")
Back to Journals » Journal of Inflammation Research » Volume 15
α-Lipoic Acid Reduces Ceramide Synthesis and Neuroinflammation in the Hypothalamus of Insulin-Resistant Rats, While in the Cerebral Cortex Diminishes the β-Amyloid Accumulation
Authors Maciejczyk M , Żebrowska E, Nesterowicz M , Supruniuk E, Choromańska B, Chabowski A, Żendzian-Piotrowska M , Zalewska A
Received 27 January 2022
Accepted for publication 29 March 2022
Published 8 April 2022 Volume 2022:15 Pages 2295—2312
DOI https://doi.org/10.2147/JIR.S358799
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Mateusz Maciejczyk,1 Ewa Żebrowska,2 Miłosz Nesterowicz,3 Elżbieta Supruniuk,2 Barbara Choromańska,4 Adrian Chabowski,2 Małgorzata Żendzian-Piotrowska,1 Anna Zalewska5
1Department of Hygiene, Epidemiology, and Ergonomics, Medical University of Bialystok, Bialystok, Poland; 2Department of Physiology, Medical University of Bialystok, Bialystok, Poland; 3Students Scientific Club “Biochemistry of Civilization Diseases” at the Department of Hygiene, Epidemiology and Ergonomics, Medical University of Bialystok, Bialystok, Poland; 4 1st Department of General and Endocrine Surgery, Medical University of Bialystok, Bialystok, Poland; 5Department of Restorative Dentistry and Experimental Dentistry Laboratory, Medical University of Bialystok, Bialystok, Poland
Correspondence: Mateusz Maciejczyk, Department of Hygiene, Epidemiology, and Ergonomics, Medical University of Bialystok, 2C Mickiewicza Street, Bialystok, Poland, Email [email protected]
Background: Oxidative stress underlies metabolic diseases and cognitive impairment; thus, the use of antioxidants may improve brain function in insulin-resistant conditions. We are the first to evaluate the effects of α-lipoic acid (ALA) on redox homeostasis, sphingolipid metabolism, neuroinflammation, apoptosis, and β-amyloid accumulation in the cerebral cortex and hypothalamus of insulin-resistant rats.
Methods: The experiment was conducted on male cmdb/outbred Wistar rats fed a high-fat diet (HFD) for 10 weeks with intragastric administration of ALA (30 mg/kg body weight) for 4 weeks. Pro-oxidant and pro-inflammatory enzymes, oxidative stress, sphingolipid metabolism, neuroinflammation, apoptosis, and β-amyloid level were assessed in the hypothalamus and cerebral cortex using colorimetric, fluorimetric, ELISA, and HPLC methods. Statistical analysis was performed using three-way ANOVA followed by the Tukey HSD test.
Results: ALA normalizes body weight, food intake, glycemia, insulinemia, and systemic insulin sensitivity in HFD-fed rats. ALA treatment reduces nicotinamide adenine dinucleotide phosphate (NADPH) and xanthine oxidase activity, increases ferric-reducing antioxidant power (FRAP) and thiol levels in the hypothalamus of insulin-resistant rats. In addition, it decreases myeloperoxidase, glucuronidase, and metalloproteinase-2 activity and pro-inflammatory cytokines (IL-1β, IL-6) levels, while in the cerebral cortex ALA reduces β-amyloid accumulation. In both brain structures, ALA diminishes ceramide synthesis and caspase-3 activity. ALA improves systemic oxidative status and reduces insulin-resistant rats’ serum cytokines, chemokines, and growth factors.
Conclusion: ALA normalizes lipid and carbohydrate metabolism in insulin-resistant rats. At the brain level, ALA primarily affects hypothalamic metabolism. ALA improves redox homeostasis by decreasing the activity of pro-oxidant enzymes, enhancing total antioxidant potential, and reducing protein and lipid oxidative damage in the hypothalamus of HFD-fed rats. ALA also reduces hypothalamic inflammation and metalloproteinases activity, and cortical β-amyloid accumulation. In both brain structures, ALA diminishes ceramide synthesis and neuronal apoptosis. Although further study is needed, ALA may be a potential treatment for patients with cerebral complications of insulin resistance.
Keywords: α-lipoic acid, insulin resistance, brain, oxidative stress, ceramide, inflammation
Introduction
Currently, one of the most severe health problems is the increased incidence of diabetes and obesity. More than 400 million people have type 2 diabetes, which is expected to double by 2040.1 Many clinical studies have shown that the leading risk factor for metabolic diseases is insulin resistance, ie, reduced sensitivity of target tissues to insulin action.2–4 At the molecular level, insulin resistance is caused by a loss/downregulation of the insulin receptor (IR) and insulin receptor substrate (IRS-1) in such organs as the liver, adipose tissue, and skeletal muscles.5–7 Under these conditions, free fatty acids accumulate in cells, inhibiting glucose uptake by serine/threonine kinases, which finally results in phosphorylation of IRS-1 and blocks the insulin signaling.8,9 Reduced insulin sensitivity and alterations in insulin release lead to disturbances in peroxisome proliferator-activated receptor (PPAR) pathways, increased formation of reactive oxygen species (ROS), adipokines, and pro-inflammatory cytokines as well as an excessive rate of cellular metabolism.10,11 Thus, insulin resistance is the first stage of a vicious circle, promoting the development of metabolic complications of diabetes.12 These include cardiovascular diseases such as cardiomyopathies, retinopathies, and nephropathies, but also neurodegeneration and cognitive disorders.13,14 Diabetic patients are about three times more likely to develop Alzheimer’s disease (AD) than those without metabolic diseases.15 Recent findings indicate that abnormalities in neuronal glucose metabolism contribute to β-amyloid accumulation in the brain, causing oxidative stress, neuroinflammation, and cognitive impairment.15,16 Therefore, AD is even referred to as type 3 diabetes.
Oxidative stress and inflammation are thought to be the common denominators underlying neurodegeneration and metabolic disorders.17–19 The brain is susceptible to oxidation since it consumes much oxygen (compared to other tissues per unit mass), has an unfavorable surface-area-to-volume ratio, low antioxidant barrier capacity, and high polyunsaturated fatty acid content.20 Overproduction of ROS results in oxidative damage to neuronal biomolecules, including enzyme inactivation, protein misfolding, lipid membrane peroxidation, and DNA strand breaks, which disrupt cerebral homeostasis and ultimately lead to cell death.21 However, not only autoxidation of neurotransmitters (eg, dopamine, norepinephrine, and hydroquinones) is responsible for free radical formation in the brain. In neurons and glial cells, the primary source of ROS is NADPH oxidase (NOX), which additionally induces a local inflammatory response. NOX activates the NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) signaling, increasing the expression of TNF-α (tumor necrosis factor α), IL-1, and IL-6, as well as brain adhesion molecules such as CAM-1 (intercellular adhesion molecule 1), VCAM-1 (vascular cell adhesion molecule 1), and MCP-1 (monocyte chemoattractant protein 1).22 It has been demonstrated that IL-1β reduces neuronal glucose transport to cells by inhibiting the expression of IRS-1, whereas TNF-α, IL-1β, and IL-6 lead to the activation of c-Jun N-terminal kinases (JNKs), decreasing insulin sensitivity. Interestingly, NOX-mediated oxidative stress also promotes ceramide accumulation in the brain, directly inducing insulin resistance at the cerebral level. Furthermore, ceramide activates JNKs, resulting in the blockage of insulin transduction pathway.22–28 Therefore, it is not surprising that the scientific community is focused on finding new therapeutic strategies to protect against oxidative brain damage.
α-Lipoic acid (ALA) is of growing interest in treating metabolic diseases.29–34 ALA is an 8-carbon, cyclic disulfide antioxidant with hydrophobic and hydrophilic properties.35,36 ALA functions as a coenzyme of multi-enzymatic complexes catalyzing the oxidative decarboxylation of α-keto acids.29 Recent studies also indicate its direct involvement in the free radical scavenging, chelation of pro-oxidant metal ions, and regeneration of other antioxidants such as reduced glutathione (GSH), coenzyme Q10, and vitamins C and E.37–39 Furthermore, ALA has anti-inflammatory effects and regulates carbohydrate and lipid metabolism.29 It was shown that ALA normalizes glycemia and insulinemia, improves peripheral insulin sensitivity, and prevents systemic oxidative stress and inflammation in insulin-resistant rats.40–43 In addition, ALA reduces total cholesterol, low-density lipoprotein (LDL), and lipoprotein a (Lp(a)), as well as inhibits LDL oxidation and adhesion of monocytes to endothelial cells.29,44,45 Nevertheless, there are limited reports on the effects of ALA on the brain function in insulin-resistant conditions.46 Given the multidirectional effects of ALA in diminishing the peripheral consequences of insulin resistance,31,47–50 we speculate that ALA may also improve brain function through several mechanisms. Importantly, ALA readily crosses the blood–brain barrier (BBB) and can be used in the pharmacotherapy of the central nervous system (CNS).51,52 In this study, we are the first to evaluate the effects of ALA on pro-oxidant and pro-inflammatory enzymes, oxidative stress, sphingolipid metabolism, neuroinflammation, apoptosis, and β-amyloid accumulation in the brain of insulin-resistant rats. We examined brain structures that regulate lipid/carbohydrate metabolism (hypothalamus) and the brain tissue responsible for cognitive function (cerebral cortex). We also evaluated the detailed inflammatory profile of serum cytokines, chemokines, and growth factors under ALA supplementation.
Materials and Methods
Animals
The experiment was performed on 40 male cmdb/outbred Wistar rats. The average initial bodyweight of the rats was 50–60 g (3-week-old). The animals were provided with standard conditions for laboratory animals with free access to drinking water and food, and a 12/12 light/darkness cycle at 21 ± 2°C. After a week’s adaptation to environmental conditions, the animals were randomly divided into four equal groups (n = 10).
Rats were fed a control diet (CD; CD ALA- and CD ALA+ groups) comprised of 10.3 kcal% fat, 24.2 kcal% protein, and 65.5% carbohydrate (Agropol, Motycz, Poland) for 10 weeks or high-fat diet (HFD; HFD ALA- and HFD ALA+ groups) containing 60 kcal% fat, 20 kcal% carbohydrates, and 20 kcal% proteins (D12492; Research Diets, Inc. New Brunswick, USA) also for 10 weeks. Additionally, in ALA- groups after 6 weeks of the experiment, the rats received intragastric saline solution for 4 weeks, whereas in ALA+ groups, the animals received intragastrically ALA solution at a dose of 30 mg/kg body weight. The ALA dose (30 mg/kg body weight) was selected based on the literature analysis.53–55 It was observed that the applied dose of ALA shows good antioxidant activity without any toxic symptoms. The saline and ALA solutions (2 mL/kg body weight) were administered daily at the same time by one experienced technician. Administration of ALA by the intragastric route ensures that the animal receives the total dose of the drug. Moreover, the intragastric route of administration did not cause chronic stress and unusual behavior (compulsive behavior, changes in food and water intake, nervousness, changes in response to external stimuli, and mutilation).
Anthropometric measurements were performed as described in detail by Novelli et al.56 The specific rate of body mass gain was calculated as the difference between final and initial body weight divided by initial weight. Body mass index (BMI) was calculated using the formula: BMI = body weight (g)/length2 (cm2). It was assumed that BMI over 0.65 g/cm2 indicates obesity. Based on food and caloric intake, energy intake (food consumption × dietary metabolizable energy) and feed efficiency (body weight gain × 100)/energy intake) were also calculated. The Lee index was estimated using the formula: Lee index = the cubic root of body weight (g)/length (cm).
In the 10th week of the experiment, all animals were weighed, anesthetized with an intraperitoneal injection of phenobarbital (at a dose of 80 mg/kg body weight), and sacrificed. Immediately after collecting blood from the abdominal aorta, the hypothalamus and cerebral cortex were taken by the same technician. The brain tissues were purified from blood and fat and then precooled in liquid nitrogen. The samples were stored at −80°C until final determinations.
Fasting plasma glucose and insulin were assayed by commercial ELISA kits (EIAab, Wuhan, China). HOMA-IR index (homeostatic model assessment of β-cell function and insulin resistance) was calculated using the formula: HOMA-IR index = fasting insulin (U/mL) × fasting glucose (mM)/22.5.57 The plasma-free fatty acids level was measured by colorimetric assay using a commercial reagent kit (Sigma-Aldrich; St. Louis, MO, USA).
The present study followed the guidelines stated in the “Guide for the Care and Use of Laboratory Animals” (National Academies Press, Washington, 2011). The experimental procedures were approved by the Institutional Committee for Ethics Use of Animals in Olsztyn, Poland (approval no. 21/2017).
Biochemical Assays
Unless otherwise indicated, all the reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA) or Fluka (Buchs, Switzerland). The absorbance/fluorescence was measured using Infinite M200 PRO (Multimode Microplate Reader, Tecan). Sphingolipid metabolism was assessed using high-performance liquid chromatography (HPLC; Varian ProStar, Agilent Technologies) with a fluorescence detector and a C18 reversed-phase column. All results were standardized to 1 mg of total protein determined by the bicinchoninic acid method (Thermo Scientific, Rockford, IL, USA).
Brain Pro-Oxidant Enzymes, Antioxidant Status, and Oxidative Stress
The activity of NADPH oxidase (NOX, EC 1.6.3.1) was evaluated colorimetrically using lucigenin as an electron acceptor.58 One nmol of superoxide radical per minute was assumed as one unit of NOX activity. The xanthine oxidase (XO, EC 1.17.3.2.) activity was assayed colorimetrically at 290 nm.59 One unit of XO activity was required to release 1 μmol of UA per 1 minute.
Ferric-reducing antioxidant power (FRAP) was used to assess the antioxidant status. FRAP level was measured colorimetrically at 592 nm based on the reduction of Fe2+ to Fe3+ in an acidic environment, resulting in a colorful ferrous tripyridyltriazine (Fe3+-TPTZ) complex.60
Protein and lipid oxidation products were used to assess the severity of oxidative stress. The concentration of total thiols was analyzed colorimetrically at 420 nm using Ellman’s reagent.61 The reduced glutathione (GSH) was used as a standard. The content of thiobarbituric acid reactive substances (TBARS) was measured using the colorimetric method described by Buege and Austa.62 1,1,3,3-Tetramethoxypropane was used as a standard. The absorbance was measured at 535 nm.
Brain Sphingolipid Metabolism
The level of sphingosine (SFO), sphinganine (SFA), sphingosine-1-phosphate (S1P), sphinganine-1-phosphate (SFA1P), and ceramide (CER) was determined using the HPLC method. In the presence of C17-SFO as a standard, lipids were extracted from ultrasonicated samples of the brain tissue. An obtained lipid extract was transferred to a tube containing N-palmitoyl-D-erythro-SFO (C17 base). The released free SFO and SFA were converted to their o-phthalaldehyde derivatives and then analyzed by the HPLC system. Neutral sphingomyelinase (nSMase) activity was measured by colorimetric assay using a commercial reagent kit (Sigma-Aldrich; St. Louis, MO, USA).
Brain Pro-Inflammatory Enzymes and Cytokines
The activity of myeloperoxidase (MPO) was analyzed by the spectrophotometric method at 450 nm. The determinations were made using hexadecyltrimethylammonium sulfanilamide, ortho-dianisidine dihydrochloride, and hydrogen peroxide.63 The activity of β-glucuronidase (GLU, EC 3.2.1.31) was assayed colorimetrically at 405 nm, measuring the intensity of the released 4-nitrophenol. 4-Nitrophenyl-β-D-glucuronide was used as a substrate.64
The concentrations of interleukin 1 beta (IL-1β) and interleukin 6 (IL-6) were determined using commercial ELISA kits (EIAab, Wuhan, China) according to the manufacturer’s manual.
Brain Apoptosis
The activity of caspase-3 (CAS-3, EC 3.4.22.56) was analyzed by colorimetric method using Ac-Asp-Glu-Val-Asp p-nitroanilide as a substrate. The absorbance was measured at a 405 nm wavelength.65 The activity of caspase-8 (CAS-8, EC 3.4.22.61) was measured using Ac-Val-Glu-Thr-Asp-AMC as a substrate.
Brain Metalloproteinases
The activity of metalloproteinase-2 (MMP-2) and −9 (MMP-9) was measured fluorometrically using the substrate MCA-Pro-Leu-Gly-Leu-Dpa(Dnp)-Ala-Arg-NH2, where MCA is the 7-methoxycoumarin-4-yl)acetate. The fluorescence was measured at 325/393 nm.
Brain β-Amyloid
The formation of the amyloid cross‐β structure was measured colorimetrically at 435/485 nm using thioflavin T.66
Serum Cytokines, Chemokines, and Growth Factors
According to the manufacturer’s instructions, serum concentrations of 23 cytokines were assessed using ELISA Bio-Plex Pro™ Rat Cytokine 23-Plex Assay (Bio Rad Laboratories, Hercules, CA, USA). Briefly, samples (1:4 serum dilution) were added to the appropriate wells in a 96-well plate and incubated successively with couple beads and detection antibodies. Following the addition of streptavidin–phycoerythrin conjugate and a series of washes, Bio-Plex 200 System (Bio-Rad Laboratories, Hercules, CA, USA) was used for the read-outs.
Statistical Analysis
GraphPad Prism 9 (GraphPad Software, La Jolla, CA, USA) for MacOS was used for statistical data analysis. Homogeneity of variance was checked by Levene’s test, while normality of distribution was by the Shapiro–Wilk test. Values are presented as means ± SD. Three-way ANOVA and one-way ANOVA followed by a post hoc Tukey HSD test were performed. In the absence of a normal distribution, the ANOVA Kruskal Wallis test followed by Dunn’s post hoc test was used, and results were presented as median, minimum and maximum. The significance level was set at p ≤ 0.05.
Results
General Characteristics
HFD induced obesity and insulin resistance, whereas ALA treatment effectively reduced body weight, specific rate of body mass gain, and BMI compared to HFD ALA-rats. ALA increased food consumption and energy intake, as well as decreased feed efficiency and Lee index in insulin-resistant rats compared to HFD ALA-. ALA normalized systemic carbohydrate and lipid metabolism by lowering plasma glucose, insulin, free fatty acids, and HOMA-IR in the HFD ALA+ group compared to HFD ALA- (Figure S1).
ALA supplementation enhanced plasma FRAP and total thiol concentrations as well as reduced TBARS levels in the HFD ALA+ group compared to HFD ALA- (Figure S2).
ALA decreased serum levels of pro-inflammatory cytokines (IL-1α, IL-2, IL-6, TNF-α; Figure S3) and chemokines (CCL-15; Figure S4) as well as increased anti-inflammatory IL-10 (Figure S5) in the HFD ALA- group compared with HFD ALA+. ALA did not affect serum growth factor concentrations (Figure S6).
Brain Pro-Oxidant Enzymes, Antioxidant Status, and Oxidative Stress
NOX activity of HFD-fed ALA- animals was increased compared to CD ALA- both in the hypothalamus (+22%) and in the cerebral cortex (+30%). Moreover, in the HFD group, hypothalamic NOX activity after ALA administration was lower (−27%) (Figure 1A).
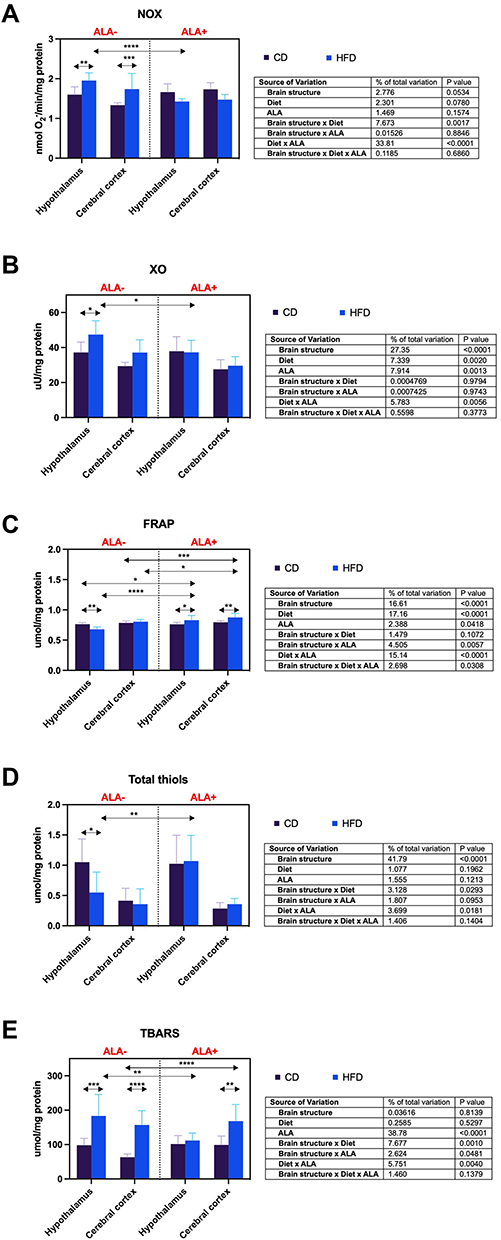 |
Figure 1 Effects of α-lipoic acid (ALA) on brain pro-oxidant enzymes [NADPH oxidase (NOX), (A) xanthine oxidase (XO), (B)], antioxidant status [ferric-reducing antioxidant power (FRAP), (C)], and oxidative stress [total thiols, (D) thiobarbituric acid reactive substances (TBARS), (E)] in the hypothalamus and cerebral cortex of rats fed a control (CD) and high-fat diet (HFD). Values are presented as mean ± SD. Three-way ANOVA followed by post hoc Tukey HSD test was performed. *p < 0.05, **p < 0.005, ***p < 0.0005, ****p < 0.0001. |
The activity of XO was enhanced (+28%) in the hypothalamus of HFD animals without ALA supplementation compared to the CD group. Additionally, this parameter in the hypothalamus of HFD ALA+ animals decreased (−21%) compared to the HFD ALA- group (Figure 1B).
Hypothalamic FRAP was reduced (−11%) in HFD compared to CD ALA- group. In ALA+ animals, FRAP was higher in HFD than in the CD group in the hypothalamus and cerebral cortex (+9% and +10%, respectively). Hypothalamic FRAP in HFD-fed ALA+ animals was increased when compared to both CD (+9%) and HFD (+22%) ALA-. Furthermore, cortical FRAP was higher in HFD ALA+ than HFD and CD groups without ALA administration (+12% and +9%, respectively) (Figure 1C).
The concentration of total thiols in the HFD group was lower (−48%) than CD in the hypothalamus of ALA-treated animals. Hypothalamic total thiols content was enhanced (+95%) by ALA supplementation in the HFD group (Figure 1D).
TBARS level of HFD animals was higher than CD ALA- both in the hypothalamus (+87%) and cerebral cortex (+148%). However, only the cortical concentration of TBARS was increased (+70%) in HFD compared to the CD group of ALA+ animals. Due to ALA administration, the TBARS content of HFD in the hypothalamus was reduced (−39%). Furthermore, the biomarker’s level in the cerebral cortex was enhanced (+166%) in HFD animals compared to CD after ALA treatment (Figure 1E).
Brain Sphingolipid Metabolism
The concentration of hypothalamic SFO was reduced in the HFD ALA+ group compared to CD ALA- (−38%) and HFD ALA- (−47%) animals (Figure 2A).
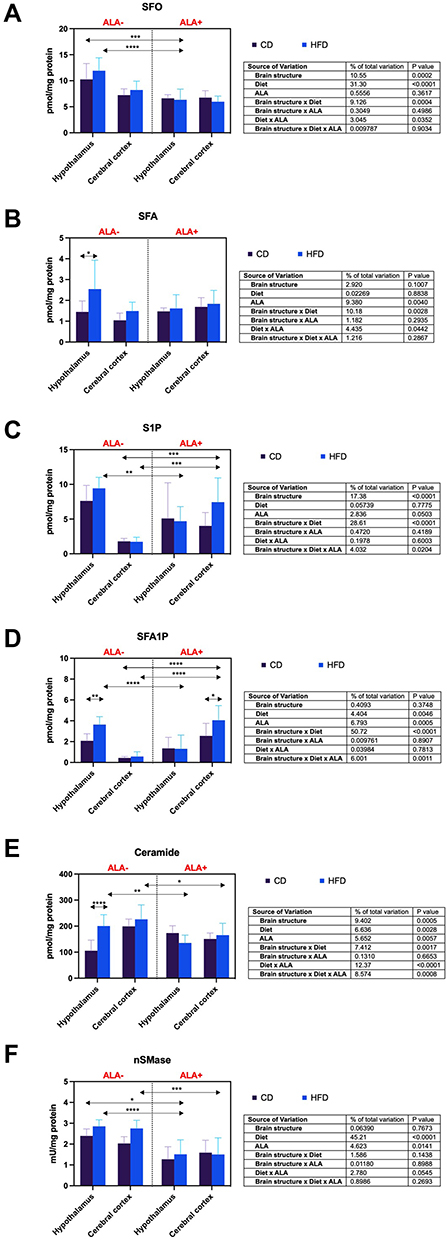 |
Figure 2 Effects of α-lipoic acid (ALA) on brain sphingolipid metabolism [sphingosine (SFO), (A) sphinganine (SFA), (B) sphingosine-1-phosphate (S1P), (C) sphinganine-1-phosphate (SFA1P), (D) ceramide (CER), (E) neutral sphingomyelinase (nSMase), Figure 2F] in the hypothalamus and cerebral cortex of rats fed a control (CD) and high-fat diet (HFD). Values are presented as mean ± SD. Three-way ANOVA followed by post hoc Tukey HSD test was performed. *p < 0.05, **p < 0.005, ***p < 0.0005, ****p < 0.0001. |
SFA content in the hypothalamus of HFD ALA- was increased compared to CD-fed ALA- animals (+76%) (Figure 2B).
The hypothalamic level of S1P was lower (−50%) in the HFD group after administration of ALA. ALA also increased cortical S1P concentration in CD and HFD groups (+316% and +330%, respectively) (Figure 2C).
SFA1P concentration in the hypothalamus of HFD was enhanced (+75%) in comparison to CD ALA-. In ALA-treated animals, this parameter was higher (+59%) in the cerebral cortex in HFD when compared to the CD group. After ALA supplementation, SFA1P was lower (−64%) in the HFD group in the hypothalamus. However, it was higher in HFD ALA+ animals compared to CD (+830%) and HFD (+606%) ALA- rats in the cerebral cortex (Figure 2D).
The hypothalamic ceramide content in HFD ALA- group was increased (+90%) compared to CD ALA-. Additionally, the level of ceramide of HFD animals was decreased by ALA administration both in the hypothalamus and the cerebral cortex (−32% and −27%, respectively) (Figure 2E).
nSMase activity in the hypothalamus of HFD ALA+ was higher than CD (−37%) and HFD (−47%) ALA- animals. After ALA treatment, the activity of nSMase was enhanced (−45%) in the cerebral cortex of the HFD group (Figure 2F).
Brain Pro-Inflammatory Enzymes and Cytokines
The cortical activity of MPO of HFD animals was higher (+124%) than CD ALA+. Hypothalamic MPO activity in the HFD group was reduced (−25%) due to ALA administration (Figure 3A).
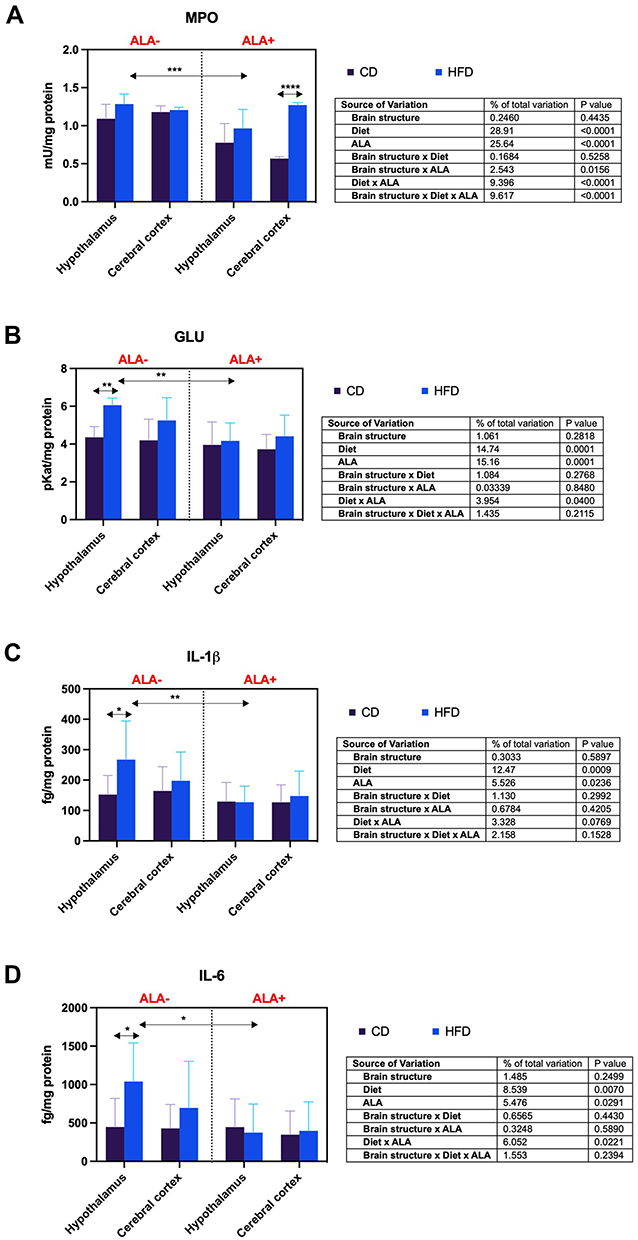 |
Figure 3 Effects of α-lipoic acid (ALA) on brain pro-inflammatory enzymes [myeloperoxidase (MPO), (A) β-glucuronidase (GLU), (B)] and cytokines [interleukin 1 beta (IL-1β), (C) interleukin 6 (IL-6), (D)] in the hypothalamus and cerebral cortex of rats fed a control (CD) and high-fat diet (HFD). Values are presented as mean ± SD. Three-way ANOVA followed by post hoc Tukey HSD test was performed. *p < 0.05, ** < 0.005, ***p < 0.0005, ****p < 0.0001. |
GLU activity in the hypothalamus of HFD-fed animals was enhanced (+39%) in comparison to CD ALA-. However, ALA treatment lowered (−31%) its activity in the HFD group after ALA treatment (Figure 3B).
The hypothalamic content of IL-1β in HFD was increased (+75%) compared to CD animals not supplemented with ALA. Administration of ALA decreased (−52%) the hypothalamic level of this biomarker in HFD animals (Figure 3C).
In the hypothalamus, the concentration of IL-6 of HFD was higher (+132%) than CD ALA- animals. Moreover, the hypothalamic IL-6 level was reduced (−64%) by ALA in the HFD group (Figure 3D).
Brain Apoptosis
The activity of CAS-3 of HFD animals was increased compared to CD ALA- animals in the hypothalamus (+76%) and the cerebral cortex (+93%). However, after ALA treatment, hypothalamic and cortical CAS-3 activity in the HFD group was reduced (−53% and −56%, respectively) (Figure 4A). ALA does not affect brain CAS-8 activity (Figure 4B).
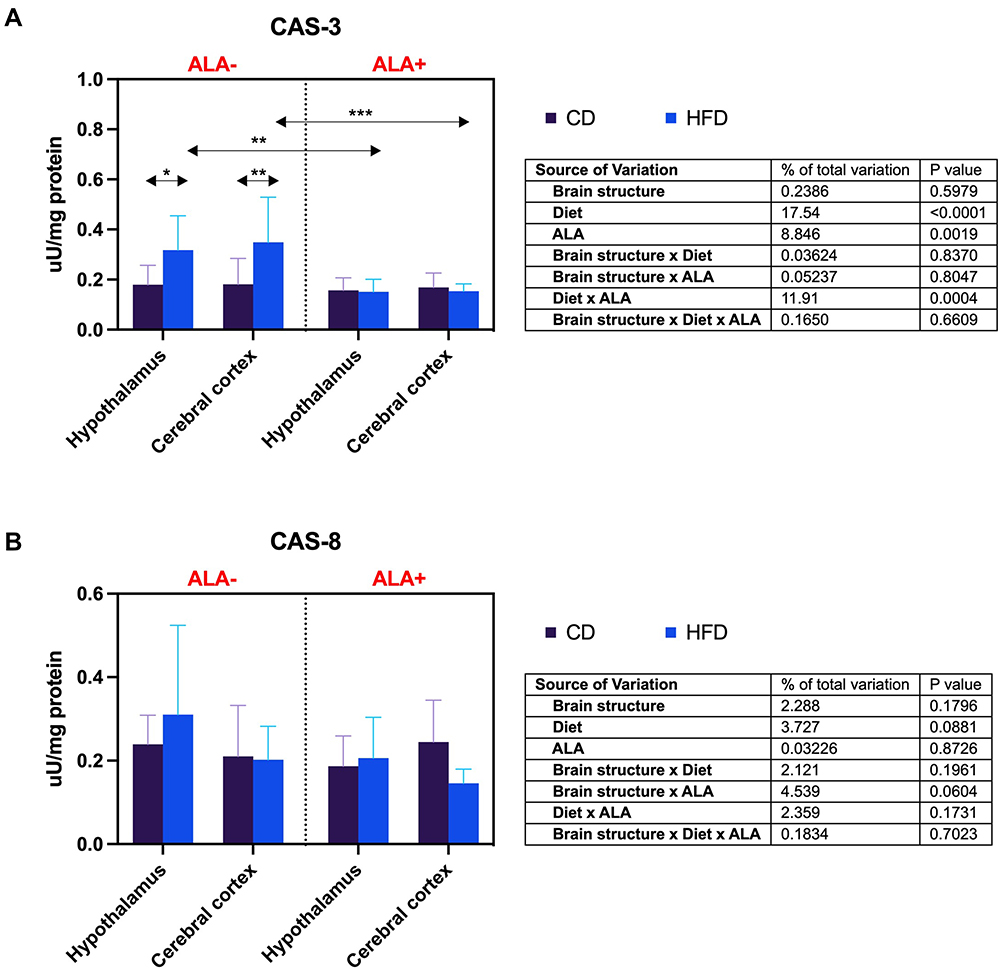 |
Figure 4 Effects of α-lipoic acid (ALA) brain apoptosis [caspase-3 (CAS-3), (A) caspase-8 (CAS-8), (B)] in the hypothalamus and cerebral cortex of rats fed a control (CD) and high-fat diet (HFD). Values are presented as mean ± SD. Three-way ANOVA followed by post hoc Tukey HSD test was performed. *p < 0.05, **p < 0.005, ***p < 0.0005. |
Brain Metalloproteinases
MMP-2 activity in HFD ALA- was enhanced compared to CD ALA- group both in the hypothalamus (+101%) and the cerebral cortex (+71%). ALA decreased (−40%) only the hypothalamic activity of MMP-2 of HFD animals (Figure 5A). ALA does not affect brain MMP-9 (Figure 5B) and MMP-2/MMP-9 activity (Figure 5C).
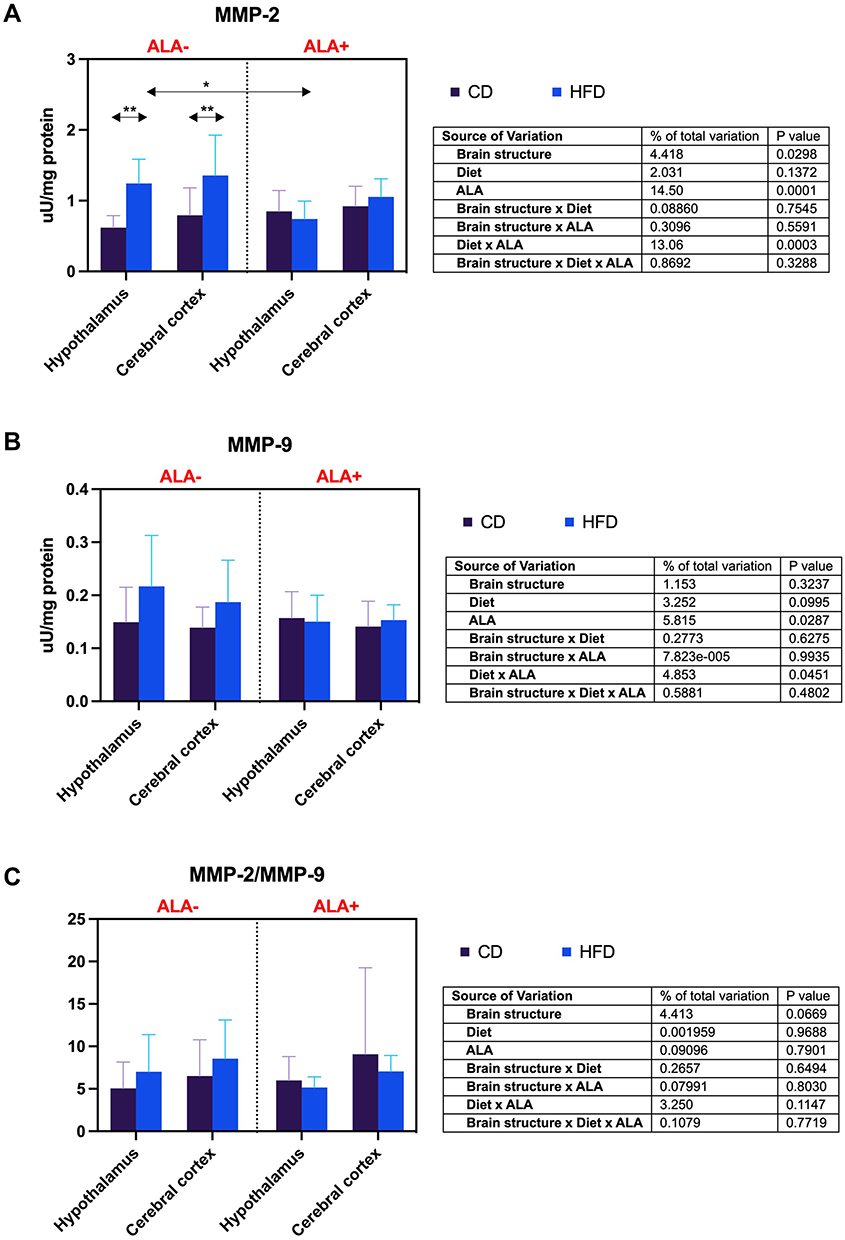 |
Figure 5 Effects of α-lipoic acid (ALA) on brain metalloproteinases [metalloproteinase-2 (MMP-2), (A) metalloproteinase-9 (MMP-9), (B)] and their ratio [MMP-2/MMP-9 ratio (MMP-2/MMP-9), (C)] in the hypothalamus and cerebral cortex of rats fed a control (CD) and high-fat diet (HFD). Values are presented as mean ± SD. Three-way ANOVA followed by post hoc Tukey HSD test was performed. * p < 0.05, ** p < 0.005. |
Brain β-Amyloid Accumulation
The cortical concentration of β-amyloid in HFD was higher (+26%) than CD in ALA-, but lower (−23%) when compared HFD to CD in the ALA+ group. β-Amyloid level in the cerebral cortex of HFD ALA+ was reduced compared to both CD and HFD ALA- animals (−26% and −41%, respectively) (Figure 6).
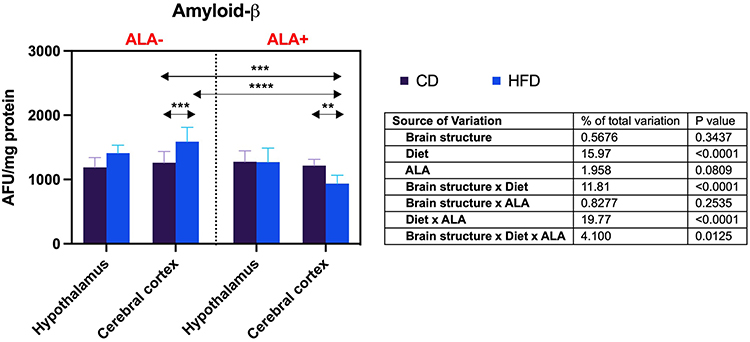 |
Figure 6 Effects of α-lipoic acid (ALA) on brain β-amyloid level in the hypothalamus and cerebral cortex of rats fed a control (CD) and high-fat diet (HFD). Values are presented as mean ± SD. Three-way ANOVA followed by post hoc Tukey HSD test was performed. **p < 0.005, ***p < 0.0005, ****p < 0.0001. |
Discussion
Many epidemiological studies indicate that antioxidants offset the detrimental effects of insulin resistance by restoring physiological redox balance.67–69 Despite reports of the positive impact of antioxidants in eliminating diabetes at the systemic level, only a few studies have evaluated the effects of antioxidants on the cerebral manifestations of insulin resistance.46 This fact should not be surprising since the primary factor limiting the antioxidants in clinical practice is their low bioavailability or inability to pass through the BBB.70 These problems do not apply to α-lipoic acid.51,52 ALA (6,8-dithiooctanoic acid) is a saturated fatty acid with potent antioxidant properties. The carbon atoms 6, 7, and 8 with two sulfur atoms form a dithiol ring that can be reduced to dihydro-lipoic acid (DHLA).35 The molecule’s chemical structure determines its ability to pass through biological membranes and the BBB.51,52 Both ALA and DHLA contain a non-polar aliphatic chain whose presence favors solubility in a lipophilic environment. In contrast, the short length of the carbon chain and the occurrence of a polar carboxyl group make ALA and DHLA also soluble in water.35,36,71 ALA is therefore well absorbed from the gastrointestinal tract and quickly removed from the bloodstream due to its intense liver metabolism. The compound’s half-life is about 30 minutes, making it safe to use and virtually free of any side effects.35,36,71
Our study confirms previous reports of the protective properties of ALA at the systemic level.40–43 ALA normalizes carbohydrate (↓fasting plasma glucose and insulin) and lipid (↓plasma-free fatty acids) metabolism, as well as diminishes systemic insulin sensitivity (↓HOMA-IR) and inflammation (serum ↓IL-1α, ↓IL-2, ↓IL-6, ↓TNF-α, ↑IL-10, and ↓CCL-15) in insulin-resistant rats. ALA also improves energy homeostasis (↓body weight, ↓specific rate of body mass gain, ↓BMI, ↓feed efficiency, ↑food consumption, ↑energy intake), and reduces the risk of cardiometabolic complications noted as a decrease in Lee’s index. It is well known that ALA inhibits intracellular fatty acid accumulation in diabetic patients, increasing insulin sensitivity and thus promoting glucose uptake by the liver, adipose tissue, and skeletal muscles.44,72–74 The hypoglycemic effects of ALA are also related to reducing triacylglycerol (TAG) accumulation in the pancreas and the stimulation of free fatty acid oxidation in mitochondria.75 ALA is a known cofactor of many mitochondrial dehydrogenases involved in cellular respiration and the regulation of cell metabolism.29 During the reduction of ALA to DHLA, this compound inhibits gluconeogenesis by decreasing pyruvate carboxylase (PC) activity and stimulates glycolysis by increasing pyruvate dehydrogenase (PDC) NAD+/NADH ratio. In insulin resistance and diabetes, intracellular increase in NAD+ results in respiratory chain overload and mitochondrial ROS overproduction.29,76 An essential source of free radicals is also hyperglycemia accompanied by polyol pathway activation, non-enzymatic glycation of proteins, glucose autoxidation, as well as induction of protein kinase C (PKC).77,78 Under these conditions, nuclear factor NF-κB is stimulated, increasing inflammation and further enhancing ROS formation. ALA inhibits NF-κB activation, thereby reducing the expression of pro-inflammatory genes, lowering oxidative and carbonyl stress, as well as normalizing lipid-carbohydrate metabolism.26,28,79
The beneficial effects of ALA are also evident at the cerebral level. We noted that ALA improves brain redox homeostasis by decreasing the activity of pro-oxidant enzymes (↓NOX, ↓XO), strengthening the antioxidant barrier (↑FRAP), and effectively preventing oxidation of proteins (↑total thiols) and lipids (↓TBARS) in the hypothalamus of insulin-resistant rats. The antioxidant effects of ALA may result from multiple mechanisms. ALA can directly act as a free radical scavenger, regenerator of low-molecular-weight antioxidants (eg, vitamins C and E), a modulator of gene expression (↓NF-κB), as well as metal ion chelator (eg, Fe2+, Cu2+).37–39 Numerous studies have shown that ALA is capable of scavenging superoxide anions (O2−∙), hydroxyl radicals (∙OH), and peroxynitrite (ONOO−). When reacting with radical and non-radical species, ALA converts to the radical cation ALA+∙ (eg, ALA + ∙OH → ALA+∙ + OH−), which is much less reactive and does not pose a significant risk to the cell.80–82 ALA and DHLA create a potent redox couple, with an oxidation-reduction potential higher than the reduced glutathione (GSH)/oxidized glutathione (GSSG) system. Therefore, DHLA guarantees more effective protection than GSH and can reconstitute the reduced form of other antioxidants.52–54,83,84 ALA may also prevent the consequences of metal-induced neuronal damage.85–87 It is well known that the brain has a low capacity of the antioxidant barrier with a relatively high content of pro-oxidant metals. In the Fenton reaction, these compounds catalyze the formation of the highly reactive hydroxyl radical (eg, Fe2+ + H2O2 → Fe3+ + ∙OH + OH−), which may induce DNA oxidation and neuronal apoptosis. Although we did not evaluate the brain antioxidants separately, the increase in FRAP level may be due to the improved GSH biosynthesis. Indeed, thiol groups have a high contribution to the total antioxidant potential in the FRAP assay, and their most important source in the brain is GSH.88 Thus, it is not surprising that the total thiols in the hypothalamus of HFD ALA+ rats are increased. GSH is the most important cerebral antioxidant. It scavenges free radicals and acts as a neurotransmitter or neuromodulator of synaptic transmission.89 Previous studies have shown that ALA increases GSH biosynthesis by inducing nuclear factor (erythroid 2)-like 2 (Nrf2) or enhancing glutathione reductase (GR) that converts GSSG to GSH.90,91 ALA also increases the availability of neuronal NADPH necessary for GSH synthesis and GSSG reduction.92–94 However, the ALA-mediated inhibition of NOX activity is of particular clinical significance. It was shown that NOX-derived free radicals play an essential role in the intracellular signal transduction leading to activation of the brain inflammatory response. Several studies demonstrated that NOX mediates NF-κB activation by affecting MAPK kinases (p38MAPK, JNK, and ERK). It is also involved in angiotensin II–induced overexpression of ICAM-1, VCAM-1, and MCP-1. NOX-derived ROS also enhances the synthesis of MMP-2 by activating NF-κB signaling.22–28 Therefore, it is not surprising that we observed high expression of IL-1β, IL-6, and MMP-2 in the hypothalamus of HFD-fed animals. Interestingly, more and more studies indicate the involvement of pro-inflammatory cytokines in the modulation of energy homeostasis.95–100 The hypothalamus, where the hunger and satiety centers are located, coordinates food intake behavior.101 Nevertheless, both peripherally produced cytokines and those derived from the blood may change hypothalamic neurotransmission.102 It has been proven that IL-1β and IL-6 act as appetite regulators, while its pharmacological blockade effectively reduces weight gain in HFD animals. The results of our study may support this hypothesis.103 ALA inhibits inflammation at the central (↓MPO, ↓GLU, ↓IL-1β, and ↓IL-6) and systemic levels (↓IL-1α, ↓IL-2, ↓IL-6, ↓TNF-α, ↑IL-10, ↓CCL-15), reduces food intake and promotes weight loss in insulin-resistant rats.3,45,104–108 Of particular note is the inhibition of MPO activity released by neutrophils and monocytes during inflammatory cell activation. MPO significantly exacerbates oxidative stress and metalloproteinases activity, as well as initiates and sustains acute and chronic inflammation.109 Recently, the involvement of matrix metalloproteinases has been increasingly emphasized in neurodegenerative diseases. Studies in a mouse model of multiple sclerosis indicate that ALA inhibits neuronal demyelination by reducing metalloproteinases activity.110 MMP-2 promotes the passage of blood lymphocytes into the CNS; thus, its pharmacological blockade may have important clinical implications.
ALA also affects brain sphingolipid metabolism. Sphingolipids belong to a large group of biologically active lipids that build cell membranes of neurons and glial cells. In addition to the structural functions, sphingolipids regulate these cells’ growth, differentiation, proliferation, and apoptosis.111,112 The central place in the sphingomyelin transduction pathway is occupied by ceramide that can be formed from sphingomyelin using sphingomyelinases (SMases) or as result of de novo condensation of serine with palmitoyl-CoA. Recent studies have shown increased ceramide production in patients with type 2 diabetes. Since ceramide mediates insulin resistance and may also cross the BBB, there is a relationship between peripheral insulin resistance and neurodegeneration.113 It has been demonstrated that ceramide activates JNKs and inhibits IRS-1, resulting in blockage of the insulin transduction pathway.114–116 In our study, ALA reduces ceramide accumulation in both the hypothalamus and cerebral cortex of HFD rats, which may occur through decreased de novo synthesis (↓plasma-free fatty acids), salvage pathway (↓SFO) and sphingomyelinase pathway (↓nSMase). Several studies demonstrated that ROS enhance SMase activity, indicating a direct relationship between the rate of ceramide formation and oxidative-reductive status of the cell.117,118 Increased levels of both acidic and neutral SMases were also observed in the brains of AD patients, which is related to the synaptic dysregulation and progression of neurotoxicity.119 In the in vitro models, exposure to short-chain ceramides results in neuronal insulin resistance, mitochondrial dysfunction, oxidative damage to DNA and lipids, and increased formation of β-amyloid precursor protein (APP).3,120,121 In our study, ALA reduces β-amyloid accumulation in the cerebral cortex of insulin-resistant rats. Although we did not perform behavioral studies, this may suggest an improved cognitive function in HFD-fed animals. We also showed that ALA could inhibit cerebral apoptosis (↓CAS-3) by reducing ceramide accumulation and limiting ROS formation. Previous studies have demonstrated that increased ceramide level intensifies ROS production, enhancing CAS-3 and CAS-8 activity and inducing neuronal apoptosis. Therefore, as in neurodegenerative diseases, ceramide-mediated apoptosis may cause cerebral complications of insulin resistance.3,122 Unfortunately, due to the very small mass of brain tissue, we could not assess insulin signaling in the studied animals. Further studies are needed to evaluate the effects of ALA on insulin sensitivity and brain metabolism.
Conclusions
Summarizing, ALA normalizes body weight as well as systemic lipid and carbohydrate metabolism in insulin-resistant rats. ALA improves systemic oxidative stress and reduces serum pro-inflammatory cytokines, chemokines, and growth factors in insulin-resistant rats. We also showed that ALA improves hypothalamic redox homeostasis by decreasing the activity of pro-oxidant enzymes, enhancing total antioxidant potential, and reducing protein and lipid oxidation. Generally, ALA does not affect the redox balance of the cerebral cortex. In insulin-resistant rats, ALA treatment reduces hypothalamic inflammation and metalloproteinases activity as well as cortical β-amyloid accumulation. In both brain structures, ALA diminishes ceramide synthesis and neuronal apoptosis. ALA may be a potential treatment for patients with cerebral complications of insulin resistance. Further studies are needed to elucidate the molecular mechanisms of ALA action in the brain.
Data Sharing Statement
The datasets generated for this study are available at reasonable request to the corresponding author.
Ethics Statement
The investigation was approved by the Institutional Committee for Ethics Use of Animals in Olsztyn, Poland (approval no. 21/2017).
Funding
This work was supported by the National Science Centre, Poland (grant no. 2018/29/N/NZ4/02011), and by the Medical University of Bialystok, Poland (grant number: SUB/1/DN/21/002/1209).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Zheng Y, Ley SH, Hu FB. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat Rev Endocrinol. 2018;14(2):88–98. doi:10.1038/nrendo.2017.151
2. Arnold SE, Arvanitakis Z, Macauley-Rambach SL, et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: concepts and conundrums. Nat Rev Neurol. 2018;14(3):168–181. doi:10.1038/nrneurol.2017.185
3. Maciejczyk M, Żebrowska E, Chabowski A. Insulin resistance and oxidative stress in the brain: what’s new? Int J Mol Sci. 2019;20(4):874. doi:10.3390/ijms20040874
4. Kellar D, Craft S. Brain insulin resistance in Alzheimer’s disease and related disorders: mechanisms and therapeutic approaches. Lancet Neurol. 2020;19(9):758–766. doi:10.1016/S1474-4422(20)30231-3
5. Vijan S. Type 2 Diabetes. Ann Intern Med. 2019;171(9):ITC65–ITC80. doi:10.7326/AITC201911050
6. Sacerdote A, Dave P, Lokshin V, Bahtiyar G. Type 2 diabetes mellitus, insulin resistance, and vitamin D. Curr Diab Rep. 2019;19(10):101. doi:10.1007/s11892-019-1201-y
7. Czech MP. Insulin action and resistance in obesity and type 2 diabetes. Nat Med. 2017;23(7):804–814. doi:10.1038/nm.4350
8. Kawanami D, Matoba K, Utsunomiya K. Signaling pathways in diabetic nephropathy. Histol Histopathol. 2016;31(10):1059–1067. doi:10.14670/HH-11-777
9. Eichmann TO, Lass A. DAG tales: the multiple faces of diacylglycerol—stereochemistry, metabolism, and signaling. Cell Mol Life Sci. 2015;72(20):3931–3952. doi:10.1007/s00018-015-1982-3
10. Yaribeygi H, Farrokhi FR, Butler AE, Sahebkar A. Insulin resistance: review of the underlying molecular mechanisms. J Cell Physiol. 2019;234(6):8152–8161. doi:10.1002/jcp.27603
11. Petersen MC, Shulman GI. Mechanisms of insulin action and insulin resistance. Physiol Rev. 2018;98(4):2133–2223. doi:10.1152/physrev.00063.2017
12. Gluvic Z, Zaric B, Resanovic I, et al. Link between metabolic syndrome and insulin resistance. Curr Vasc Pharmacol. 2016;15(1):30–39. doi:10.2174/1570161114666161007164510
13. Petrie JR, Guzik TJ, Touyz RM. Diabetes, hypertension, and cardiovascular disease: clinical insights and vascular mechanisms. Can J Cardiol. 2018;34(5):575–584. doi:10.1016/j.cjca.2017.12.005
14. Avogaro A, Fadini GP. Microvascular complications in diabetes: a growing concern for cardiologists. Int J Cardiol. 2019;291:29–35. doi:10.1016/j.ijcard.2019.02.030
15. Tumminia A, Vinciguerra F, Parisi M, Frittitta L. Type 2 diabetes mellitus and Alzheimer’s disease: role of insulin signalling and therapeutic implications. Int J Mol Sci. 2018;19(11):3306. doi:10.3390/ijms19113306
16. Sun Y, Ma C, Sun H, et al. Metabolism: a novel shared link between diabetes mellitus and Alzheimer’s disease. J Diabetes Res. 2020;2020:1–12. doi:10.1155/2020/4981814
17. Maciejczyk M, Żebrowska E, Zalewska A, Chabowski A. Redox balance, antioxidant defense, and oxidative damage in the hypothalamus and cerebral cortex of rats with high fat diet-induced insulin resistance. Oxid Med Cell Longev. 2018;2018:1–11. doi:10.1155/2018/6940515
18. Żebrowska E, Maciejczyk M, Żendzian-Piotrowska M, Zalewska A, Chabowski A. High protein diet induces oxidative stress in rat cerebral cortex and hypothalamus. Int J Mol Sci. 2019;20(7):1547. doi:10.3390/ijms20071547
19. Żebrowska E, Chabowski A, Zalewska A, Maciejczyk M. High-sugar diet disrupts hypothalamic but not cerebral cortex redox homeostasis. Nutrients. 2020;12(10):3181. doi:10.3390/nu12103181
20. Cobley JN, Fiorello ML, Bailey DM. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018;15:490–503. doi:10.1016/j.redox.2018.01.008
21. Saleem U, Sabir S, Niazi SG, Naeem M, Ahmad B. Role of oxidative stress and antioxidant defense biomarkers in neurodegenerative diseases. Crit Rev Eukaryot Gene Expr. 2020;30(4):311–322. doi:10.1615/CritRevEukaryotGeneExpr.2020029202
22. Ma MW, Wang J, Zhang Q, et al. NADPH oxidase in brain injury and neurodegenerative disorders. Mol Neurodegener. 2017;12(1):7. doi:10.1186/s13024-017-0150-7
23. Barua S, Kim JY, Yenari MA, Lee JE. The role of NOX inhibitors in neurodegenerative diseases. IBRO Rep. 2019;7:59–69. doi:10.1016/j.ibror.2019.07.1721
24. Sun GY, Horrocks LA, Farooqui AA. The roles of NADPH oxidase and phospholipases A 2 in oxidative and inflammatory responses in neurodegenerative diseases. J Neurochem. 2007;070611013409004. doi:10.1111/j.1471-4159.2007.04670.x
25. Zhang X, Dong F, Ren J, Driscoll M, Culver B. High dietary fat induces NADPH oxidase-associated oxidative stress and inflammation in rat cerebral cortex. Exp Neurol. 2005;191(2):318–325. doi:10.1016/j.expneurol.2004.10.011
26. Morgan MJ, Liu Z. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011;21(1):103–115. doi:10.1038/cr.2010.178
27. Verdile G, Keane KN, Cruzat VF, et al. Inflammation and oxidative stress: the molecular connectivity between insulin resistance, obesity, and Alzheimer’s disease. Mediators Inflamm. 2015;2015:1–17. doi:10.1155/2015/105828
28. Kopitar-Jerala N. Innate immune response in brain, NF-Kappa B signaling and cystatins. Front Mol Neurosci. 2015;8. doi:10.3389/fnmol.2015.00073.
29. Salehi B, Berkay Yılmaz Y, Antika G, et al. Insights on the use of α-lipoic acid for therapeutic purposes. Biomolecules. 2019;9(8):356. doi:10.3390/biom9080356
30. Papanas N, Ziegler D. Efficacy of α-lipoic acid in diabetic neuropathy. Expert Opin Pharmacother. 2014;15(18):2721–2731. doi:10.1517/14656566.2014.972935
31. Rochette L, Ghibu S, Muresan A, Vergely C. Alpha-lipoic acid: molecular mechanisms and therapeutic potential in diabetes. Can J Physiol Pharmacol. 2015;93(12):1021–1027. doi:10.1139/cjpp-2014-0353
32. Han Y, Wang M, Shen J, et al. Differential efficacy of methylcobalamin and alpha-lipoic acid treatment on symptoms of diabetic peripheral neuropathy. Minerva Endocrinol. 2018;43:1. doi:10.23736/S0391-1977.16.02505-0
33. Ziegler D, Gries FA. α-Lipoic acid in the treatment of diabetic peripheral and cardiac autonomic neuropathy. Diabetes. 1997;46(Supplement_2):S62–S66. doi:10.2337/diab.46.2.S62
34. Ziegler D, Hanefeld M, Ruhnau KJ, et al. Treatment of symptomatic diabetic peripheral neuropathy with the anti-oxidant alpha-lipoic acid. Diabetologia. 1995;38(12):1425–1433. doi:10.1007/BF00400603
35. Packer L, Witt EH, Tritschler HJ. Alpha-lipoic acid as a biological antioxidant. Free Radic Biol Med. 1995;19(2):227–250. doi:10.1016/0891-5849(95)00017-R
36. Gu L, Li S, Bai J, Zhang Q, Han Z. α‐Lipoic acid protects against microcystin‐LR induced hepatotoxicity through regeneration of glutathione via activation of Nrf2. Environ Toxicol. 2020;35(7):738–746. doi:10.1002/tox.22908
37. Thaakur S, Himabindhu G. Effect of alpha lipoic acid on the tardive dyskinesia and oxidative stress induced by haloperidol in rats. J Neural Transm. 2009;116(7):807–814. doi:10.1007/s00702-009-0232-y
38. Myzak MC, Carr AC. Myeloperoxidase-dependent caspase-3 activation and apoptosis in HL-60 cells: protection by the antioxidants ascorbate and (dihydro)lipoic acid. Redox Rep. 2002;7(1):47–53. doi:10.1179/135100002125000181
39. Marsh SA, Pat BK, Gobe GC, Coombes JS. Evidence for a non-antioxidant, dose-dependent role of α -lipoic acid in caspase-3 and ERK2 activation in endothelial cells. Apoptosis. 2005;10(3):657–665. doi:10.1007/s10495-005-1901-4
40. Karafakioğlu YS. Effects of α lipoic acid on noise induced oxidative stress in rats. Saudi J Biol Sci. 2019;26(5):989–994. doi:10.1016/j.sjbs.2018.08.008
41. Feng B, Yan X-F, Xue J-L, Xu L, Wang H. The protective effects of α-lipoic acid on kidneys in type 2 diabetic Goto-Kakisaki rats via reducing oxidative stress. Int J Mol Sci. 2013;14(4):6746–6756. doi:10.3390/ijms14046746
42. Wang L, Wu C-G, Fang C-Q, et al. The protective effect of α-Lipoic acid on mitochondria in the kidney of diabetic rats. Int J Clin Exp Med. 2013;6(2):90–97.
43. Bitar MS, Wahid S, Pilcher CW, Al-Saleh E, Al-Mulla F. α-lipoic acid mitigates insulin resistance in goto-kakizaki rats. Horm Metab Res. 2004;36(8):542–549. doi:10.1055/s-2004-825760
44. Erickson N, Zafron M, Harding SV, Marinangeli CPF, Rideout TC. Evaluating the lipid-lowering effects of α-lipoic acid supplementation: a systematic review. J Diet Suppl. 2020;17(6):753–767. doi:10.1080/19390211.2019.1651436
45. Fiedler SE, Spain RI, Kim E, Salinthone S. Lipoic acid modulates inflammatory responses of monocytes and monocyte‐derived macrophages from healthy and relapsing‐remitting multiple sclerosis patients. Immunol Cell Biol. 2021;99(1):107–115. doi:10.1111/imcb.12392
46. Ahuja S, Uniyal A, Akhtar A, Sah SP. Alpha lipoic acid and metformin alleviates experimentally induced insulin resistance and cognitive deficit by modulation of TLR2 signalling. Pharmacol Rep. 2019;71(4):614–623. doi:10.1016/j.pharep.2019.02.016
47. Henriksen EJ. Exercise training and the antioxidant α-lipoic acid in the treatment of insulin resistance and type 2 diabetes. Free Radic Biol Med. 2006;40(1):3–12. doi:10.1016/j.freeradbiomed.2005.04.002
48. Weinstein RB, Tritschler HJ, Henriksen EJ. Antioxidant alpha-lipoic acid and protein turnover in insulin-resistant rat muscle. Free Radic Biol Med. 2001;30(4):383–388. doi:10.1016/S0891-5849(00)00489-5
49. Castro MC, Villagarcía HG, Massa ML, Francini F. Alpha-lipoic acid and its protective role in fructose induced endocrine-metabolic disturbances. Food Funct. 2019;10(1):16–25. doi:10.1039/C8FO01856A
50. Lei L, Zhu Y, Gao W, et al. Alpha-lipoic acid attenuates endoplasmic reticulum stress-induced insulin resistance by improving mitochondrial function in HepG2 cells. Cell Signal. 2016;28(10):1441–1450. doi:10.1016/j.cellsig.2016.06.024
51. Della Giustina A, Goldim MP, Danielski LG, et al. Alpha-lipoic acid attenuates acute neuroinflammation and long-term cognitive impairment after polymicrobial sepsis. Neurochem Int. 2017;108:436–447. doi:10.1016/j.neuint.2017.06.003
52. Zhao L, Hu F-X. α-Lipoic acid treatment of aged type 2 diabetes mellitus complicated with acute cerebral infarction. Eur Rev Med Pharmacol Sci. 2014;18(23):3715–3719.
53. Karkabounas S, Papadopoulos N, Anastasiadou C, et al. Effects of α -lipoic acid, carnosine, and thiamine supplementation in obese patients with type 2 diabetes mellitus: a randomized, double-blind study. J Med Food. 2018;21(12):1197–1203. doi:10.1089/jmf.2018.0007
54. Altunina NV, Lizogub VG, Bondarchuk OM. Alpha-lipoic acid as a means of influence on systemic inflammation in type 2 diabetes mellitus patients with prior myocardial infarction. J Med Life. 2020;13(1):32–36. doi:10.25122/jml-2020-0018
55. Yang Y, Li W, Liu Y, Li Y, Gao L, Zhao J. Alpha-lipoic acid attenuates insulin resistance and improves glucose metabolism in high fat diet-fed mice. Acta Pharmacol Sin. 2014;35(10):1285–1292. doi:10.1038/aps.2014.64
56. Novelli ELB, Diniz YS, Galhardi CM, et al. Anthropometrical parameters and markers of obesity in rats. Lab Anim. 2007;41(1):111–119. doi:10.1258/002367707779399518
57. Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and β-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28(7):412–419. doi:10.1007/BF00280883
58. Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res. 1994;74(6):1141–1148. doi:10.1161/01.RES.74.6.1141
59. Prajda N, Weber G. Malignant transformation-linked imbalance: decreased xanthine oxidase activity in hepatomas. FEBS Lett. 1975;59(2):245–249. doi:10.1016/0014-5793(75)80385-1
60. Benzie IFF, Strain JJ. The ferric reducing ability of plasma (FRAP) as a measure of “antioxidant power”: the FRAP assay. Anal Biochem. 1996;239(1):70–76. doi:10.1006/abio.1996.0292
61. Ellman GL. Tissue sulfhydryl groups. Arch Biochem Biophys. 1959;82(1):70–77. doi:10.1016/0003-9861(59)90090-6
62. Buege JA, Aust SD. Microsomal lipid peroxidation. Methods Enzymol. 1978. doi:10.1016/S0076-6879(78)52032-6
63. Kruidenier L, Kuiper I, van Duijn W, et al. Imbalanced secondary mucosal antioxidant response in inflammatory bowel disease. J Pathol. 2003;201(1):17–27. doi:10.1002/path.1408
64. Borys J, Maciejczyk M, Antonowicz B, Sidun J, Świderska M, Zalewska A. Free radical production, inflammation and apoptosis in patients treated with titanium mandibular fixations—an observational study. Front Immunol. 2019;10:2662. doi:10.3389/fimmu.2019.02662
65. Meki ARMA, Esmail EEDF, Hussein AA, Hassanein HM. Caspase-3 and heat shock protein-70 in rat liver treated with aflatoxin B1: effect of melatonin. Toxicon. 2004;43(1):93–100. doi:10.1016/j.toxicon.2003.10.026
66. LeVine H. Quantification of beta-sheet amyloid fibril structures with thioflavin T. Methods Enzym. 1999;309:274–284. doi:10.1016/S0076-6879(99
67. Evans JL. Antioxidants: do they have a role in the treatment of insulin resistance? Indian J Med Res. 2007;125(3):355–372. doi:10.1007/s00125-019-4937-7
68. Anna Z, Joanna K, Sara Z, et al. N-acetylcysteine supplementation did not reverse mitochondrial oxidative stress, apoptosis, and inflammation in the salivary glands of hyperglycemic rats. Nutr Diabetes. 2021;11(1):35. doi:10.1038/s41387-021-00177-w
69. Drygalski K, Fereniec E, Zalewska A, Krętowski A, Żendzian-Piotrowska M, Maciejczyk M. Phloroglucinol prevents albumin glycation as well as diminishes ROS production, glycooxidative damage, nitrosative stress and inflammation in hepatocytes treated with high glucose. Biomed Pharmacother. 2021;142:111958. doi:10.1016/j.biopha.2021.111958
70. Aoyama K. Glutathione in the Brain. Int J Mol Sci. 2021;22(9):5010. doi:10.3390/ijms22095010
71. Ghibu S, Richard C, Vergely C, Zeller M, Cottin Y, Rochette L. Antioxidant properties of an endogenous thiol: alpha-lipoic acid, useful in the prevention of cardiovascular diseases. J Cardiovasc Pharmacol. 2009;54(5):391–398. doi:10.1097/FJC.0b013e3181be7554
72. Genazzani AD, Shefer K, Della Casa D, et al. Modulatory effects of alpha-lipoic acid (ALA) administration on insulin sensitivity in obese PCOS patients. J Endocrinol Invest. 2018;41(5):583–590. doi:10.1007/s40618-017-0782-z
73. Kamenova P. Improvement of insulin sensitivity in patients with type 2 diabetes mellitus after oral administration of alpha-lipoic acid. Hormones. 2006;5(4):251–258. doi:10.14310/horm.2002.11191
74. Evans JL, Goldfine ID. α-Lipoic acid: a multifunctional antioxidant that improves insulin sensitivity in patients with type 2 diabetes. Diabetes Technol Ther. 2000;2(3):401–413. doi:10.1089/15209150050194279
75. Huong DTT, Ide T. Dietary lipoic acid-dependent changes in the activity and mRNA levels of hepatic lipogenic enzymes in rats. Br J Nutr. 2008;100(1):79–87. doi:10.1017/S0007114507876227
76. Zempleni J, Trusty TA, Mock DM. Lipoic acid reduces the activities of biotin-dependent carboxylases in rat liver. J Nutr. 1997;127(9):1776–1781. doi:10.1093/jn/127.9.1776
77. Mizuno M, Packer L. Effects of α-lipoic acid and dihydrolipoic acid on expression of proto-oncogene c-fos. Biochem Biophys Res Commun. 1994;200(2):1136–1142. doi:10.1006/bbrc.1994.1569
78. Román-Pintos LM, Villegas-Rivera G, Rodríguez-Carrizalez AD, Miranda-Díaz AG, Cardona-Muñoz EG. Diabetic polyneuropathy in type 2 diabetes mellitus: inflammation, oxidative stress, and mitochondrial function. J Diabetes Res. 2016;2016:1–16. doi:10.1155/2016/3425617
79. Solinas G, Karin M. JNK1 and IKKβ: molecular links between obesity and metabolic dysfunction. FASEB J. 2010;24(8):2596–2611. doi:10.1096/fj.09-151340
80. Matsugo S, Konishi T, Matsuo D, Tritschler HJ, Packer L. Reevaluation of superoxide scavenging activity of dihydrolipoic acid and its analogues by chemiluminescent method using 2-methyl-6-[p-methoxyphenyl]-3,7-dihydroimidazo-[1,2-a]pyrazine-3-one (MCLA) as a superoxide probe. Biochem Biophys Res Commun. 1996;227(1):216–220. doi:10.1006/bbrc.1996.1492
81. Zhao F, Liu Z-Q. Comparison of antioxidant effectiveness of lipoic acid and dihydrolipoic acid. J Biochem Mol Toxicol. 2011;25(4):216–223. doi:10.1002/jbt.20378
82. Packer L, Kraemer K, Rimbach G. Molecular aspects of lipoic acid in the prevention of diabetes complications. Nutrition. 2001;17(10):888–895. doi:10.1016/S0899-9007(01)00658-X
83. Dworacka M, Chukanova G, Iskakova S, et al. New arguments for beneficial effects of alpha-lipoic acid on the cardiovascular system in the course of type 2 diabetes. Eur J Pharm Sci. 2018;117:41–47. doi:10.1016/j.ejps.2018.02.009
84. Ibrahimpasic K. Alpha lipoic acid and glycaemic control in diabetic neuropathies at type 2 diabetes treatment. Med Arch. 2013;67(1):7. doi:10.5455/medarh.2013.67.7-9
85. Ou P, Tritschler HJ, Wolff SP. Thioctic (lipoic) acid: a therapeutic metal-chelating antioxidant? Biochem Pharmacol. 1995;50(1):123–126. doi:10.1016/0006-2952(95)00116-H
86. Suh JH, Zhu B-Z, DeSzoeke E, Frei B, Hagen TM. Dihydrolipoic acid lowers the redox activity of transition metal ions but does not remove them from the active site of enzymes. Redox Rep. 2004;9(1):57–61. doi:10.1179/135100004225003923
87. Shay KP, Moreau RF, Smith EJ, Smith AR, Hagen TM. Alpha-lipoic acid as a dietary supplement: molecular mechanisms and therapeutic potential. Biochim Biophys Acta - Gen Subj. 2009;1790(10):1149–1160. doi:10.1016/j.bbagen.2009.07.026
88. Huang S-F, Othman A, Koshkin A, et al. Astrocyte glutathione maintains endothelial barrier stability. Redox Biol. 2020;34:101576. doi:10.1016/j.redox.2020.101576
89. Bottino F, Lucignani M, Napolitano A, et al. In vivo brain GSH: MRS methods and clinical applications. Antioxidants. 2021;10(9):1407. doi:10.3390/antiox10091407
90. Bian H, Wang G, Huang J, et al. Dihydrolipoic acid protects against lipopolysaccharide-induced behavioral deficits and neuroinflammation via regulation of Nrf2/HO-1/NLRP3 signaling in rat. J Neuroinflammation. 2020;17(1):166. doi:10.1186/s12974-020-01836-y
91. Alam S-I, Kim M-W, Shah FA, Saeed K, Ullah R, Kim M-O. Alpha-linolenic acid impedes cadmium-induced oxidative stress, neuroinflammation, and neurodegeneration in mouse brain. Cells. 2021;10(9):2274. doi:10.3390/cells10092274
92. O’Neill HC, Rancourt RC, White CW. Lipoic acid suppression of neutrophil respiratory burst: effect of NADPH. Antioxid Redox Signal. 2008;10(2):277–286. doi:10.1089/ars.2007.1890
93. Cavdar Z, Oktan MA, Ural C, et al. Alpha lipoic acid attenuates iron induced oxidative acute kidney injury in rats. Biotech Histochem. 2021;96(6):409–417. doi:10.1080/10520295.2020.1812001
94. Biewenga GP, Haenen GRMM, Bast A. The pharmacology of the antioxidant lipoic acid. Gen Pharmacol Vasc Syst. 1997;29(3):315–331. doi:10.1016/S0306-3623(96)00474-0
95. Kim YA, Keogh JB, Clifton PM. Probiotics, prebiotics, synbiotics and insulin sensitivity. Nutr Res Rev. 2018;31(1):35–51. doi:10.1017/S095442241700018X
96. Orihuela R, McPherson CA, Harry GJ. Microglial M1/M2 polarization and metabolic states. Br J Pharmacol. 2016;173(4):649–665. doi:10.1111/bph.13139
97. Barra NG, Henriksbo BD, Anhê FF, Schertzer JD. The NLRP3 inflammasome regulates adipose tissue metabolism. Biochem J. 2020;477(6):1089–1107. doi:10.1042/BCJ20190472
98. Zhao P, Wong K, Sun X, et al. TBK1 at the crossroads of inflammation and energy homeostasis in adipose tissue. Cell. 2018;172(4):731–743.e12. doi:10.1016/j.cell.2018.01.007
99. Omran F, Christian M. Inflammatory signaling and brown fat activity. Front Endocrinol (Lausanne). 2020;11. doi:10.3389/fendo.2020.00156.
100. Phillips C, Shivappa N, Hébert J, Perry I. Dietary inflammatory index and biomarkers of lipoprotein metabolism, inflammation and glucose homeostasis in adults. Nutrients. 2018;10(8):1033. doi:10.3390/nu10081033
101. Challet E. The circadian regulation of food intake. Nat Rev Endocrinol. 2019;15(7):393–405. doi:10.1038/s41574-019-0210-x
102. Hori T, Nakashima T, Take S, Kaizuka Y, Mori T, Katafuchi T. Immune cytokines and regulation of body temperature, food intake and cellular immunity. Brain Res Bull. 1991;27(3–4):309–313. doi:10.1016/0361-9230(91
103. Shirazi R, Palsdottir V, Collander J, et al. Glucagon-like peptide 1 receptor induced suppression of food intake, and body weight is mediated by central IL-1 and IL-6. Proc Natl Acad Sci. 2013;110(40):16199–16204. doi:10.1073/pnas.1306799110
104. Salinthone S, Yadav V, Schillace RV, Bourdette DN, Carr DW. Lipoic acid attenuates inflammation via cAMP and protein kinase A signaling. PLoS One. 2010;5(9):e13058. doi:10.1371/journal.pone.0013058
105. Matsumoto J, Dohgu S, Takata F, et al. TNF-α-sensitive brain pericytes activate microglia by releasing IL-6 through cooperation between IκB-NFκB and JAK-STAT3 pathways. Brain Res. 2018;1692:34–44. doi:10.1016/j.brainres.2018.04.023
106. Petronilho F, Florentino D, Danielski LG, et al. Alpha-lipoic acid attenuates oxidative damage in organs after sepsis. Inflammation. 2016;39(1):357–365. doi:10.1007/s10753-015-0256-4
107. Selvakumar E, Prahalathan C, Sudharsan PT, Varalakshmi P. Protective effect of lipoic acid on cyclophosphamide-induced testicular toxicity. Clin Chim Acta. 2006;367(1–2):114–119. doi:10.1016/j.cca.2005.11.034
108. Doggrell SA. α-Lipoic acid, an anti-obesity agent? Expert Opin Investig Drugs. 2004;13(12):1641–1643. doi:10.1517/13543784.13.12.1641
109. Chen S, Chen H, Du Q, Shen J. Targeting Myeloperoxidase (MPO) mediated oxidative stress and inflammation for reducing brain ischemia injury: potential application of natural compounds. Front Physiol. 2020;11. doi:10.3389/fphys.2020.00433.
110. Xie H, Yang X, Cao Y, Long X, Shang H, Jia Z. Role of lipoic acid in multiple sclerosis. CNS Neurosci Ther. 2021. doi:10.1111/cns.13793
111. van Echten-deckert G, Herget T. Sphingolipid metabolism in neural cells. Biochim Biophys Acta. 2006;1758(12):1978–1994. doi:10.1016/j.bbamem.2006.06.009
112. Giussani P, Prinetti A, Tringali C. The role of Sphingolipids in myelination and myelin stability and their involvement in childhood and adult demyelinating disorders. J Neurochem. 2021;156(4):403–414. doi:10.1111/jnc.15133
113. De La Monte SM, Lyn-Cook LE, Lawton M, et al. Hepatic ceramide may mediate brain insulin resistance and neurodegeneration in type 2 diabetes and non-alcoholic steatohepatitis. Adv Alzheimers Dis. 2011;1:179–199. doi:10.3233/978-1-60750-733-8-179
114. Mandal N, Grambergs R, Mondal K, Basu SK, Tahia F, Dagogo-Jack S. Role of ceramides in the pathogenesis of diabetes mellitus and its complications. J Diabetes Complications. 2021;35(2):107734. doi:10.1016/j.jdiacomp.2020.107734
115. Parveen F, Bender D, Law S-H, Mishra VK, Chen -C-C, Ke L-Y. Role of ceramidases in sphingolipid metabolism and human diseases. Cells. 2019;8(12):1573. doi:10.3390/cells8121573
116. Yaribeygi H, Bo S, Ruscica M, Sahebkar A. Ceramides and diabetes mellitus: an update on the potential molecular relationships. Diabet Med. 2020;37(1):11–19. doi:10.1111/dme.13943
117. Kitatani K, Akiba S, Sato T. Ceramide-induced enhancement of secretory phospholipase A2 expression via generation of reactive oxygen species in tumor necrosis factor-α-stimulated mesangial cells. Cell Signal. 2004;16(8):967–974. doi:10.1016/j.cellsig.2004.02.003
118. Cataldi S, Borrelli A, Ceccarini MR, et al. Neutral sphingomyelinase modulation in the protective/preventive role of rMnSOD from radiation-induced damage in the brain. Int J Mol Sci. 2019;20(21):5431. doi:10.3390/ijms20215431
119. Askari VR, Shafiee-Nick R. The protective effects of β-caryophyllene on LPS-induced primary microglia M1/M2 imbalance: a mechanistic evaluation. Life Sci. 2019;219:40–73. doi:10.1016/j.lfs.2018.12.059
120. Jazvinšćak Jembrek M, Hof PR, Šimić G. Ceramides in Alzheimer’s disease: key mediators of neuronal apoptosis induced by oxidative stress and Aβ accumulation. Oxid Med Cell Longev. 2015;2015:1–17. doi:10.1155/2015/346783
121. Jung J-S, Choi M-J, Ko H-M, Kim H-S. Short-chain C2 ceramide induces heme oxygenase-1 expression by upregulating AMPK and MAPK signaling pathways in rat primary astrocytes. Neurochem Int. 2016;94:39–47. doi:10.1016/j.neuint.2016.02.004
122. Movsesyan VA, Yakovlev AG, Dabaghyan EA, Stoica BA, Faden AI. Ceramide induces neuronal apoptosis through the caspase-9/caspase-3 pathway. Biochem Biophys Res Commun. 2002;299(2):201–207. doi:10.1016/S0006-291X(02)02593-7
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.