaining cell identity by affecting gene expression. 5-Aza-CdR, a DNMT inhibitor, inhibits DNMT and demethylates DNA by incorporation into DNA,51 degradation of DNMT,52 downregulation of DNMT mRNA and protein levels,53,54 or repression of DNMT enzymatic activity,55 leading to changes in gene reactivation.
Typically, methylation in the promoter region of a gene is associated with repression. A high frequency of aberrant methylation of the gene for p16Ink4a has been shown in cases of non-small-cell lung cancer,56 heavy smokers, and advanced and poorly differentiated small adecocarcinoma.57 Breuer et al showed that loss of p16Ink4a expression by promoter hypermethylation is inconsistent and occurs late in the carcinogenic process at the level of severe dysplasia.58 Another intriguing observation is that DNA-methylation levels within gene bodies are also dynamic in relation to gene expression.59 In the present study, increased p16Ink4a expression in CSE-induced emphysematous mice was partly suppressed by 5-Aza-CdR. Lung morphological changes and apoptosis in emphysematous mice induced by CSE were also partly reversed by 5-Aza-CdR. The molecular mechanism of active demethylation in mammalian cells is not well understood, but seems to be linked to DNA-repair machinery.60 We noticed that in this study, there was no statistical difference in lung function between the CSE group and CSE + 5-Aza-CdR group, despite little change in the numbers. The possible reason may lie in lung-function tests being less sensitive than morphometry.61
Since methylation is reversible, it is an interesting target for intervention with specific inhibitors of DNA methylation. The antitumor effect or auxiliary-therapy effect of 5-Aza-CdR has been investigated and confirmed by many studies.62–65 It could be assumed that 5-Aza-CdR at lower concentrations might be applied in the attenuation of emphysema.
In summary, the present study indicated that p16Ink4a expression was increased in EPCs and lung tissue in CSE-induced emphysematous mice, and contributed to alterations in lung function, histomorphological changes, and apoptosis in emphysematous lung tissue. 5-Aza-CdR partly reversed the structural emphysematous outcomes resulted from CSE stimulation, which in turn suggested that DNA methylation may be involved in the pathogenesis of emphysema with regard to epigenetic modifications in terms of hypermethylation of genes and EPC senescence. DNA methyltransferase inhibitors might help potentially in clinical treatment of emphysema. Future study is expected to elucidate the exact mechanism of regulation of p16Ink4a on EPC senescence.
Acknowledgments
This study was supported by the Natural Science Foundation of China (81070039, 81400032) and Hunan province science and technology project (2015JC3033).
Disclosure
The authors report no conflicts of interest in this work.
References
Ito K, Barnes PJ. COPD as a disease of accelerated lung aging. Chest. 2009;135:173–180. | ||
Lee J, Sandford A, Man P, Sin DD. Is the aging process accelerated in chronic obstructive pulmonary disease? Curr Opin Pulm Med. 2011;17:90–97. | ||
Yao HW, Chung SW, Hwang JW, et al. Sirt1 protects against emphysema via FoxO3-mediated reduction of premature senescence in mice. J Clin Invest. 2012;122:2032–2045. | ||
Liu HJ, Fergusson MM, Castilho RM, et al. Augmented Wnt signaling in a mammalian model of accelerated aging. Science. 2007;317:803–806. | ||
Kopp HG, Ramos CA, Rafii S. Contribution of endothelial progenitors and proangiogenic hematopoietic cells to vascularization of tumor and ischemic tissue. Curr Opin Hematol. 2006;13:175–181. | ||
Krenning G, van Luyn MJ, Harmsen MC. Endothelial progenitor cell-based neovascularization: implications for therapy. Trends Mol Med. 2009;15:180–189. | ||
Yang Z, von Ballmoos MW, Faessler D, et al. Paracrine factors secreted by endothelial progenitor cells prevent oxidative stress-induced apoptosis of mature endothelial cells. Atherosclerosis. 2010;211: 103–109. | ||
Santos S, Peinado VI, Ramirez J, et al. Characterization of pulmonary vascular remodelling in smokers and patients with mild COPD. Eur Respir J. 2002;19:632–638. | ||
Loebinger MR, Aguilar S, Janes SM. Therapeutic potential of stem cells in lung disease: progress and pitfalls. Clin Sci (Lond). 2008;114:99–108. | ||
Critser PJ, Yoder MC. Endothelial colony-forming cell role in neoangiogenesis and tissue repair. Curr Opin Organ Transplant. 2010;15:68–72. | ||
Yang Y, Gan Y, Cao J, et al. Decreased and dysfunctional circulating endothelial progenitor cells in patients with chronic obstructive pulmonary disease. Chin Med J (Engl). 2013;126:3222–3227. | ||
Kamb A, Gruis NA, Weaver-Feldhaus J, et al. A cell cycle regulator potentially involved in genesis of many tumor types. Science. 1994;264:436–440. | ||
Stein GH, Drullinger LF, Soulard A, Dulić V. Differential roles for cyclin-dependent kinase inhibitors p21 and p16 in the mechanisms of senescence and differentiation in human fibroblasts. Mol Cell Biol. 1999;19:2109–2117. | ||
Duan J, Zhang Z, Tong T. Senescence delay of human diploid fibroblast induced by anti-sense p16Ink4a expression. J Biol Chem. 2001;276:48325–48331. | ||
Aoshiba K, Zhou F, Tsuji T, Nagai A. DNA damage as a molecular link in the pathogenesis of COPD in smokers. Eur Respir J. 2012;39:1368–1376. | ||
Soria JC, Rodriguez M, Liu DD, Lee JJ, Hong WK, Mao L. Aberrant promoter methylation of multiple genes in bronchial brush samples from former cigarette smokers. Cancer Res. 2002;62:351–355. | ||
Kikuchi S, Yamada D, Fukami T, et al. Hypermethylation of the TSLC1/IGSF4 promoter is associated with tobacco smoking and a poor prognosis in primary nonsmall cell lung carcinoma. Cancer. 2006;106:1751–1758. | ||
Plimack ER, Kantarjian HM, Issa JP. Decitabine and its role in the treatment of hematopoietic malignancies. Leuk Lymphoma. 2007;48:1472–1481. | ||
Chen Y, Hanaoka M, Chen P, Droma Y, Voelkel NF, Kubo K. Protective effect of beraprost sodium, a stable prostacyclin analog, in the development of cigarette smoke extract-induced emphysema. Am J Physiol Lung Cell Mol Physiol. 2009;296:L648–L656. | ||
Zhang Y, Cao J, Chen Y, Chen P, Peng H, et al. Intraperitoneal injection of cigarette smoke extract induced emphysema, and injury of cardiac and skeletal muscles in BALB/c mice. Exp Lung Res. 2013;39:18–31. | ||
Rafat N, Hanusch C, Brinkkoetter PT, et al. Increased circulating endothelial progenitor cells in septic patients: correlation with survival. Crit Care Med. 2007;35:1677–1684. | ||
Purhonen S, Palm J, Rossi D, et al. Bone marrow-derived circulating endothelial precursors do not contribute to vascular endothelium and are not needed for tumor growth. Proc Natl Acad Sci U S A. 2008;105:6620–6625. | ||
Bartsch T, Brehm M, Zeus T, Kögler G, Wernet P, Strauer BE. Transplantation of autologous mononuclear bone marrow stem cells in patients with peripheral arterial disease (the TAM-PAD study). Clin Res Cardiol. 2007;96:891–899. | ||
Chen JZ, Zhu JH, Wang XX, Zhu JH, Xie XD, et al. Effects of homocysteine on number and activity of endothelial progenitor cells from peripheral blood. J Mol Cell Cardiol. 2004;36:233–239. | ||
Kojima K, Ohhashi R, Fujita Y, et al. A role for Sirt1 in cell growth and chemoresistance in prostate cancer PC3 and DU145 cells. Biochem Biophys Res Commun. 2008;373:423–428. | ||
Jeong YY, Park HJ, Cho YW, et al. Aged red garlic extract reduces cigarette smoke extract-induced cell death in human bronchial smooth muscle cells by increasing intracellular glutathione levels. Phytother Res. 2012;26:18–25. | ||
DeMarini DM. Genotoxicity of tobacco smoke and tobacco smoke condensate: a review. Mutat Res. 2004;567:447–474. | ||
Jeong SH, Park JH, Kim JN, et al. Up-regulation of TNF-alpha secretion by cigarette smoke is mediated by Egr-1 in HaCaT human keratinocytes. Exp Dermatol. 2010;19:e206–e212. | ||
Preciado D, Kuo E, Ashktorab S, Manes P, Rose M. Cigarette smoke activates NFκB-mediated TNF-α release from mouse middle ear cells. Laryngoscope. 2010;120:2508–2515. | ||
Matthews JB, Chen FM, Milward MR, Ling MR, Chapple IL. Neutrophil superoxide production in the presence of cigarette smoke extract, nicotine and cotinine. J Clin Periodontol. 2012;39:626–634. | ||
Nyunoya T, Monick MM, Klingelhutz A, Yarovinsky TO, Cagley JR, Hunninghake GW. Cigarette smoke induces cellular senescence. Am J Respir Cell Mol Biol. 2006;35:681–688. | ||
Amsellem V, Gary-Bobo G, Marcos E, et al. Telomere dysfunction causes sustained inflammation in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2011;184:1358–1366. | ||
Vassallo PF, Simoncini S, Ligi I, Chateau AL, Bachelier R, et al. Accelerated senescence of cord blood endothelial progenitor cells in premature neonates is driven by Sirt1 decreased expression. Blood. 2014;123:2116–2126. | ||
MacNee W. Aging, inflammation, and emphysema. Am J Respir Crit Care Med. 2011;184:1327–1329. | ||
Tuder RM, Kern JA, Miller YE. Senescence in chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2012;9:62–63. | ||
Aoshiba K, Nagai A. Senescence hypothesis for the pathogenetic mechanism of chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2009;6:596–601. | ||
Johnson TE. Recent results: biomarkers of aging. Exp Gerontol. 2006;41:1243–1246. | ||
He Z, Chen Y, Chen P, Wu G, Cai S. Local inflammation occurs before systemic inflammation in patients with COPD. Respirology. 2010;15:478–484. | ||
Nana-Sinkam SP, Lee JD, Sotto-Santiago S, Stearman RS, Keith RL, et al. Prostacyclin prevents pulmonary endothelial cell apoptosis induced by cigarette smoke. Am J Respir Crit Care Med. 2007;175:676–685. | ||
Zampetaki A, Kirton JP, Xu Q. Vascular repair by endothelial progenitor cells. Cardiovasc Res. 2008;78:413–421. | ||
Devanesan AJ, Laughlan KA, Girn HR, Homer-Vanniasinkam S. Endothelial progenitor cells as a therapeutic option in peripheral arterial disease. Eur J Vasc Endovasc Surg. 2009;38:475–481. | ||
He ZH, Chen P, Chen Y, Zhu YQ, He SD, et al. Dual effects of cigarette smoke extract on proliferation of endothelial progenitor cells and the protective effect of 5-aza-2′-deoxycytidine on EPCs against the damage caused by CSE. Biomed Res Int. 2014;2014:640752. | ||
Kähler CM, Wechselberger J, Hilbe W, Gschwendtner A, Colleselli D, et al. Peripheral infusion of rat bone marrow derived endothelial progenitor cells leads to homing in acute lung injury. Respir Res. 2007;8:50. | ||
Cao JP, He XY, Xu HT, Zou Z, Shi XY. Autologous transplantation of peripheral blood-derived circulating endothelial progenitor cells attenuates endotoxin-induced acute lung injury in rabbits by direct endothelial repair and indirect immunomodulation. Anesthesiology. 2012;116:1278–1287. | ||
Yang Q, Underwood MJ, Hsin MK, Liu XC, He GW. Dysfunction of pulmonary vascular endothelium in chronic obstructive pulmonary disease: basic considerations for future drug development. Curr Drug Metab. 2008;9:661–667. | ||
Demedts IK, Demoor T, Bracke KR, Joos GF, Brusselle GG. Role of apoptosis in the pathogenesis of COPD and pulmonary emphysema. Respir Res. 2006;7:53. | ||
Pierrou S, Broberg P, O’Donnell RA, et al. Expression of genes involved in oxidative stress responses in airway epithelial cells of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2007;175:577–586. | ||
Tzortzaki EG, Papi A, Neofytou E, Soulitzis N, Siafakas NM. Immune and genetic mechanisms in COPD: possible targets for therapeutic interventions. Curr Drug Targets. 2013;14:141–148. | ||
Ohtani K, Vlachojannis GJ, Koyanagi M, Boeckel JN, Urbich C, et al. Epigenetic regulation of endothelial lineage committed genes in pro-angiogenic hematopoietic and endothelial progenitor cells. Circ Res. 2011;109:1219–1229. | ||
Garcia-Dominguez P, dell’Aversana C, Alvarez R, Altucci L, de Lera AR. Synthetic approaches to DNMT inhibitor SGI-1027 and effects on the U937 leukemia cell line. Bioorg Med Chem Lett. 2013;23:1631–1635. | ||
Jüttermann R, Li E, Jaenisch R. Toxicity of 5-aza-2′-deoxycytidine to mammalian cells is mediated primarily by covalent trapping of DNA methyltransferase rather than DNA demethylation. Proc Natl Acad Sci U S A. 1994;91:11797–11801. | ||
Ghoshal K, Datta J, Majumder S, Bai S, Kutay H, et al. 5-Aza-deoxycytidine induces selective degradation of DNA methyltransferase 1 by a proteasomal pathway that requires the KEN box, bromo-adjacent homology domain, and nuclear localization signal. Mol Cell Biol. 2005;25:4727–4741. | ||
Deng T, Zhang Y. 5-Aza-2′-deoxycytidine reactivates expression of RUNX3 by deletion of DNA methyltransferases leading to caspase independent apoptosis in colorectal cancer Lovo cells. Biomed Pharmacother. 2009;63:492–500. | ||
Yu JN, Xue CY, Wang XG, et al. 5-Aza-2′-deoxycytidine (5-aza-CdR) leads to down-regulation of Dnmt1o and gene expression in preimplantation mouse embryos. Zygote. 2009;17:137–145. | ||
Benbrahim-Tallaa L, Waterland RA, Dill AL, Webber MM, Waalkes MP. Tumor suppressor gene inactivation during cadmium-induced malignant transformation of human prostate cells correlates with overexpression of de novo DNA methyltransferase. Environ Health Perspect. 2007;115:1454–1459. | ||
Ota N, Kawakami K, Okuda T, Takehara A, Hiranuma C, et al. Prognostic significance of p16Ink4a hypermethylation in non-small cell lung cancer is evident by quantitative DNA methylation analysis. Anticancer Res. 2006;26:3729–3732. | ||
Wang D, Wang J, Li Y, He Z, Zhang Y. The influence of anthracosis and p16 Ink4a gene aberrant methylation on small-sized pulmonary adenocarcinoma. Exp Mol Pathol. 2011;90:131–136. | ||
Breuer RH, Snijders PJ, Sutedja GT, et al. Expression of the p16Ink4a gene product, methylation of the p16Ink4a promoter region and expression of the polycomb-group gene BMI-1 in squamous cell lung carcinoma and premalignant endobronchial lesions. Lung Cancer. 2005;48:299–306. | ||
Auclair G, Weber M. Mechanisms of DNA methylation and demethylation in mammals. Biochimie. 2012;94:2202–2211. | ||
Niehrs C. Active DNA demethylation and DNA repair. Differentiation. 2009;77:1–11. | ||
Wright JL, Cosio M, Churg A. Animal models of chronic obstructive pulmonary disease. Am J Physiol Lung Cell Mol Physiol. 2008;295:L1–L15. | ||
Joeckel TE, Lubbert M. Clinical results with the DNA hypomethylating agent 5-aza-2′-deoxycytidine (decitabine) in patients with myelodysplastic syndromes: an update. Semin Hematol. 2012;49: 330–341. | ||
Chu BF, Karpenko MJ, Liu Z, et al. Phase I study of 5-aza-2′-deoxycytidine in combination with valproic acid in non-small-cell lung cancer. Cancer Chemother Pharmacol. 2013;71:115–121. | ||
Wang L, Zhang Y, Li R, et al. 5-Aza-2′-deoxycytidine enhances the radiosensitivity of breast cancer cells. Cancer Biother Radiopharm. 2013;28:34–44. | ||
Tao SF, Zhang CS, Guo XL, et al. Anti-tumor effect of 5-aza-2′-deoxycytidine by inhibiting telomerase activity in hepatocellular carcinoma cells. World J Gastroenterol. 2012;18:2334–2343. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.