")
Back to Journals » Drug Design, Development and Therapy » Volume 13
15-Deoxy-Δ12,14-prostaglandin J2 as a potential regulator of bone metabolism via PPARγ-dependent and independent pathways: a review
Authors Xiong Z , Luo P , Zhou J , Tan M
Received 25 February 2019
Accepted for publication 10 May 2019
Published 30 May 2019 Volume 2019:13 Pages 1879—1888
DOI https://doi.org/10.2147/DDDT.S206695
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Manfred Ogris
Zhencheng Xiong,1,2 Pan Luo,1 Jun Zhou,2,3 Mingsheng Tan1–3
1Graduate School of Peking Union Medical College, Chinese Academy of Medical Sciences, Beijing, People’s Republic of China; 2Department of Spine Surgery, China-Japan Friendship Hospital, Beijing, People’s Republic of China; 3School of Clinical Medicine, Graduate School of Beijing University of Chinese Medicine, Beijing, People’s Republic of China
Abstract: Bone metabolism is a complex physiological process that primarily involves osteoblast-mediated bone formation and osteoclast-mediated bone resorption, both of which are regulated by a variety of biological factors. There is increasing evidence that peroxisome proliferator-activated receptor γ (PPARγ) is a member of the nuclear receptor superfamily and plays an important role in lipid metabolism and bone metabolism. Through the PPARγ-dependent pathway, 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) promotes the formation of marrow adipocytes and inhibits the formation of osteoblasts, resulting in bone loss and increasing the risk of fracture and osteoporosis. Recent studies have found that through the PPARγ-independent pathway, 15d-PGJ2 plays a regulatory role in bone metastasis of breast cancer, which can inhibit osteoclastogenesis and reduce bone destruction. The purpose of our review is to summarize the recent progress in elucidating the mechanisms and effects of 15d-PGJ2 in bone metabolism, which can serve as a novel therapeutic target for bone tumors, osteoporosis, rheumatoid arthritis (RA), and other bone diseases.
Keywords: bone metabolism, osteoblast, adipogenesis, osteoporosis, rheumatoid arthritis, bone metastasis
Introduction
Bone is a dynamically changing tissue that constantly adapts to vertebrate life to maintain bone shape, size, and structural integrity, and bone regulates mineral homeostasis in the body.1 Bone formation and bone resorption are mediated by osteoblasts and osteoclasts, respectively, which are the two main processes of bone metabolism.2 Formation and resorption are tightly coupled under physiological conditions to maintain bone mass. In the pathological process of osteoporosis, bone resorption exceeds bone formation and can cause an imbalance in bone metabolism and a net loss of bone.3 The bone marrow stroma includes mesenchymal stem cells (MSCs) and hematopoietic stem cells (HSCs), which can differentiate into a variety of cell types common to bone.4 Two main types of cells in bone have different developmental origins: osteoblasts are derived from mesenchymal lineage,5 and osteoclasts are derived from hematopoietic lineage.6 Many biological factors regulate osteoblast differentiation and osteoclast differentiation, thus affecting bone formation and bone resorption and playing a role in bone metabolism.
Prostaglandins (PGs) are a class of bioactive compounds produced by arachidonic acid (AA) that have a variety of regulatory functions in the human body.7 Different types of PGs have complex functions in different target cells.8 15d-PGJ2 is an endogenous ligand for PPARγ, which is produced by the cyclooxygenase (COX)-mediated AA metabolism pathway.8–10 Studies have shown that ligand-activated PPARγ alters the fate of bone marrow MSCs by promoting the differentiation of adipocytes and inhibiting the differentiation of osteoblasts.11–13 Numerous studies have confirmed that 15d-PGJ2 promotes the differentiation of adipocytes and inhibits the differentiation of osteoblasts by activating PPARγ, thereby inhibiting bone formation and causing bone loss.9,10 Recently, studies have shown that through a PPARγ-independent pathway, 15d-PGJ2 inhibits bone destruction caused by bone metastasis in breast cancer by suppressing osteoclast differentiation and bone resorption.14 Treatment with 15d-PGJ2 also inhibits bone loss caused by estrogen deficiency.14 15d-PGJ2 may be a potential regulator of bone metabolism, and drugs targeting 15d-PGJ2 may offer new prospects for metabolic bone diseases. To further explore these prospects, this review was performed.
The biosynthesis and bioactivity of 15d-PGJ2
The biosynthesis of 15d-PGJ2 is based on the continuous action of several enzymes, the general pathway of which is illustrated in Figure 1.15 Under the action of enzyme phospholipase A2 (PLA2), AA is released by membrane phospholipids as the first step of this metabolic pathway.8,15 Under the action of COX-1 or COX-2, AA first produces PGG2 and then PGH2.8,16 PGH2 is an unstable intermediate that can be converted into a series of stable prostaglandins by their specific prostaglandin synthases, including PGD2, PGE2, PGF2α, PGI2, and thromboxane A2.16,17 PGD2 is synthesized under the action of prostaglandin D synthase (PTGDS, including H-PTGDS and L-PTGDS).18,19 Subsequently, PGD2 readily undergoes chemical dehydration, which in turn forms the cyclopentenone prostaglandin PGJ2.9,20 Both 15d-PGJ2 and Δ12-PGJ2 are produced by PGJ2, but the latter requires the participation of albumin.21
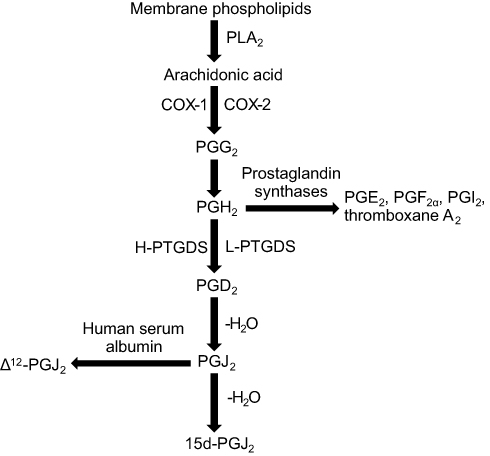 | Figure 1 Biosynthesis of 15d-PGJ2. In the first step, membrane phospholipids are catalyzed by the action of the PLA2 enzyme to release AA. In the second step, AA is sequentially metabolized to PGG2 and then to PGH2 by COX-1 or COX-2. Subsequently, PGD2, PGE2, PGF2α, PGI2, and thromboxane A2 were converted from PGH2 by their respective prostaglandin synthetase. The rate-limiting enzyme used to synthesize PGD2 is PTGDS, both H-PTGDS and L-PTGDS. PGD2 readily undergoes chemical dehydration, losing water to form the cyclopentenone prostaglandin PGJ2. In the final step, 15d-PGJ2 and Δ12-PGJ2 are produced from PGJ2 by albumin-independent and albumin-dependent reactions, respectively. Abbreviations: 15d-PGJ2, 15-deoxy-Δ12,14-prostaglandin J2; PLA2, phospholipase A2; AA, arachidonic acid; PG, prostaglandin; COX, cyclooxygenase; PTGDS, prostaglandin D synthase. |
Unlike other types of PGs, 15d-PGJ2 contains a cyclopentenone ring structure.8 The cyclopentenone ring has an electrophilic α, β-unsaturated ketone moiety that provides a unique bioactivity spectrum for 15d-PGJ2.22 In 1995, Forman and Tontonoz9 examined the ability of AA metabolites to act as ligands to activate PPARγ and identified 15d-PGJ2 as its natural ligand. Although 15d-PGJ2 has a lower affinity for PPARγ than steroid hormones for its homologous intracellular receptors, it is the highest affinity natural ligand of PPARγ identified to date.8 Due to its specific cyclopentenone ring structure and ligand activity that activates PPARγ, 15d-PGJ2 plays a regulatory role in many physiological and disease processes. Studies have shown that 15d-PGJ2 promotes apoptosis23,24 and exerts anti-inflammatory,16 anti-angiogenic,24,25 anti-metastatic,14 and even anticancer25–27 effects in animals.
15d-PGJ2 regulates adipocyte differentiation and osteoblast differentiation via a PPARγ-dependent pathway
MSCs in the bone marrow are capable of differentiating into a variety of cell types, such as osteoblasts, adipocytes, chondrocytes, fibroblasts, or myocytes.5 Each phenotypic transition requires a rigid, uninterrupted, and asynchronous program of gene expression.3 Osteoblasts are involved in the construction of the organic and inorganic components of bone.28 Recent gene deletion studies have shown that osteoblast differentiation requires a complex sequence of processes to activate transcription factors involved in osteoblastogenesis (including Wnt/β-catenin, Runt-related transcription factor 2 (Runx2), and Osterix) and to suppress transcription factors involved in adipogenesis (including PPARγ and CCAAT/enhancer binding protein (C/EBP)).11,29 The key factors in the process of MSC adipogenic and osteogenic lineage differentiation are PPARγ and Wnt, respectively.11
PPARγ, which acts as a nuclear receptor, contains a central DNA-binding domain and a C-terminal ligand-binding domain.30 Under the strict regulation of many transcription factors such as PPARγ and C/EBPα/β/δ, adipogenesis is achieved.31 Numerous studies have shown that after 15d-PGJ2 binds to PPARγ, transcription begins with the involvement of coregulators called corepressors and coactivators.9,10 When 15d-PGJ2 is absent, PPARγ forms a protein complex with corepressors such as silencing mediator for retinoid or thyroid-hormone receptors (SMRT), NCoR, and histone deacetylases, which transcriptionally silence PPARγ.32 Upon 15d-PGJ2 binding, corepressors dissociate from the heterodimer of PPARγ and retinoid X receptor (RXR) that were previously bound to DNA sequences in the promoter regions (called PPAR-response elements (PPRE)), thereby allowing PPARγ to recruit coactivators and initiate the transcription of adipocyte genes.33 With or without ligand binding, PPARγ still binds to target genes (Figure 2).34 The process of PPARγ regulation of target gene expression is regulated by histone modification.35 Research has shown that the transcriptional activity of PPARγ is inhibited by phosphorylation of serine residue 112 on the N terminus31 and SUMOylation of lysine 107.36 Nocturnin is a circadian-regulated protein that has been shown to control preadipocyte differentiation and regulate lipid metabolism through the modulation of PPARγ activity.37 15d-PGJ2 is an endogenous natural ligand for PPARγ that regulates adipocyte differentiation by activating PPARγ, and coregulators that affect the expression of PPARγ may also regulate this process. These studies indirectly indicate that 15d-PGJ2 is a potential regulator of bone marrow adipocyte differentiation via a PPARγ-dependent pathway.
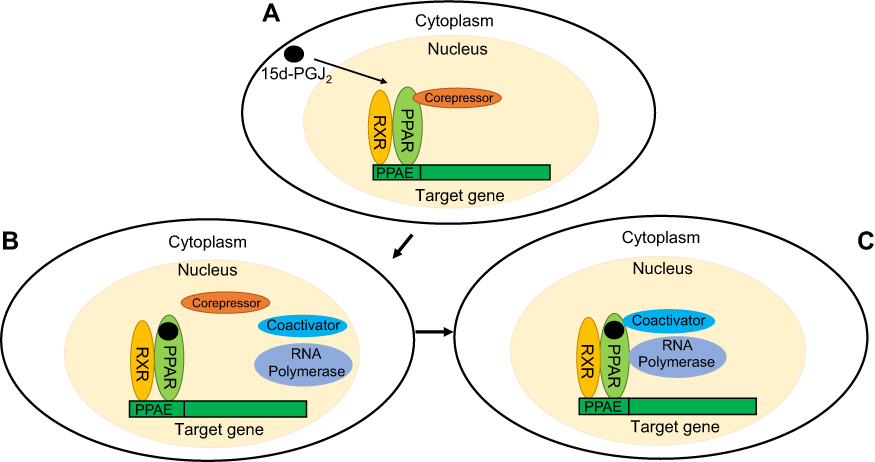 | Figure 2 Regulation of target gene expression by binding of 15d-PGJ2 to PPARγ. (A) PPARγ forms a heterodimer with RXR to recognize PPRE in the promoter region of target genes. When the PPARγ ligand is absent, PPARγ forms a protein complex with corepressors, thus transcriptionally silencing PPARγ. (B) 15d-PGJ2 is an endogenous ligand for PPARγ that binds to PPARγ resulting in PPARγ dissociation from a corepressor. (C) Upon binding of 15d-PGJ2 to the natural ligand, PPARγ can recruit coactivators and RNA polymerase to stimulate transcription of target genes. Abbreviations: RXR, retinoid X receptor; PPRE, PPAR-response elements. |
Emerging evidence suggests that Wnt ligands play a role in promoting osteoblastogenesis through canonical and noncanonical signaling pathways.13,38 The Wnt/β-catenin signaling pathway, commonly known as the canonical pathway, inhibits the formation of adipocytes by reducing the expression of PPARγ and C/EBPα mRNA.39,40 Wnt5a acts as a noncanonical Wnt ligand to suppresses adipogenesis by inhibiting the transcriptional activity of PPARγ and subsequently activating the histone methyltransferase SETDB1.11 Studies have also found that PPARγ deficiency induces osteoblastogenesis resulting in an increase in bone mass.41 Adipocyte differentiation and osteoblast differentiation are affected by both the PPARγ and Wnt pathways, and 15d-PGJ2, an endogenous ligand for PPARγ, is also involved in this regulatory process (Figure 3). These findings indicate that 15d-PGJ2 promotes bone marrow adipogenesis and inhibits osteoblastogenesis via a PPARγ-dependent pathway, thereby reducing bone formation in bone metabolism.
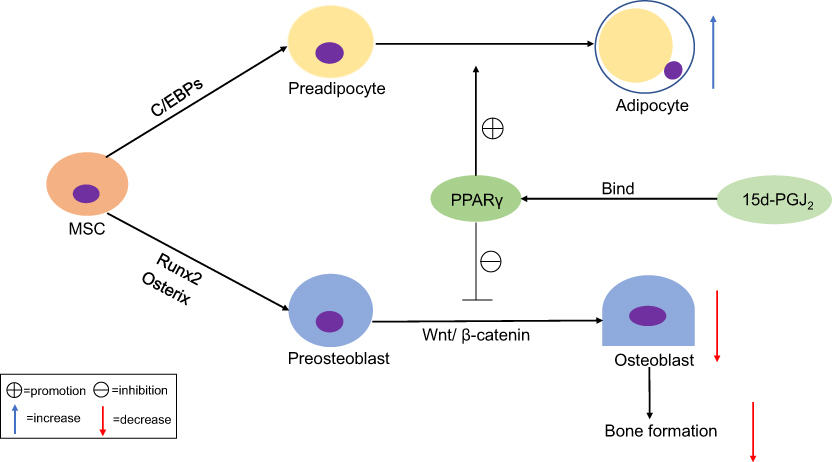 | Figure 3 15d-PGJ2 regulates adipocyte differentiation and osteoblast differentiation via a PPARγ-dependent pathway. MSCs have the potential to differentiate towards adipocytes and osteoblasts, through multiple factors and extracellular signaling pathways. Adipogenesis occurs under the strict regulation of multiple transcription factors, including PPARγ and C/EBPs, and osteoblastogenesis occurs under the regulation of Runx2, Osterix, and Wnt/β-catenin. Activation of PPARγ by 15d-PGJ2 and other synthetic ligands can promote adipogenesis but suppress osteoblastogenesis, resulting in the inhibition of bone formation. Abbreviations: MSCs, mesenchymal stem cells; C/EBPs, CCAAT/enhancer binding proteins; Runx2, runt-related transcription factor 2. |
Role of 15d-PGJ2 in osteoporosis
Osteoporosis is a serious social problem whose underlying mechanism is an imbalance between bone formation and bone resorption that increases the propensity of fragility fractures.42 Osteoporosis has multiple risk factors, including old age, menopause in women, long-term use of glucocorticoids, a history of fragility fractures, and a history of smoking.42,43 The imbalance between adipogenesis and osteoblastogenesis in the pathogenesis of primary or secondary osteoporosis may be related to the activation of PPARγ.41,44–46
Studies have shown that the expression of PPARγ in the bone marrow microenvironment increases with age.46 Between 20 and 65 years of age, the trabecular volume decreased by 10%, while the volume of adipose tissue increased by 45%.47 In the third decade of human life, adipose tissue occupies the femoral cavity, and in the seventh or eighth decade, fat can occupy the vertebrae.48 An increasing number of studies have confirmed that the application of glucocorticoids produces significant intramedullary fatty infiltration.49 Research has shown that glucocorticoids stimulate the differentiation of MSCs into adipocytes and promote the accumulation of fat by decreasing the expression of type I collagen and osteocalcin mRNA, thereby inhibiting osteoblast differentiation.50 Li et al51 demonstrated that glucocorticoids reduced Cbfa1/Runx2 gene expression by 50–60%, while PPARγ gene expression was increased by 200%.
In one study, Guo et al52 demonstrated that estrogen inhibited the formation of osteoclasts and bone resorption via microRNA‐27a targeting of PPARγ. This study also showed that in postmenopausal women, estrogen deficiency reduced microRNA-27a expression and increased PPARγ expression, which in turn leads to loss of bone mass and even osteoporosis.52 These studies indicate that PPARγ is highly expressed in age-related osteoporosis, glucocorticoid-induced osteoporosis, and postmenopausal osteoporosis, and its activators promote adipogenesis and inhibit osteoblastogenesis.46,51,52
Bisphosphonate is an important drug for the prevention and treatment of osteoporosis in clinical applications.53 Bisphosphonates not only inhibit bone resorption by directly inhibiting mineral dissolution but also inhibit cell viability by directly acting on osteoclasts and interfering with specific cellular biochemical processes.53 Studies have shown that ligand-activated PPARγ leads to osteoporosis by promoting adipocyte differentiation and inhibiting osteoblast differentiation.51,52 In vivo, 15d-PGJ2, as an endogenous ligand for PPARγ, may have an important regulatory role in the pathogenesis of osteoporosis in the absence of PPARγ synthetic agonists. However, research on the therapeutic advantages of 15d-PGJ2 compared with traditional drugs is limited.
15d-PGJ2 regulates osteoclast differentiation via PPARγ-independent and/or PPARγ-dependent pathways
Osteoclasts are involved in the removal of organic and inorganic bone components.29 Receptor activator of NF-κB ligand (RANKL) regulates the activity of osteoclasts by binding to receptor activator of NF-κB (RANK) on its surface.54 Osteoprotegerin (OPG) is a decoy receptor for RANKL and belongs to the tumor necrosis factor (TNF) receptor superfamily, which blocks the effects of RANKL.54 Differentiation of osteoclasts requires coordinated regulation of transcription factors such as c-fos, c-jun and nuclear factor of activated T-cells, cytoplasmic 1 (NFATc1).1 Studies have shown that OPG does not inhibit the process by which TNF-α promotes the formation of osteoclasts in humans.55 In one study, Hounoki et al56 found that 15d-PGJ2 inhibited osteoclast differentiation induced by TNF-α through the PPARγ-independent pathway. 15d-PGJ2 and ciglitazone, both of which are PPARγ agonists, were found to inhibit TNF-mediated osteoclast differentiation. The addition of the PPARγ antagonist, GW9662, to the culture rescued the inhibition induced by ciglitazone, but did not affect the inhibition induced by 15d-PGJ2.56 RANKL-induced monocyte chemoattractant protein-1 (MCP-1) has been shown to play an important role in osteoclast differentiation.57 In this study, 15d-PGJ2 reduced MCP-1, thereby inhibiting osteoclast differentiation induced by TNF-α.57 Considering the pivotal role of TNF-α in inflammatory joints, 15d-PGJ2 appears to be a promising targeting factor for the treatment of inflammatory bone resorption diseases such as rheumatoid arthritis (RA).58 A series of studies have shown that 15d-PGJ2 may play an important role in osteoclast differentiation via a PPARγ-independent pathway, whereas ciglitazone acts via a PPARγ-dependent pathway.
15d-PGJ2 is an important inflammatory mediator in RA
RA is a chronic disease affecting multiple joints that is accompanied by inflammation, massive synovial membranes, and neovascularization.59 PGE2 plays an important regulatory role in RA by inducing joint erosion and synovial inflammation.60 15d-PGJ2 is also a key negative regulator of the AA metabolism pathway and exerts an anti-inflammatory effect.15 An earlier clinical study in 2001 showed that 15d-PGJ2 inhibited the synthesis of PGE2 induced by interleukin-1 (IL)-1β in rheumatoid synovial fibroblasts by downregulating COX-2 and cPLA2 expression.61 15d-PGJ2 has been suggested to be an important inflammatory mediator with potential for the treatment of RA.23 The accumulated data suggest that 15d-PGJ2 and a portion of synthetic PPARγ agonists inhibit inflammation in models of arthritis,23 inflammatory bowel disease,62 ischemia–reperfusion injury,63 lupus nephritis,64 and Alzheimer’s disease.65
The anti-inflammatory activity of 15d-PGJ2 has been proven in many studies, but some studies have also found it to have pro-inflammatory properties.15,66,67 Glucocorticoids have also had a crucial role in the regression of inflammation through the intracellular glucocorticoid receptor (GR).68 Recent studies have shown that 15d-PGJ2 transiently attenuates GR signaling in monocytes/macrophages through a mechanism that relies on the interaction of the cyclopentenone ring in 15d-PGJ2 with a cysteine residue in the components of the GR activation pathway.66 The regulation of GR sensitivity by 15d-PGJ2 requires the participation of SUMOylation.67 The effect of the pro-inflammatory properties of 15d-PGJ2 on the therapeutic effect of AA still requires further study.
Role of 15d-PGJ2 in bone tumors
Bone metastasis is a type of cancer metastasis that results from primary tumor invasion to the bone, mainly osteolytic metastasis, which leads to an imbalance in bone metabolism.69 Breast cancer can metastasize to the bone, causing bone damage and ultimately leading to bone loss.70 In one study, Kim et al14 found that through a PPARγ-independent pathway, 15d-PGJ2 dose-dependently inhibited the RANKL/OPG ratio and osteoclast differentiation, which in turn reduced the formation of absorbed pits by inhibiting the activities of cathepsin K and matrix metalloproteinase (MMP)-2/9 (Figure 4). Osteolytic factors derived from breast cancer cells include parathyroid hormone-related protein (PTHrP) and some interleukins (mainly IL-6, IL-8).69,71 PTHrP enhances osteoclastogenesis and the activity of mature osteoclasts by upregulating RANKL and downregulating OPG in osteoblasts.69,72 OPG regulates bone resorption by inhibiting osteoclast activation and final differentiation and by inducing apoptosis in osteoclasts.73 Previous studies showed that transforming growth factor β (TGF-β) produced by the bone matrix increased the expression of PTHrP via Smad-dependent or Smad-independent pathways and was responsible for osteolytic lesions in breast cancer.74,75 Studies have also shown that application of 15d-PGJ2 inhibited significant enhancement of Smad2 phosphorylation as well as the nuclear levels of pSmad2 in TGF-β-stimulated cells.14,76 The addition of GW9662 to the culture media did not rescue the inhibition of PTHrP by 15d-PGJ2 regardless of TGF stimulation.14 These results indicate that 15d-PGJ2 inhibits PTHrP production via PPARγ-independent and Smad2-dependent pathways, thereby regulating osteolytic metastasis.
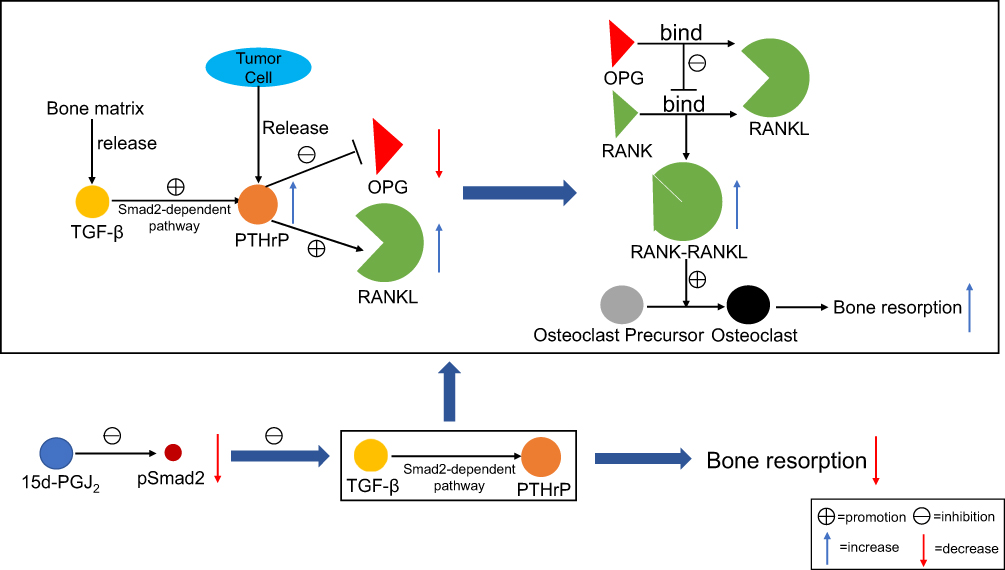 | Figure 4 Regulation of 15d-PGJ2 in bone metastasis of breast cancer. TGF-β is released from the bone matrix. PTHrP is an osteolytic factor derived from breast cancer cells. TGF-β increases the expression of PTHrP via Smad-dependent or Smad-independent pathways. PTHrP enhances osteoclastogenesis by upregulating RANKL and downregulating OPG in osteoblasts. Studies have shown that 15d-PGJ2 inhibits PTHrP production via PPARγ-independent and Smad2-dependent pathways, thereby regulating osteolytic metastasis. This reduces the RANKL/OPG ratio and is beneficial for increasing the formation of osteoclasts. OPG is a decoy receptor for RANKL, which inhibits the association of RANK and RANKL, thereby inhibiting the production of RANK-RANKL. 15d-PGJ2 reduces the production of RANK-RANKL, thereby inhibiting the differentiation of osteoclast progenitor cells into osteoclasts and, ultimately, reducing bone resorption. Abbreviations: TGF-β, transforming growth factor β; PTHrP, parathyroid hormone-related protein; RANKL, receptor activator of NF-κB ligand; OPG, osteoprotegerin. |
sCommon therapeutic agents for bone metastasis include denosumab and bisphosphonates.77 Denosumab is a fully human anti-RANKL IgG2 antibody that inhibits the interaction between RANKL and RANK, thereby reducing the maturation and activity of osteoclasts.78 Bisphosphonates are defined as “nitrogen-containing” (N-BPs: zoledronate, ibandronate) or “non-nitrogen containing” (non-N-BPs: clodronate, etidronate).77 The former is essential for the survival and activity of osteoclasts, while the latter’s metabolites induce osteoclast apoptosis.78 15d-PGJ2 affects osteoclastogenesis by reducing the production of PTHrP via a PPARγ-independent pathway. Denosumab and bisphosphonates act through anti-RANKL and target osteoclasts, respectively.77,78 However, there is limited research on the therapeutic advantages of 15d-PGJ2 compared with traditional drugs.
Osteosarcoma is a type of malignant tumor with a low survival that is uncommon and usually affects young people.79 A low patient survival rate may be associated with the high metastatic potential of cancer.80 However, the specific mechanisms that influence the progression and development of osteosarcoma are largely unknown. COX-2, which has pro-inflammatory activity, is highly expressed in primary osteosarcoma.81 Recent studies have shown that COX-2 promotes cell migration, invasion, and proliferation in human osteosarcoma cells.81,82 In one study, Kitz et al80 found that 15d-PGJ2 stimulates the expression of COX-2 via both the p38 and p42/44 mitogen-activated protein kinase (MAPK) and PPARγ-independent signaling pathways. Polo-like kinase 1 (PLK1) is an important cell cycle regulator and a potential target for osteosarcoma.83 In another study, Yen et al84 found that 15d-PGJ2 promotes apoptosis through reactive oxygen species (ROS)-mediated JNK activation, which may downregulate p-Akt and protein kinase A (PKA)–PLK1 pathways. Thus, 15d-PGJ2 is a regulator of osteosarcoma and exerts cytotoxic effects through AKT and PKA-PLK1 inhibition.84 From these studies, it was concluded that 15d-PGJ2 is a potential regulator of osteosarcoma through the PPARγ-dependent pathway.
Conclusion
Bone metabolism is an uninterrupted process that continuously removes old bone tissue and forms new bone tissue.28 Inhibition of osteoblast differentiation or enhancement of osteoclast differentiation will lead to osteoporosis, which in turn can lead to osteopetrosis.3 As an endogenous ligand for PPARγ, the cyclopentenone prostaglandin 15d-PGJ2 plays an important regulatory role in metabolic bone diseases, RA and bone tumors through PPARγ-dependent and independent pathways.9,14,23 It has been shown that through a PPARγ-dependent pathway, 15d-PGJ2 stimulates the differentiation of preadipocytes into adipocytes in a cell culture model system, thereby inhibiting osteoblast differentiation and affecting bone formation in bone metabolism.9,10 Many drugs with lipid-lowering effects have been used to treat the 15d-PGJ2 pathway for bone metabolism. For example, Li et al85 found that lovastatin decreased the expression of PPARγ but increased Cbfa1/Runx2 gene expression, causing transformation of the unformed osteoprogenitor cells from the adipocyte differentiation pathway to the osteoblast differentiation pathway. In one study, Jiang et al86 demonstrated that pravastatin alleviated steroid-induced osteonecrosis in rats by activating the Wnt signaling pathway and inhibiting PPARγ expression. Studies have shown that tanshinol reduces bone formation damage by inhibiting adipogenesis via PPARγ signaling in glucocorticoid-induced osteoporosis rats.87 This approach may be an attractive strategy to target osteoblastic cells by specific PPARγ inhibitors to treat bone diseases. The effect of 15d-PGJ2 in promoting adipocyte differentiation and inhibiting osteoclast differentiation through a PPARγ-dependent pathway is also affected by these PPARγ inhibitors. The above studies conclude that 15d-PGJ2 plays an important regulatory role in RA, osteosarcoma, and bone metastases via a PPARγ-independent pathway.14,23,80 Understanding the underlying mechanisms responsible for controlling the balance between osteoclastogenesis and osteoblastogenesis in human is important for regulating bone metabolism. Finally, this review summarizes the advances in the potential regulation of 15d-PGJ2 in bone metabolism and provides new therapeutic targets for osteoporosis, bone tumors, and other bone diseases.
Author contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work. Zhencheng Xiong and Pan Luo are co-first authors.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Raggatt LJ, Partridge NC. Cellular and molecular mechanisms of bone remodeling. J Biol Chem. 2010;285:25103–25108. doi:10.1074/jbc.R109.041087
2. Wan Y. PPARgamma in bone homeostasis. Trends Endocrinol Metab. 2010;21:722–728. doi:10.1016/j.tem.2010.08.006
3. Zaidi M. Skeletal remodeling in health and disease. Nat Med. 2007;13:791–801. doi:10.1038/nm1593
4. Ali D, Abuelreich S, Alkeraishan N, et al. Multiple intracellular signaling pathways orchestrate adipocytic differentiation of human bone marrow stromal stem cells. Biosci Rep. 2018;38(1). doi:10.1042/BSR20171252.
5. Abdallah BM, Alzahrani AM, Kassem M. Secreted clusterin protein inhibits osteoblast differentiation of bone marrow mesenchymal stem cells by suppressing ERK1/2 signaling pathway. Bone. 2018;110:221–229. doi:10.1016/j.bone.2018.02.018
6. Boyce BF. Advances in the regulation of osteoclasts and osteoclast functions. J Dent Res. 2013;92:860–867. doi:10.1177/0022034513500306
7. Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011;31:986–1000. doi:10.1161/ATVBAHA.110.207449
8. Straus DS, Glass CK. Cyclopentenone prostaglandins: new insights on biological activities and cellular targets. Med Res Rev. 2001;21:185–210.
9. Forman BM, Tontonoz P, Chen J, et al. 15-deoxy-delta 12, 14-prostaglandin J2 is a ligand for the adipocyte determination factor PPAR gamma. Cell. 1995;83:803–812.
10. Kliewer SA, Lenhard JM, Willson TM, et al. A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor gamma and promotes adipocyte differentiation. Cell. 1995;83:813–819.
11. Takada I, Kouzmenko AP, Kato S. Wnt and PPARgamma signaling in osteoblastogenesis and adipogenesis. Nat Rev Rheumatol. 2009;5:442–447. doi:10.1038/nrrheum.2009.137
12. Viccica G, Francucci CM, Marcocci C. The role of PPARgamma for the osteoblastic differentiation. J Endocrinol Invest. 2010;33:9–12.
13. Kawai M. Adipose tissue and bone: role of PPARgamma in adipogenesis and osteogenesis. Horm Mol Biol Clin Investig. 2013;15:105–113. doi:10.1515/hmbci-2013-0036
14. Kim KR, Kim HJ, Lee SK, et al. 15-deoxy-delta12,14-prostaglandin j2 inhibits osteolytic breast cancer bone metastasis and estrogen deficiency-induced bone loss. PLoS One. 2015;10:e0122764. doi:10.1371/journal.pone.0122764
15. Scher JU, Pillinger MH. 15d-PGJ2: the anti-inflammatory prostaglandin? Clin Immunol. 2005;114:100–109. doi:10.1016/j.clim.2004.09.008
16. Ueno N, Murakami M, Tanioka T, et al. Coupling between cyclooxygenase, terminal prostanoid synthase, and phospholipase A2. J Biol Chem. 2001;276:34918–34927. doi:10.1074/jbc.M100429200
17. Kunz T, Marklund N, Hillered L, et al. Cyclooxygenase-2, prostaglandin synthases, and prostaglandin H2 metabolism in traumatic brain injury in the rat. J Neurotrauma. 2002;19:1051–1064. doi:10.1089/089771502760341965
18. Urade Y, Eguchi N. Lipocalin-type and hematopoietic prostaglandin D synthases as a novel example of functional convergence. Prostaglandins Other Lipid Mediat. 2002;68–69:375–382.
19. Bie Q, Dong H, Jin C, et al. 15d-PGJ2 is a new hope for controlling tumor growth. Am J Transl Res. 2018;10:648–658.
20. Zhu F, Wang P, Kontrogianni-Konstantopoulos A, et al. Prostaglandin (PG)D(2) and 15-deoxy-Delta(12,14)-PGJ(2), but not PGE(2), mediate shear-induced chondrocyte apoptosis via protein kinase A-dependent regulation of polo-like kinases. Cell Death Differ. 2010;17:1325–1334. doi:10.1038/cdd.2010.13
21. Shibata T, Kondo M, Osawa T, et al. 15-deoxy-delta 12,14-prostaglandin J2. A prostaglandin D2 metabolite generated during inflammatory processes. J Biol Chem. 2002;277:10459–10466. doi:10.1074/jbc.M110314200
22. Shibata T. 15-deoxy-delta(1)(2),(1)(4)-prostaglandin J(2) as an electrophilic mediator. Biosci Biotechnol Biochem. 2015;79:1044–1049. doi:10.1080/09168451.2015.1012149
23. Kawahito Y, Kondo M, Tsubouchi Y, et al. 15-deoxy-delta(12,14)-PGJ(2) induces synoviocyte apoptosis and suppresses adjuvant-induced arthritis in rats. J Clin Invest. 2000;106:189–197. doi:10.1172/JCI9652
24. Ho TC, Chen SL, Yang YC, et al. 15-deoxy-delta(12,14)-prostaglandin J2 induces vascular endothelial cell apoptosis through the sequential activation of MAPKS and p53. J Biol Chem. 2008;283:30273–30288. doi:10.1074/jbc.M804196200
25. Kim EH, Surh YJ. The role of 15-deoxy-delta(12,14)-prostaglandin J(2), an endogenous ligand of peroxisome proliferator-activated receptor gamma, in tumor angiogenesis. Biochem Pharmacol. 2008;76:1544–1553. doi:10.1016/j.bcp.2008.07.043
26. Fu YG, Sung JJ, Wu KC, et al. Inhibition of gastric cancer cells associated angiogenesis by 15d-prostaglandin J2 through the downregulation of angiopoietin-1. Cancer Lett. 2006;243:246–254. doi:10.1016/j.canlet.2005.11.039
27. Murata T, Aritake K, Matsumoto S, et al. Prostagladin D2 is a mast cell-derived antiangiogenic factor in lung carcinoma. Proc Natl Acad Sci U S A. 2011;108:19802–19807. doi:10.1073/pnas.1110011108
28. Hadjidakis DJ, Androulakis II. Bone remodeling. Ann N Y Acad Sci. 2006;1092:385–396. doi:10.1196/annals.1365.035
29. Muruganandan S, Roman AA, Sinal CJ. Adipocyte differentiation of bone marrow-derived mesenchymal stem cells: cross talk with the osteoblastogenic program. Cell Mol Life Sci. 2009;66:236–253. doi:10.1007/s00018-008-8429-z
30. Rosen ED, Spiegelman BM. PPARgamma: a nuclear regulator of metabolism, differentiation, and cell growth. J Biochem. 2001;276:37731–37734. doi:10.1074/jbc.R100034200
31. Ge C, Zhao G, Li B, et al. Genetic inhibition of PPARγ S112 phosphorylation reduces bone formation and stimulates marrow adipogenesis. Bone. 2018;107:1–9. doi:10.1016/j.bone.2017.10.023
32. Guan HP, Ishizuka T, Chui PC, et al. Corepressors selectively control the transcriptional activity of PPARgamma in adipocytes. Genes Dev. 2005;19:453–461. doi:10.1101/gad.1263305
33. Zhu Y, Kan L, Qi C, et al. Isolation and characterization of peroxisome proliferator-activated receptor (PPAR) interacting protein (PRIP) as a coactivator for PPAR. J Biochem. 2000;275:13510–13516. doi:10.1074/jbc.275.18.13510
34. Lefterova MI, Lazar MA. New developments in adipogenesis. Trends Endocrinol Metab. 2009;20:107–114. doi:10.1016/j.tem.2008.11.005
35. Pascual G, Fong AL, Ogawa S, et al. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature. 2005;437:759–763. doi:10.1038/nature03988
36. Yamashita D, Yamaguchi T, Shimizu M, et al. The transactivating function of peroxisome proliferator-activated receptor gamma is negatively regulated by SUMO conjugation in the amino-terminal domain. Genes Cells. 2004;9:1017–1029. doi:10.1111/j.1365-2443.2004.00786.x
37. Kawai M, Green CB, Lecka-Czernik B, et al. A circadian-regulated gene, nocturnin, promotes adipogenesis by stimulating PPAR-gamma nuclear translocation. Proc Natl Acad Sci U S A. 2010;107:10508–10513. doi:10.1073/pnas.1000788107
38. Baron R, Rawadi G. Targeting the Wnt/beta-catenin pathway to regulate bone formation in the adult skeleton. Endocrinology. 2007;148:2635–2643. doi:10.1210/en.2007-0270
39. Westendorf JJ, Kahler RA, Schroeder TM. Wnt signaling in osteoblasts and bone diseases. Gene. 2004;341:19–39. doi:10.1016/j.gene.2004.06.044
40. Bennett CN, Longo KA, Wright WS, et al. Regulation of osteoblastogenesis and bone mass by Wnt10b. Proc Natl Acad Sci U S A. 2005;102:3324–3329. doi:10.1073/pnas.0408742102
41. Akune T, Ohba S, Kamekura S, et al. PPAR γ insufficiency enhances osteogenesis through osteoblast formation from bone marrow progenitors. J Clin Invest. 2004;113:846–855. doi:10.1172/JCI200419900
42. Rachner TD, Khosla S, Hofbauer LC. Osteoporosis: now and the future. Lancet (London, England). 2011;377:1276–1287. doi:10.1016/S0140-6736(10)62349-5
43. Kanis JA. Diagnosis of osteoporosis and assessment of fracture risk. Lancet (London, England). 2002;359:1929–1936. doi:10.1016/S0140-6736(02)08761-5
44. Kim SW, Her SJ, Kim SY, et al. Ectopic overexpression of adipogenic transcription factors induces transdifferentiation of MC3T3-E1 osteoblasts. Biochem Biophys Res Commun. 2005;327:811–819. doi:10.1016/j.bbrc.2004.12.076
45. Raisz LG. Pathogenesis of osteoporosis: concepts, conflicts, and prospects. J Clin Invest. 2005;115:3318–3325. doi:10.1172/JCI27071
46. Moerman EJ, Teng K, Lipschitz DA, et al. Aging activates adipogenic and suppresses osteogenic programs in mesenchymal marrow stroma/stem cells: the role of PPAR-gamma2 transcription factor and TGF-beta/BMP signaling pathways. Aging Cell. 2004;3:379–389. doi:10.1111/j.1474-9728.2004.00127.x
47. Muruganandan S, Govindarajan R, Sinal CJ. Bone marrow adipose tissue and skeletal health. Curr Osteoporos Rep. 2018;16(4):434–442. doi:10.1007/s11914-018-0451-y
48. Tontonoz P, Spiegelman BM. Fat and beyond: the diverse biology of PPARgamma. Annu Rev Biochem. 2008;77:289–312. doi:10.1146/annurev.biochem.77.061307.091829
49. Miyanishi K, Yamamoto T, Irisa T, et al. Bone marrow fat cell enlargement and a rise in intraosseous pressure in steroid-treated rabbits with osteonecrosis. Bone. 2002;30:185–190.
50. Yin L, Li YB, Wang YS. Dexamethasone-induced adipogenesis in primary marrow stromal cell cultures: mechanism of steroid-induced osteonecrosis. Chin Med J. 2006;119:581–588.
51. Li X, Jin L, Cui Q, et al. Steroid effects on osteogenesis through mesenchymal cell gene expression. Osteoporos Int. 2005;16:101–108. doi:10.1007/s00198-004-1649-7
52. Guo L, Chen K, Yuan J, et al. Estrogen inhibits osteoclasts formation and bone resorption via microRNA-27a targeting PPARgamma and APC. J Cell Physiol. 2018;234:581–594. doi:10.1002/jcp.26788
53. Russell RG, Xia Z, Dunford JE, et al. Bisphosphonates: an update on mechanisms of action and how these relate to clinical efficacy. Ann N Y Acad Sci. 2007;1117:209–257. doi:10.1196/annals.1402.089
54. Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423:337–342. doi:10.1038/nature01658
55. Kwak HB, Jin HM, Ha H, et al. Tumor necrosis factor-alpha induces differentiation of human peripheral blood mononuclear cells into osteoclasts through the induction of p21(WAF1/Cip1). Biochem Biophys Res Commun. 2005;330:1080–1086. doi:10.1016/j.bbrc.2005.03.085
56. Hounoki H, Sugiyama E, Mohamed SG, et al. Activation of peroxisome proliferator-activated receptor gamma inhibits TNF-alpha-mediated osteoclast differentiation in human peripheral monocytes in part via suppression of monocyte chemoattractant protein-1 expression. Bone. 2008;42:765–774. doi:10.1016/j.bone.2007.11.016
57. Cappellen D, Luong-Nguyen NH, Bongiovanni S, et al. Transcriptional program of mouse osteoclast differentiation governed by the macrophage colony-stimulating factor and the ligand for the receptor activator of NFkappa B. J Biochem. 2002;277:21971–21982. doi:10.1074/jbc.M200434200
58. Giri S, Rattan R, Singh AK, Singh I. The 15-deoxy-delta12,14-prostaglandin J2 inhibits the inflammatory response in primary rat astrocytes via down-regulating multiple steps in phosphatidylinositol 3-kinase-Akt-NF-kappaB-p300 pathway independent of peroxisome proliferator-activated receptor gamma. J Immunol. 2004;173:5196–5208.
59. Firestein GS. Evolving concepts of rheumatoid arthritis. Nature. 2003;423:356–361. doi:10.1038/nature01661
60. Park JY, Pillinger MH, Abramson SB. Prostaglandin E2 synthesis and secretion: the role of PGE2 synthases. Clin Immunol. 2006;119(3):229–240. doi:10.1016/j.clim.2006.01.016
61. Tsubouchi Y, Kawahito Y, Kohno M, et al. Feedback control of the arachidonate cascade in rheumatoid synoviocytes by 15-deoxy-delta(12,14)-prostaglandin J2. Biochem Biophys Res Commun. 2001;283:750–755. doi:10.1006/bbrc.2001.4847
62. Wada K, Nakajima A, Blumberg RS. PPARgamma and inflammatory bowel disease: a new therapeutic target for ulcerative colitis and Crohn’s disease. Trends Mol Med. 2001;7:329–331.
63. Nakajima A, Wada K, Miki H, et al. Endogenous PPAR gamma mediates anti-inflammatory activity in murine ischemia-reperfusion injury. Gastroenterology. 2001;120:460–469.
64. Reilly CM, Oates JC, Cook JA, et al. Inhibition of mesangial cell nitric oxide in MRL/lpr mice by prostaglandin J2 and proliferator activation receptor-gamma agonists. J Immunol. 2000;164:1498–1504.
65. Combs CK, Johnson DE, Karlo JC, et al. Inflammatory mechanisms in Alzheimer’s disease: inhibition of beta-amyloid-stimulated proinflammatory responses and neurotoxicity by PPARgamma agonists. J Neurosci. 2000;20:558–567.
66. Cheron A, Peltier J, Perez J, et al. 15-deoxy-delta12,14-prostaglandin J2 inhibits glucocorticoid binding and signaling in macrophages through a peroxisome proliferator-activated receptor gamma-independent process. J Immunol. 2004;172:7677–7683.
67. Paakinaho V, Kaikkonen S, Levonen AL, et al. Electrophilic lipid mediator 15-deoxy-delta12,14-prostaglandin j2 modifies glucocorticoid signaling via receptor SUMOylation. Mol Cell Biol. 2014;34:3202–3213. doi:10.1128/MCB.00748-14
68. Oh KS, Patel H, Gottschalk RA, et al. Anti-inflammatory chromatinscape suggests alternative mechanisms of glucocorticoid receptor action. Immunity. 2017;47(2):298–309.e295. doi:10.1016/j.immuni.2017.07.012
69. Guise TA. Molecular mechanisms of osteolytic bone metastases. Cancer. 2000;88:2892–2898.
70. Suva LJ, Washam C, Nicholas RW, et al. Bone metastasis: mechanisms and therapeutic opportunities. Nat Rev Endocrinol. 2011;7:208–218. doi:10.1038/nrendo.2010.227
71. Bendre M, Gaddy D, Nicholas RW. et al. Breast cancer metastasis to bone: it is not all about PTHrP. Clin Orthop Relat Res.2003:S39–S45. doi:10.1097/01.blo.0000093844.72468.f4
72. Karaplis AC, Goltzman D. PTH and PTHrP effects on the skeleton. Rev Endocr Metab Disord. 2000;1:331–341. doi:10.1023/A:1026526703898
73. Trouvin AP, Goëb V. Receptor activator of nuclear factor-κB ligand and osteoprotegerin: maintaining the balance to prevent bone loss. Clin Interv Aging. 2010;5:345–354. doi:10.2147/CIA.S10153
74. Kakonen SM, Selander KS, Chirgwin JM, et al. Transforming growth factor-beta stimulates parathyroid hormone-related protein and osteolytic metastases via Smad and mitogen-activated protein kinase signaling pathways. J Biol Chem. 2002;277:24571–24578. doi:10.1074/jbc.M202561200
75. Safina A, Sotomayor P, Limoge M, Morrison C, Bakin AV. TAK1-TAB2 signaling contributes to bone destruction by breast carcinoma cells. Mol Cancer Res. 2011;9:1042–1053. doi:10.1158/1541-7786.MCR-10-0196
76. Vaamonde-Garcia C, Malaise O, Charlier E, et al. 15-deoxy-Δ-12, 14-prostaglandin J2 acts cooperatively with prednisolone to reduce TGF-β-induced pro-fibrotic pathways in human osteoarthritis fibroblasts. Biochem Pharmacol. 2019. doi:10.1016/j.bcp.2019.03.039
77. D’Oronzo S, Coleman R, Brown J, Silvestris F. Metastatic bone disease: pathogenesis and therapeutic options: up-date on bone metastasis management. J Bone Oncol. 2019;15:004. doi:10.1016/j.jbo.2018.10.004
78. Sousa S, Clézardin P. Bone-targeted therapies in cancer-induced bone disease. Calcif Tissue Int. 2018;102:227–250. doi:10.1007/s00223-017-0353-5
79. Kansara M, Thomas DM. Molecular pathogenesis of osteosarcoma. DNA Cell Biol. 2007;26:1–18. doi:10.1089/dna.2006.0505
80. Kitz K, Windischhofer W, Leis HJ, et al. 15-deoxy-delta12,14-prostaglandin J2 induces cox-2 expression in human osteosarcoma cells through MAPK and EGFR activation involving reactive oxygen species. Free Radic Biol Med. 2011;50:854–865. doi:10.1016/j.freeradbiomed.2010.12.039
81. Masi L, Recenti R, Silvestri S, et al. Expression of cyclooxygenase-2 in osteosarcoma of bone. Appl Immunohistochem Mol Morphol. 2007;15:70–76. doi:10.1097/01.pai.0000213131.63417.fa
82. Lee EJ, Choi EM, Kim SR, et al. Cyclooxygenase-2 promotes cell proliferation, migration and invasion in U2OS human osteosarcoma cells. Exp Mol Med. 2007;39:469–476. doi:10.1038/emm.2007.51
83. Cheng L, Wang C, Jing J. Polo-like kinase 1 as a potential therapeutic target for osteosarcoma. Curr Pharm Des. 2015;21:1347–1350.
84. Yen CC, Hsiao CD, Chen WM, et al. Cytotoxic effects of 15d-PGJ2 against osteosarcoma through ROS-mediated AKT and cell cycle inhibition. Oncotarget. 2014;5:716–725. doi:10.18632/oncotarget.1704
85. Li X, Cui Q, Kao C, et al. Lovastatin inhibits adipogenic and stimulates osteogenic differentiation by suppressing PPARgamma2 and increasing Cbfa1/Runx2 expression in bone marrow mesenchymal cell cultures. Bone. 2003;33:652–659.
86. Jiang Y, Zhang Y, Zhang H, et al. Pravastatin prevents steroid-induced osteonecrosis in rats by suppressing PPARgamma expression and activating Wnt signaling pathway. Exp Biol Med (Maywood, NJ). 2014;239:347–355. doi:10.1177/1535370213519215
87. Yang YJ, Zhu Z, Wang DT, et al. Tanshinol alleviates impaired bone formation by inhibiting adipogenesis via KLF15/PPARgamma2 signaling in GIO rats. Acta Pharmacol Sin. 2018;39:633–641. doi:10.1038/aps.2017.134
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.