")
Back to Journals » OncoTargets and Therapy » Volume 12
Estradiol promotes the progression of ER+ breast cancer through methylation-mediated RSK4 inactivation
Authors Li Q, Gao H, Yang H, Wei W, Jiang Y
Received 16 March 2019
Accepted for publication 22 June 2019
Published 22 July 2019 Volume 2019:12 Pages 5907—5916
DOI https://doi.org/10.2147/OTT.S208988
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Arseniy Yuzhalin
Qiuyun Li,1,* Hui Gao,2,* Huawei Yang,1 Wei Wei,1 Yi Jiang1
1Department of Breast Surgery, The Affiliated Tumor Hospital of Guangxi Medical University, Nanning, Guangxi 530021, People’s Republic of China; 2Department of Breast Surgery, Ruikang Hospital Affiliated to Guangxi University of Chinese Medicine, Nanning, Guangxi 530001, People’s Republic of China
*These authors contributed equally to this work
Background: It’s well documented that estrogen plays a vital role in breast cancer progression, but the molecular mechanisms underlying it still remains incompletely clear. This study aimed to explore whether ribosomal protein S6 kinase 4 (RSK4) was involved in estrogen-induced breast cancer development and its underlying mechanism.
Methods: The expressions of estrogen receptor (ER) and RSK4 were assessed by immunohistochemistry, RT-PCR and Western blotting. The methylation of RSK4 promoter was evaluated by bisulfite genomic sequencing. MTT, clone formation, flow cytometry and Transwell chamber assays were performed to detect cell proliferation, clone formation, apoptosis and invasion abilities. Luciferase gene reporter assay was used to detect the transcriptional activity of RSK4. The expressions of methyltransferases, such as DNMT1, DNMT3A and DNMT3B were tested by Western blotting.
Results: ER expression and RSK4 methylation were significantly elevated in ER positive (ER+) breast cancer tissues and MCF-7 cells, whereas RSK4 expression was reduced. 17β-estradiol (E2) treatment obviously decreased RSK4 expression and transcriptional activity, as well as promoted cell proliferation, clone formation and invasion and reduced cell apoptosis in ER+ MCF-7 cells. Furthermore, E2 reduced RSK4 expression through promoting DNMT3B-induced RSK4 methylation.
Conclusion: In summary, the present study reveals that estrogen promotes the progression of breast cancer through methylation-mediated RSK4 inactivation in ER+ breast cancer. Our study might provide a novel target for the treatment of ER+ breast cancer.
Keywords: estrogen, RSK4, DNMT3B, ER positive, methylation
Introduction
Breast cancer is the most frequently diagnosed malignant tumor and remains the leading cause of cancer-related death in women worldwide.1 In China, it is estimated that the incidence of breast cancer is increasing at an annual rate of 3%, with the fastest growing mortality in urban area.2 Nowadays, breast cancer is unambiguously divided into four subtypes, Luminal A [ER+/PR+/HER2-, grade 1 or grade 2], Luminal B (ER+/PR+/HER2+, grade 3), HER2 overexpression (ER-/PR-/HER2+) and Triple Negative Breast Cancer (TNBC, ER-/PR-/HER2-).3 Among them, ER+ subtype accounts for more than 67% of all kinds of breast cancer cases, and the anti-estrogen adjuvant therapy is the preferred treatment approach for these patients, with a reduced 15-year mortality of 33%.4,5 However, the prognosis of patients with ER+ breast cancer is unsatisfactory. Therefore, it is required to further explore the molecular mechanisms of estrogen in the progression of breast cancer.
Ribosomal protein S6 kinase 4 (RSK4) is a member of p90 ribosomal S6 kinase (RSK) family, which consists of both the activating and inhibiting isoforms of MAPK/ERK pathway.6,7 On the contrary of other RSK family members, such as RSK1-3, RSK4 has been identified to be constitutively activated in serum-starved cells and then inhibits cell growth.8 RSK4 has been shown to be downregulated in breast cancer tissues with increased DNA methylated modification in its promoter region.9,10 In addition, RSK4 was reported to serve as a tumor suppressor in breast cancer, as RSK4 overexpression significantly inhibited cell proliferation, invasion, migration and tumorigenesis.9,11
Specificity protein (SP1), the first identified transcription factor, is frequently overexpressed in multiple kinds of cancers,12,13 including breast cancer.14 SP1 recognizes G-rich motifs and modulates gene expression via 3 C2H2-type zinc fingers located at the C-terminus, leading to tumorigeneis.15 However, whether SP1 interacts with RSK4 promoter and regulates RSK4 expression remains unclear.
In the present study, we explored the effects and mechanisms of estrogen in the progression of breast cancer. Our results showed that E2 negatively regulated RSK4 expression through repressing its interaction with SP1 and enhancing its DNA methylation, which then accelerated the progression of breast cancer.
Materials and methods
Ethics statement
For experiments involving human samples, the protocols have been performed in accordance with the Helsinki Declaration and were approved by the Ethics Committee of The Affiliated Tumor Hospital of Guangxi Medical University. All participators had signed the informed consents before this study.
Tissue samples
Seventeen paired breast cancer tissues and the adjoining normal breast tissues were obtained from patients with ER+ breast cancer. All patients have accepted adenomammectomy as the first treatment.
Immunohistochemistry (IHC)
IHC analysis was carried to test RSK4 and ER expression profiles in human breast cancer and normal tissues. The tissues were embedded by paraffin and cut into 5-μm slides. Then, the slides were dewaxed with xylene, and rehydrated with an ethanol gradient, followed by 3% H2O2 incubation to block endogenous peroxidase activity. Subsequently, the slides were blocked with 5% goat serum (Solarbio, Beijing, China), and incubated with anti‑RSK4 (1:150 dilution; No.ab76117, Abcam, MA, USA) or anti‑ER (1:100 dilution; No. ab3575, Abcam) antibody at 4 °C overnight. After washing with phosphate-buffered saline (PBS), the slides were incubated with peroxidase‑conjugated secondary antibodies (Abcam) for 1 h at room temperature. Protein expression were enhanced with diaminobenzene (DAB) staining.
Cell culture and treatment
Human breast epithelial HBL-100 cells incorporating SV40 gene and breast cancer ER+ cell line MCF-7, ER-negative (ER-) cell lines MDA-MB-231 and MDA-MB-453 were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). MCF-7 cells were cultured in Eagle’s Minimum Essential Medium (Gibco, MA, USA); MDA-MB-231 and MDA-MB-453 cells were cultured in Leibovitz’s L-15 Medium (Gibco); HBL-100 cells were maintained in RPMI-1640 medium (Gibco). To make the complete growth medium, 10% fetal bovine serum (FBS; Gibco) was added to all kinds of cell culture mediums.
For E2 treatment, MCF-7 cells were incubated with 10, 50 or 100 nM E2 (Sigma-Aldrich Corp, MO, USA) dissolved in 0.1% (vol/vol) DMSO for indicated times.
Cell transfection
SP1 overexpressing vector (SP1-Vector) and its negative control vector (Vector-NC) were obtained from OriGene (Beijing, China), and were transfected into cells using Lipofectamine 2000 (Thermo Fisher Scientific, MA, USA) according to the manufacturer’s recommendation. Short hair RNAs (shRNAs) used to silence DNMT3A (sh-DNMT3A), DNMT1 (sh-DNMT1), and DNMT3B (sh-DNMT3B) in breast cancer cells and their negative control lentivirus vectors were designed and synthesized by GenePharma (Shanghai, China).
Real-time PCR (RT-PCR)
Total RNA was extracted from tissues and cells using TRIzol reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions. After the quality and quantity of total RNA determination by a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, USA), 1 μg of total RNA was reversely transcribed into cDNA using PrimeScript First Strand cDNA Synthesis Kit (Transgen, Beijing, China).The RT-PCR was then performed on ABI Prism 7900 HT using TransScript Green Two-Step qRT-PCR SuperMix (Transgen). The expressions of mRNA were normalized to GAPDH level. All primers used in this study were listed in Table 1.
 |
Table 1 Sequences used in RT-PCR |
Western blotting assay
Total protein extraction from cultured breast cancer cells were performed using RIPA lysis buffer containing 1X protease inhibitor cocktail. After protein concentrations measurement using the BCA method, equal protein (about 20–30 μg) from different samples were separated by 10% SDS‑PAGE and subsequently transferred into the polyvinylidene difluoride (PVDF) membranes (Millipore Corp., Billerica, MA, USA). Next, the membranes were immersed into 5% non-fat milk diluted in TBST for 1 h at room temperature, followed by incubation with the indicated primary antibodies overnight at 4 °C, including anti‑RSK4 (1:2000 dilution; No.ab76117, Abcam), anti‑SP1 (1:1000 dilution; No. ab227383, Abcam), anti-DNMT1 (1:2000 dilution; No. #5032, Cell Signaling Technology, MA, USA), anti-DNMT3A (1:2000 dilution; No. #2160, Cell Signaling Technology), anti-DNMT3B (1:2000 dilution; No. #67,259, Cell Signaling Technology), anti-Ub (1:1000 dilution; No. #3933, Cell Signaling Technology) and anti-GAPDH (No. ab181602, Abcam). After being washed with Tris Buffered Saline with Tween-20 (TBST) for 3 times, the membranes were probed with the corresponding secondary antibodies at room temperature for 1 h. Band intensity was measured using chemiluminescent reagents (Millipore) and quantified by ImageJ software (verson1.48, National Institutes of Health, MD, USA).
Immunoprecipitation (IP)
The ubiquitination of DNMT3B was assessed by IP assay. Briefly, breast cancer cells were lysed using 5 ml of lysis buffer which consists of 50 mM Tris–HCl (pH 7.5), 200 mM NaCl, 0.5% Nonidet P40 and protease inhibitor cocktail. After 1 h of incubation with 50 μl of protein A, the supernatants were incubated with antibody against DNMT3B (No. #67,259, Cell Signaling Technology) at 4 °C overnight. Subsequently, the beads were washed with Western/IP lysis buffer for five times, followed by being resuspended in SDS-PAGE loading buffer (Beyotime), and evaluated by Western blotting analysis with antibodies against Ub (No. No. #3933, Cell Signaling Technology).
MTT assay for cell proliferation detection
MTT assay was carried out to determine cell proliferation. Briefly, cells were seeded in 96-well plates and cultured overnight, followed by E2 treatment for 1, 2, 3, 4 or 5 days. Then, the medium was removed and 20 μl MTT solution (5 mg/ml) was added into each well. After incubation for 4 h, 150 μl DMSO was added into each well and cultured for 10 min with shock. The absorbance was measured at OD =590 nm.
Colony formation assay
Two hundred breast cancer cells with E2 treatment or not were inoculated into 6-well plate and cultured for 14 days. The cells were then incubated with 0.2% crystal violet (Solarbio, Beijing, China) for 10 min. The number of colonies (containing ≥50 cells) was counted under a microscope.
Flow cytometry assay
After incubation with E2 for 48 h, MCF-7 cells were collected and washed with PBS once, followed by incubation with the Annexin V-FITC and PI using a cell apoptosis detection kit (BD, USA). The fluorescent signal was measured by flow cytometry within 1 h of staining. FITC−/PI− quadrant represents living cells, FITC+/PI− represents early apoptotic cells and FITC+/PI+ represents late apoptotic cells.
Transwell chamber assay
E2 treated or untreated MCF-7 cells were suspended in the FBS-free culture medium at 1×106/mL density. Then, 100 μL of cell suspension was added into the upper chamber of the Transwell inserts which were pre-coated with 1 mg/mL Matrigel (BD Biosciences, USA), while 600 μL medium was added into the lower well. After incubation at 37 °C for another 48 h, cells in upper surface were gently removed with swabs and cells at the bottom were fixed with cold methanol for 10 min and stained with 0.2% crystal violet for 10 min at room temperature.
Dual-luciferase reporter assay
The wild-type (WT) or mutated type (MUT) of putative SP1 binding site were cloned into the luciferase reporter vector pGL3-Basic (Promega, WI, USA). Then, MCF-7 cells were co-transfected with WT or MUT vector and Vector-SP1 or Vector-NC using lipofectamine 2000 according to the manufactory’s instructions with E2 treatment or not. After 48 h of transfection, cells were harvested and the relative luciferase activities were detected by using Dual-luciferase Assay System (Promega).
Bisulfite genomic sequencing
Bisulfite genomic DNA sequencing was performed according to previous study.10 The CpG island at the promoter region of RSK4 gene is located in −496 to +28 bp. The bisulfite-treated DNA was first subjected to PCR assay to amplify the RSK4 promoter region using the following primers, forward: 5ʹ-TTTTTGGAGAGGTAGTATAGAGTTGT-3ʹ, reverse-5ʹ- TAATAACATCTCCCCTTCAAAA-3ʹ. Then, the PCR products were cloned into the pMD18-T Vector (TaKaRa, Japan) and subsequently transformed into Escherichia coli strain DH5a. Six positive clones were analyzed using ABI 3730 DNA Sequencer (Thermo Fisher Scientific). The methylation was calculated as the number of methylated CpG dinucleotides/(5×48)×100%.
Statistical analysis
All data in this study were obtained from ≥3 individual experiments and presented as mean ± standard deviation (SD). The statistical analysis was executed with SPSS 20.0 software, and students ’ t test was used for comparison between 2 groups and one-way analysis of variance (ANOVA) was used for multiple groups (≥3 groups) comparison. The P-value <0.05 was thought to be statistically different.
Results
Different expression patterns of ER and RSK4 in breast cancer tissues and cells
To uncover whether RSK4 was involved in estrogen-mediated ER+ breast cancer progression, we first assessed the expression profiles of ER and RSK4 in breast cancer tissues and cells using immunohistochemistry, RT-PCR and Western blotting technologies. Compared with the para-cancerous normal tissues, the expression levels of RSK4 were notably reduced in ER+ breast cancer tissues (17 paired; Figure 1A), while ER expression was significantly elevated (Figure 1B), as well as the mRNA levels of ER and RSK4 detected by RT-PCR (Figure 1C and D). Besides, we also detected the methylation of RSK4 promoter in 9 paired breast cancer tissues and the surrounding normal tissues using bisulfite direct sequencing assay. The results demonstrated that the methylation level of RSK4 promoter in breast cancer tissues was significantly higher than that in normal tissues (Figure 1E). Similarly, RSK4 methylation level was obviously increased in breast cancer MCF-7, MDA-MB-231 and MDA-MB-453 cells as compared with that of the normal breast epithelial HBL-100 cells (Figure 1F), as well as a reduced expression of RSK4 (Figure 1G and H). In addition, the expression of ER was significantly increased in ER+ MCF-7 cells compared to HBL-100 cells, while it was significantly reduced in ER- MDA-MB-231 and MDA-MB-453 cells as compared with that of HBL-100 cells (Figure 1G and H). Together, these results demonstrated that ER expression was elevated in ER+ breast cancer tissues and cells, while RSK4 expression was decreased.
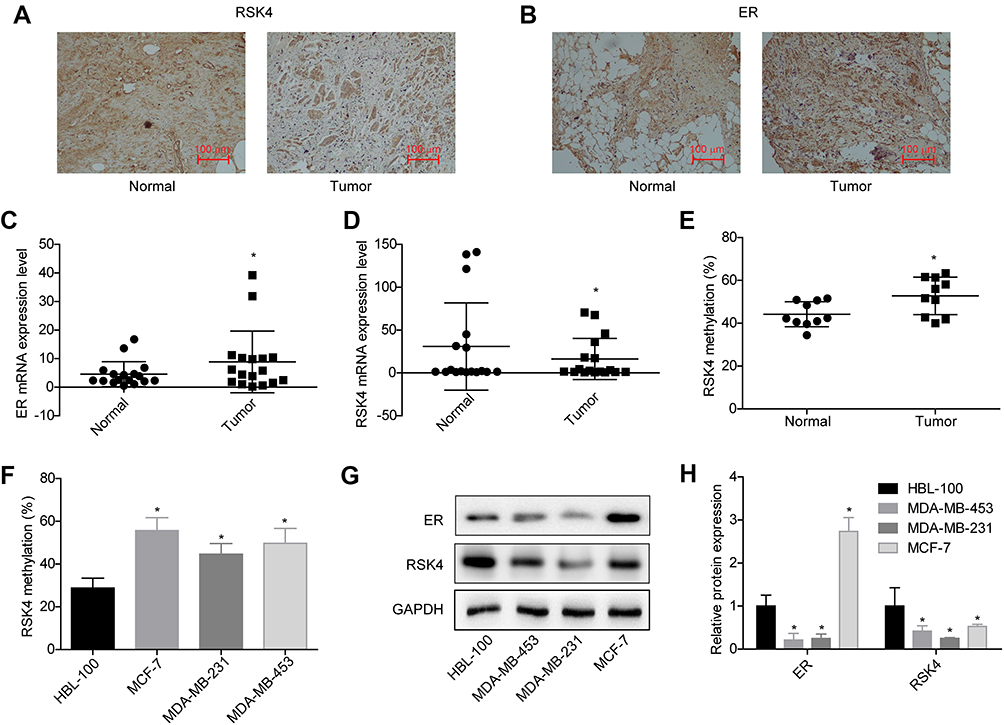 |
Figure 1 Evaluation of the expression patterns of ER and RSK4 in ER+ breast cancer tissues and cell lines. (A and B) ER and RSK4 expressions in 17 pairs of breast cancer and adjacent non-cancerous tissues were assessed by immunohistochemistry, the repressive images were shown. (C and D) The mRNA levels of ER and RSK4 in 17 paired breast cancer tissues and normal tissues were detected by RT-PCR assay. (E) Methylation levels of RSK4 promoter in 9 paired ER+ breast cancer tissues and normal tissues were determined by bisulfite direct sequencing assay. (F) Methylation levels in RSK4 promoter in HBL-100, MCF-7, MDA-MB-231 and MDA-MB-453 cell lines were determined by bisulfite direct sequencing assay. (G and H) Western blotting analysis of the protein expression levels of ER and RSK4 in HBL-100, MCF-7, MDA-MB-231 and MDA-MB-453 cells. (*P<0.05). |
E2 decreases RSK4 expression through weakening its interactions with SP1
As SP1 is a transcription factor which is requisite for gene transcription, we then investigated the effects of E2 on the expression and interaction of SP1 and RSK4. Compared with control group, E2 treatment significantly decreased RSK4 expression while increased SP1 expression (Figure 2A and B), and increased the methylation of RSK4 promoter (Figure 2C). As 10 nM E2 induced significant differences in RSK4 and SP1 expressions, we applied it in the following assays to explore the effects of low concentration of E2 on the progression of ER+ breast cancer progression. Upregulation of SP1 significantly increased the luciferase activity of RSK4, whereas it was abolished when the binding sites in RSK4 promoter were mutated (Figure 3A). Moreover, E2 treatment significantly decreased the luciferase activity (Figure 3B). These results suggested that the methylation of RSK4 promoter might lead to E2-mediated RSK4 downregulation.
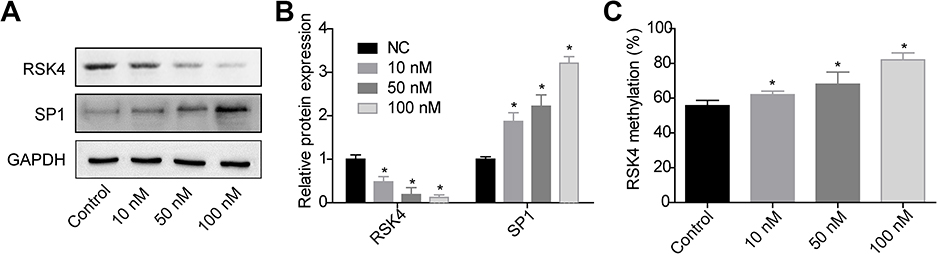 |
Figure 2 E2 decreased RSK4 expression and increased its methylation. After 48 h of treatments with 0, 10, 50 or 100 nM E2, MCF-7 cells were collected to the following assays. (A-B) Western blotting assay was performed to assess RSK4 and SP1 expressions. (C) Bisulfite direct sequencing assay was used to determine the methylation level in =RSK4 promoter (n=3, *P<0.05, compared with control group). |
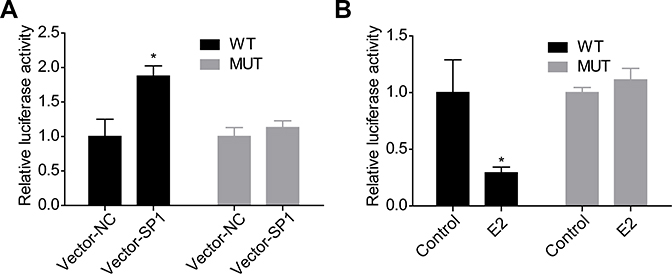 |
Figure 3 Effects of SP1 and E2 on the transcriptional activity of RSK4 in MCF-7 cells. (A) MCF-7 cells were co-transfected with Vector-NC/Vetor-SP1 and WT/MUT for 48 h, then the luciferase activity was determined by Dual-luciferase Assay System. (B) MCF-7 cells were treated with E2/DMSO (control) and WT/MUT for 48 h, then the luciferase activity was determined by Dual-luciferase Assay System (n=3, *P<0.05, compared with control group). |
E2 accelerates the malignant phenotypic transformation of breast cancer cells
Next, we explored the effects of E2 on the progression of breast cancer via carrying out in vitro experiments. The results showed that E2 treatment significantly enhanced cell proliferation (Figure 4A), clone formation (Figure 4C) and invasion (Figure 4D) abilities, whereas reduced cell apoptosis (Figure 4B) in MCF-7 cells. These results demonstrated that E2 accelerated the progression of breast cancer.
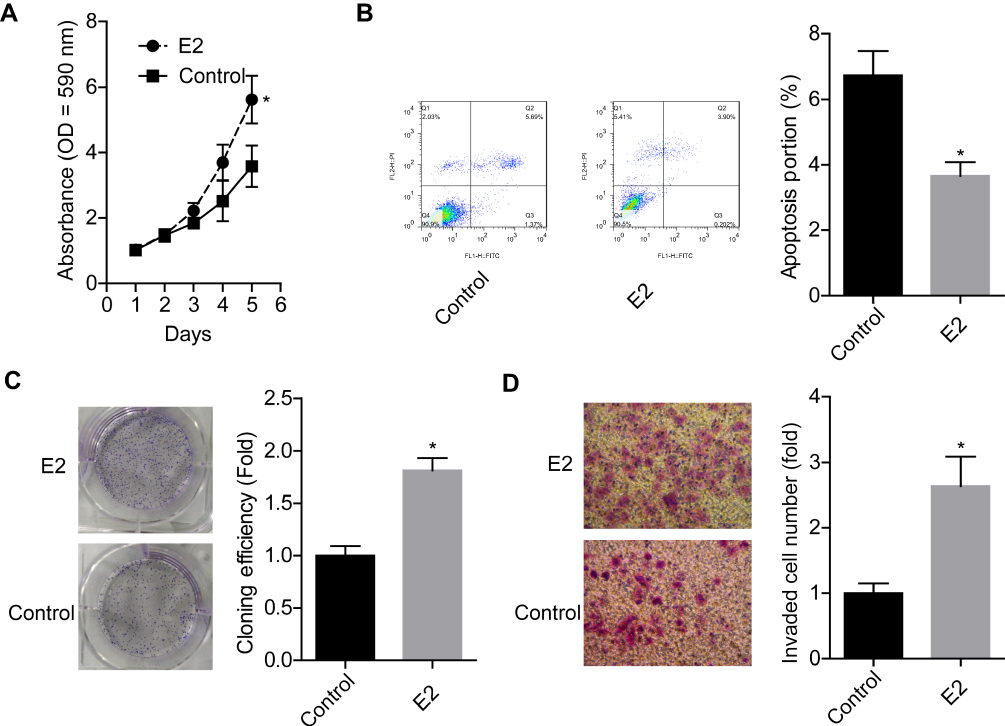 |
Figure 4 E2 treatment promoted cell growth, clone formation and invasion abilities and repressed apoptosis in MCF-7 cells. MCF-7 cells were treated with 10 nM E2, then the following assays were performed. (A) Cell proliferation was detected by MTT assay after 1, 2, 3, 4 or 5 days of the treatment. (B) Cell apoptosis was tested by flow cytometry assay after 48 h of E2 treatment. (C) Cell clone formation assay was determined by clone formation assay after 48 h of E2 treatment. (D) Cell invasion ability was assessed using Transwell chambers after 48 h of E2 treatment (n=3, *P<0.05, compared with control group). |
E2 increases DNMT3B protein expression in breast cancer cells
To explore the molecular mechanisms underlying E2 on the regulation of RSK4 expression and methylation, we then assessed the effects of E2 on the expressions of methyltransferases, such as DNMT1, DNMT3A and DNMT3B. The results showed that only the expression of DNMT3B was obviously increased when MCF-7 cells were treated with E2 in a concentration-dependent manner, while the protein expressions of DNMT1 and DNMT3A showed no obvious change (Figure 5A). However, E2 treatment had no notable influence in the mRNA level of DNMT3B (Figure 5B). Besides, E2 treatment (10 nM) significantly inhibited the degradation of DNMT3B protein (Figure 5C) and decreased its ubiquitination (Figure 5D). These results illustrated that E2 could positively regulate DNMT3B expression through repressing its ubiquitination-mediated degradation.
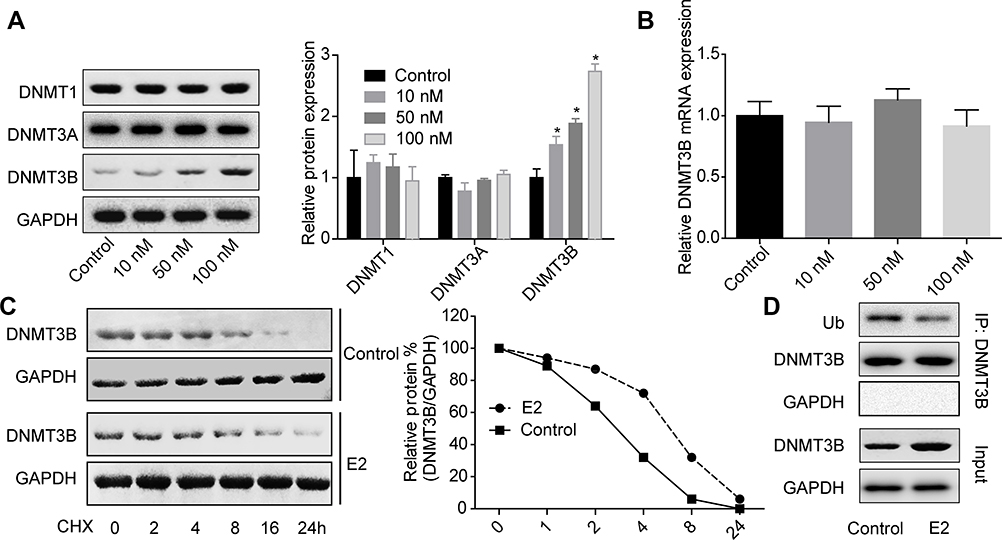 |
Figure 5 E2 treatment increased DNMT3B expression in MCF-7 cells. (A) MCF-7 cells were treated with 0, 10, 50 or 100 nM E2 for 48 h, then the cells were subjected to Western blotting assay to test DNMT1, DNMT3A and DNMT3B protein expressions. (B) MCF-7 cells were treated with 0, 10, 50 or 100 nM E2 for 48 h, then the cells were subjected to RT-PCR to test DNMT3B mRNA expression. (C) After treatment with 10 nM E2 for 24 h, MCF-7 cells were given for another 24 h of E2 treatment together with 100 μg/ml CHX (cyclohexane) for 0, 2, 4, 8, 16 and 24 h, then cells were harvested for Western blotting assay to determine DNMT3B protein expressions. (D) IP assay was used to detect the interaction between Ub and DNMT3B proteins after MCF-7 cells were treated with 10 nM E2 for 48 h(n=3, *P<0.05, compared with control group). |
E2 downregulates RSK4 expression in a dnmt3b-dependent manner
Then, we explored whether DNMT3B was involved in E2-mediated RSK4 downregulation using loss-of-function assays. Figure 6A and C showed the knockdown efficiencies of DNMT1, DNMT3A and DNMT3B, respectively. The sh-DNMT1-1, sh-DNMT3A-2 and sh-DNMT3B-1 showed the highest knockdown efficiencies of DNMT1, DNMT3A and DNMT3B in MCF-7 cells respectively, and were chosen for further study. Moreover, knockdown of DNMT3B significantly abolished E2 roles on RSK4 methylation promotion (Figure 6D) and RSK4 expression reduction (Figure 6E), with no obvious change in sh-DNMT1 and sh-DNMT3A groups (Figure 6D and E). These results further confirmed that E2 negatively regulated RSK4 expression in a DNMT3B-dependent manner.
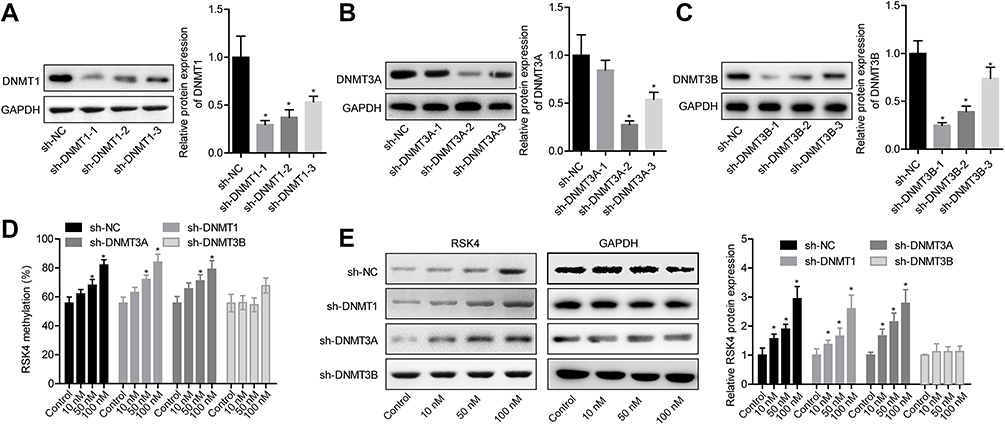 |
Figure 6 E2 decreased RSK4 expression through DNMT3B-mediated methylation of RSK4 promoter. (A–C) Western blotting assay was carried out to detect the protein expressions of DNMT1, DNMT3A and DNMT3B after MCF-7 cells were transfected with shRNAs targeting DNMT1, DNMT3A and DNMT3B, respectively. (D) After 24 h of cell transfection with sh-DNMT1, sh-DNMT3A or sh-DNMT3B, MCF-7 cells were treated with 10, 50 or 100 nM E2 for 48 h, then cells were collected for bisulfite direct sequencing assay to detect the methylation level of RSK4 promoter. (E) MCF-7 cells were collected for Western blotting assay to assess RSK4 expression after the similar treatment protocols to Figure 6D (n=3, *P<0.05, compared with NC group). |
Discussion
It’s well documented that estrogen exposure plays a key role in breast cancer initiation and progression,16 as same as our study displayed. The anti-estrogen adjuvant treatment to block ERs is the first line therapeutic schedule for patients with ER+ breast cancer.5,17 However, nearly 50% of patients are resistant to anti-estrogen therapy.18 In the present study, we focused on exploration of the molecular mechanism of estrogen in the promotion of ER+ breast cancer progression and aimed to find novel therapeutic targets. Our results showed that estrogen significantly promoted the transformation of breast cancer MCF-7 cells (ER+) to more malignant phenotype through methylation-mediated RSK4 downregulation.
To explore the molecular mechanism of E2 in the progression of ER+ breast cancer, we first determined RSK4 expression patterns in ER+ breast cancer tissues and the adjoining normal tissues. To the contrary of ER expression pattern in ER+ breast cancer tissues and cells (MCF-7), RSK4 expression was significantly decreased, implying that RSK4 may be involved in estrogen-induced breast cancer progression. Consistently, Jiang et al.9 and Thakur et al.19 reported that RSK4 was obviously lowly expressed in breast cancer tissues and cell lines, including T47D, MCF‑7, MDA‑MB‑436, ZR‑75‑1, SK‑BR‑3 and MDA‑MB‑231. Notably, Li et al.10 found that RSK4 promoter methylation was significantly enhanced in breast cancer tissues as compared to the adjacent noncancerous tissues, and revealed that a close association between the methylation and expression of RSK4. Similarly, in the present study we also detected the methylation level of RSK4 promoter and found it was significantly elevated in breast cancer tissues and cells. Moreover, RSK4 has been found to exert anti-oncogenic, anti-invasive, and anti-metastatic roles in breast cancer,9,11,20,21 suggesting a crucial role of RSK4 plays in breast cancer progression.
ER belongs to the nuclear receptor superfamily, which has three subtypes, ERα, ERβ and G protein-coupled receptor (GPR).22 Generally, ERα or ERβ binds to their ligands and then translocates to the nucleus and regulates gene expression through binding to the estrogen response elements (ERE) of their target genes or the binding sites of Fos, c-Jun, SP1 and/or DNA CpG island interaction.23,24 There are numerous SP1 binding sites in the CpG island of RSK4 promoter region, enlightening that the methylation of target gene might affect genes’ transcription via impairing their binding to transcription factors. Consistently with our hypothesis, Zhu et al.25 found that methylation in the surrounding region of p21 promoter significantly repressed the binding of SP1 to p21, thereby reducing gene transcription. Honda et al.26 pointed out that the DNA methylation of the SP1 binding site and surrounding areas in the promoter region of CLDN4 decreased the transcription of CLDN4. In the present study, we explored the relationship between SP1 and RSK4 transcription. Our results revealed that E2 stimulation significantly increased SP1 expression, which then promoted the transcription of RSK4. However, E2 stimulation decreased RSK4 expression and enhanced its methylation, which indicated that the methylation in RSK4 promoter region impaired E2-mediated SP1 upregulation on the promotion of RSK4 transcription.
We also investigated the mechanism underlying E2 on the promotion of RSK4 promoter methylation. We observed that, among all the 3 methyltransferases, DNMT1, DNMT3A and DNMT3B, only DNMT3B expression was increased under the treatment of different concentrations of E2, which was consistent with previous study.27 From the cyclohexane (CHX) chase and IP assays, we found that ubiquitylation-mediated DNMT3B protein degradation was significantly slowed down when MCF-7 cells were treated with E2 as compared with the control group. Yu et al.28 also showed that DNMT3B expression could be regulated by ubiquitination. Moreover, knockdown of DNMT3B abolished E2 roles on the methylation promotion and expression inhibition of RSK4, suggesting that E2 treatment accelerated RSK4 methylation in a DNMT3B-dependent manner.
Although the present study explored the molecular mechanisms underlying E2 on the progression of ER+ breast cancer, there are two main limitations. One is the small clinical sample size, the other is that we didn’t explore the effects of the whole E2/ER/SP1/RSK4 axis on the progression of ER+ breast cancer.
Collectively, this study reveals that E2 treatment significantly accelerates the progression of ER+ breast cancer through DNMT3B-mediated methylation of RSK4 promoter. Our study might provide a new target of RSK4 for the treatment of ER+ breast cancer patients who is resistant to anti-estrogen therapy.
Acknowledgment
This study was supported by the China National Natural Science Foundation (No. 81560431).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. doi:10.3322/caac.21262
2. Zhang ML, Huang ZZ, Zheng Y. [Estimates and prediction on incidence, mortality and prevalence of breast cancer in China, 2008]. Zhonghua Liu Xing Bing Xue Za Zhi. 2012;33(10):1049–1051.
3. Cancer Genome Atlas Network.Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. doi:10.1038/nature11412
4. Early Breast Cancer Trialists' Collaborative Group (EBCTCG).Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet. 2005;365(9472):1687–1717. doi:10.1016/S0140-6736(05)66544-0
5. Jameera Begam A, Jubie S, Nanjan MJ. Estrogen receptor agonists/antagonists in breast cancer therapy: A critical review. Bioorg Chem. 2017;71:257–274. doi:10.1016/j.bioorg.2017.02.011
6. Bjorbaek C, Zhao Y, Moller DE. Divergent functional roles for p90rsk kinase domains. J Biol Chem. 1995;270(32):18848–18852. doi:10.1074/jbc.270.32.18848
7. Frodin M, Gammeltoft S. Role and regulation of 90 kDa ribosomal S6 kinase (RSK) in signal transduction. Mol Cell Endocrinol. 1999;151(1–2):65–77.
8. Dummler BA, Hauge C, Silber J, et al. Functional characterization of human RSK4, a new 90-kDa ribosomal S6 kinase, reveals constitutive activation in most cell types. J Biol Chem. 2005;280(14):13304–13314. doi:10.1074/jbc.M408194200
9. Jiang Y, Ye X, Ji Y, et al. Aberrant expression of RSK4 in breast cancer and its role in the regulation of tumorigenicity. Int J Mol Med. 2017;40(3):883–890. doi:10.3892/ijmm.2017.3069
10. Li Q, Jiang Y, Wei W, Ji Y, Gao H, Liu J. Frequent epigenetic inactivation of RSK4 by promoter methylation in cancerous and non-cancerous tissues of breast cancer. Med Oncol. 2014;31(1):793. doi:10.1007/s12032-014-0374-0
11. Thakur A, Sun Y, Bollig A, et al. Anti-invasive and antimetastatic activities of ribosomal protein S6 kinase 4 in breast cancer cells. Clin Cancer Res. 2008;14(14):4427–4436. doi:10.1158/1078-0432.CCR-08-0458
12. Li Y, Wang Y. Bioinformatics analysis of gene expression data for the identification of critical genes in breast invasive carcinoma. Mol Med Rep. 2017;16(6):8657–8664. doi:10.3892/mmr.2017.7717
13. Guan H, Cai J, Zhang N, et al. Sp1 is upregulated in human glioma, promotes MMP-2-mediated cell invasion and predicts poor clinical outcome. Int J Cancer. 2012;130(3):593–601. doi:10.1002/ijc.26049
14. Zannetti A, Del Vecchio S, Carriero MV, et al. Coordinate up-regulation of Sp1 DNA-binding activity and urokinase receptor expression in breast carcinoma. Cancer Res. 2000;60(6):1546–1551.
15. Kadonaga JT, Courey AJ, Ladika J, Tjian R. Distinct regions of Sp1 modulate DNA binding and transcriptional activation. Science. 1988;242(4885):1566–1570. doi:10.1126/science.3059495
16. Davies C, Godwin J, Gray R, et al. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: patient-level meta-analysis of randomised trials. Lancet. 2011;378(9793):771–784. doi:10.1016/S0140-6736(11)60993-8
17. Goldhirsch A, Wood WC, Coates AS, Gelber RD, Thurlimann B, Senn HJ. Strategies for subtypes–dealing with the diversity of breast cancer: highlights of the St. Gallen international expert consensus on the primary therapy of early breast cancer 2011. Ann Oncol. 2011;22(8):1736–1747. doi:10.1093/annonc/mdr304
18. Vazquez Rodriguez G, Abrahamsson A, Jensen LDE, Dabrosin C. Estradiol promotes breast cancer cell migration via recruitment and activation of neutrophils. Cancer Immunol Res. 2017;5(3):234–247. doi:10.1158/2326-6066.CIR-16-0150
19. Thakur A, Rahman KW, Wu J, et al. Aberrant expression of X-linked genes RbAp46, Rsk4, and Cldn2 in breast cancer. Mol Cancer Res. 2007;5(2):171–181. doi:10.1158/1541-7786.MCR-06-0071
20. Zhu J, Li QY, Liu JL, Wei W, Yang HW, Tang W. RSK4 knockdown promotes proliferation, migration and metastasis of human breast adenocarcinoma cells. Oncol Rep. 2015;34(6):3156–3162. doi:10.3892/or.2015.4291
21. Sun Y, Cao S, Yang M, et al. Basic anatomy and tumor biology of the RPS6KA6 gene that encodes the p90 ribosomal S6 kinase-4. Oncogene. 2013;32(14):1794–1810. doi:10.1038/onc.2012.200
22. Andre F, Broglio K, Pusztai L, et al. Estrogen receptor expression and docetaxel efficacy in patients with metastatic breast cancer: a pooled analysis of four randomized trials. Oncologist. 2010;15(5):476–483. doi:10.1634/theoncologist.2009-0150
23. Chen GG, Zeng Q, Tse GM. Estrogen and its receptors in cancer. Med Res Rev. 2008;28(6):954–974. doi:10.1002/med.20131
24. Castro-Rivera E, Samudio I, Safe S. Estrogen regulation of cyclin D1 gene expression in ZR-75 breast cancer cells involves multiple enhancer elements. J Biol Chem. 2001;276(33):30853–30861. doi:10.1074/jbc.M103339200
25. Zhu WG, Srinivasan K, Dai Z, et al. Methylation of adjacent CpG sites affects Sp1/Sp3 binding and activity in the p21(Cip1) promoter. Mol Cell Biol. 2003;23(12):4056–4065. doi:10.1128/mcb.23.12.4056-4065.2003
26. Honda H, Pazin MJ, Ji H, Wernyj RP, Morin PJ. Crucial roles of Sp1 and epigenetic modifications in the regulation of the CLDN4 promoter in ovarian cancer cells. J Biol Chem. 2006;281(30):21433–21444. doi:10.1074/jbc.M603767200
27. Wu Q, Odwin-Dacosta S, Cao S, Yager JD, Tang WY. Estrogen down regulates COMT transcription via promoter DNA methylation in human breast cancer cells. Toxicol Appl Pharmacol. 2019;367:12–22. doi:10.1016/j.taap.2019.01.016
28. Yu J, Qin B, Moyer AM, et al. DNA methyltransferase expression in triple-negative breast cancer predicts sensitivity to decitabine. J Clin Invest. 2018;128(6):2376–2388. doi:10.1172/JCI97924
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.