")
Back to Journals » The Application of Clinical Genetics » Volume 8
The genetic landscape of X-linked adrenoleukodystrophy: inheritance, mutations, modifier genes, and diagnosis
Authors Wiesinger C, Eichler F, Berger J
Received 22 January 2015
Accepted for publication 10 March 2015
Published 2 May 2015 Volume 2015:8 Pages 109—121
DOI https://doi.org/10.2147/TACG.S49590
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Prof. Dr. Martin Maurer
Christoph Wiesinger,1 Florian S Eichler,2 Johannes Berger1
1Department of Pathobiology of the Nervous System, Center for Brain Research, Medical University of Vienna, Vienna, Austria; 2Department for Neurology, Massachusetts General Hospital, Harvard Medical School, Boston, MA, USA
Abstract: X-linked adrenoleukodystrophy (X-ALD) is caused by mutations in the ABCD1 gene encoding a peroxisomal ABC transporter. In this review, we compare estimates of incidence derived from different populations in order to provide an overview of the worldwide incidence of X-ALD. X-ALD presents with heterogeneous phenotypes ranging from adrenomyeloneuropathy (AMN) to inflammatory demyelinating cerebral ALD (CALD). A large number of different mutations has been described, providing a unique opportunity for analysis of functional domains within ABC transporters. Yet the molecular basis for the heterogeneity of clinical symptoms is still largely unresolved, as no correlation between genotype and phenotype exists in X-ALD. Beyond ABCD1, environmental triggers and other genetic factors have been suggested as modifiers of the disease course. Here, we summarize the findings of numerous reports that aimed at identifying modifier genes in X-ALD and discuss potential problems and future approaches to address this issue. Different options for prenatal diagnosis are summarized, and potential pitfalls when applying next-generation sequencing approaches are discussed. Recently, the measurement of very long-chain fatty acids in lysophosphatidylcholine for the identification of peroxisomal disorders was included in newborn screening programs.
Keywords: X-ALD, AMN, mutations, incidence, prenatal diagnosis, newborn screening
Introduction
X-linked adrenoleukodystrophy (X-ALD) is a monogenic disease caused by mutations in the ABCD1 gene located on Xq28.1 Clinically, X-ALD can present with a wide range of phenotypic manifestations.2,3 Two predominant phenotypes prevail: adrenomyeloneuropathy (AMN) and the cerebral form of X-ALD (CALD). AMN is characterized by a slowly progressive axonopathy and first symptoms in males usually appear between 20 and 30 years of age, while affected females may develop some symptoms of AMN with an average onset between 40 and 50 years. In contrast, CALD usually only affects males and presents with rapidly progressive inflammatory demyelination in the brain, leading to rapid cognitive and neurological decline.3,4 The mutation in ABCD1 is necessary but not sufficient for CALD to occur, as additional genetic or environmental factors are required to trigger this devastating brain inflammation.
The ABCD1 gene codes for the peroxisomal transporter ATP-binding cassette subfamily D member 1 (ABCD1, formerly ALDP), which mediates the import of very long-chain fatty acid (VLCFA) CoA esters across the peroxisomal membrane.5,6 The dysfunction of ABCD1 results in impaired degradation of VLCFAs in peroxisomes and consequently leads to their accumulation in various lipid species in tissues and body fluids.7 While this accumulation of VLCFAs is thought to directly contribute to the demyelinating pathology in AMN, the molecular mechanism by which VLCFAs are involved in the onset or progression of inflammation in CALD is still not completely understood. Among the biomaterial available for diagnosis, VLCFAs accumulate in plasma, leucocytes, and fibroblasts from X-ALD patients independent of phenotype. Thus, an elevated level of VLCFAs represents the standard biomarker for diagnosis of X-ALD, but does not predict the phenotype or progression of disease.
While the lifetime risk for male X-ALD patients to develop CALD is estimated to be at least 60%, the peak occurrence is between 5 and 12 years of age. In cases where brain inflammation is detected early, it can be arrested by hematopoietic stem cell transplantation (HSCT).8 In cases where no appropriate donor is available, autologous hematopoietic stem cell gene therapy may be an option and is currently being actively investigated.9 However, HSCT is thought to only arrest the inflammatory demyelination and not impact the non-inflammatory axonopathy.10 Therefore, it is not a therapeutic option for AMN patients without inflammatory involvement. Thus, no curative therapy is currently available for AMN, and treatment is limited to symptomatic therapy of adrenal or gonadal insufficiency, neuropathic pain, and spasticity.2
Inheritance and frequency of X-ALD
X-ALD is a monogenic, X-linked disorder and the risk of affected parents for transmitting the affected allele can be clearly predicted. While sons of males with X-ALD will never inherit the affected X chromosome from their father, their daughters will automatically be carriers of the disease allele. For heterozygous female carriers, the probability for transmission of the affected gene is 50% for both sons and daughters. The exceptions are parents of patients with de novo mutations and rare cases of gonosomal or gonadal mosaicism, where different rates of risk for transmission must be expected.11
While the majority of patients typically inherit the defective ABCD1 allele from one parent, between 4.1% and 19% of X-ALD cases have been reported to carry mutations acquired de novo (see “de novo mutations in ABCD1”).
Incidence and prevalence of X-ALD
The estimation of rates for incidence or prevalence of X-ALD cases faces various problems. These include clinical heterogeneity with late onset of symptoms (yet undetected), small numbers of subjects and differences in the extent of pedigree characterization. Furthermore, the lack of centralized recording in some countries and underdiagnosis of mildly affected male patients as well as heterozygous females can lead to an underestimation of frequency. It should be mentioned that the comprehensive inclusion of females heterozygous for X-ALD relies on genetic testing, as normal plasma VLCFA levels are found in at least 15%.12,13 Incidence estimates from different countries in Europe, North and South America, Asia, and Australasia are summarized in Table 1. For the estimation of incidence rates, basically two different methods were used: either the numbers of newly diagnosed patients within a certain time period were compared with the birth rates in the same period or the established patients were assigned to their respective birth years. While the former design faces the problem that during periods when routine diagnosis became readily available, the number of annually diagnosed cases might have exceeded the annual rate of affected newborns, the latter design must be expected to underestimate actual numbers, as before the widespread availability of VLCFA detection, a portion of cases was likely missed.
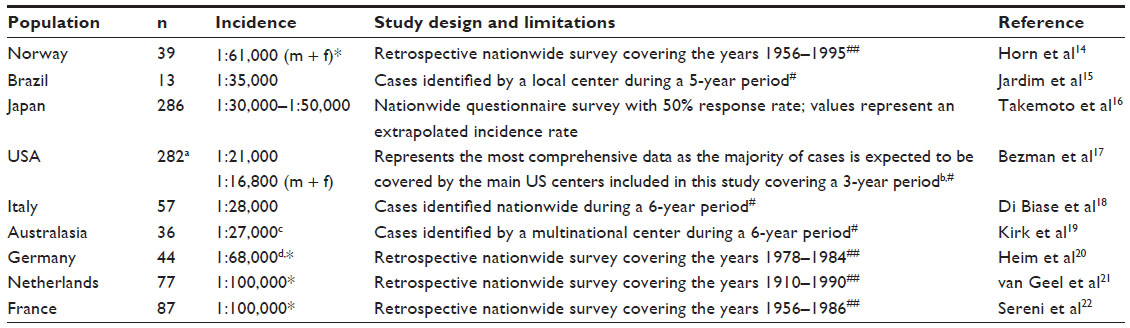 | Table 1 Summary of studies reporting incidence rates for X-linked adrenoleukodystrophy in different populations |
In summary, the data given in Table 1 suggest the average incidence rates for X-ALD in male patients to be between 1:20,000 and 1:30,000. Further, the study of Horn et al for the first time also directly addressed the number of affected females and found a combined incidence rate of 1:61,000 for males plus females in the Norwegian population.14 However, some underestimation must be assumed in this study. In contrast, the combined incidence rate of 1:16,800 reported by the thorough study of Bezman et al represents an extrapolation based on male incidence rates and therefore also includes unidentified and unaffected female carriers.17 In addition to incidence rates, prevalence rates of around 1:200,000 for males and 1:100,000 for females were estimated by thorough investigations carried out for the Dutch and the Norwegian populations.14,21 However, the implementation of newborn screening programs for X-ALD will provide a more complete coverage. Preliminary results already suggest a similar incidence rate of around 1:30,000. Overall, the data suggest that no major differences in incidence rates occur between countries across the world.
Phenotypes and their relative frequencies
While AMN represents the default manifestation of X-ALD, only a subset of patients is affected by CALD. It was recently reported that all male and possibly even most female patients will develop at least some symptoms of AMN late in life and thus AMN has a near 100% lifetime penetrance.12,14,23 The onset of AMN usually occurs between the second and fourth decade of life. A recent prospective cross-sectional cohort study including 46 heterozygous X-ALD females reported that 18% of women under the age of 40 years had symptoms but in the age group 60 years or older, 88% were symptomatic.12 Similarly, Horn et al found that no heterozygous females older than 50 years were free of neurological symptoms.24
While previously the lifetime risk for a newborn male X-ALD patient to develop the inflammatory form of X-ALD was estimated to be around 60%,3,25,26 two recent reports suggest an even higher risk to develop CALD.14,27 The risk of developing the inflammatory form of X-ALD between 5 and 12 years of age is around 35%–40% and decreases during adulthood. However, while CALD was previously considered to occur only rarely after the age of 45 years, recent reports suggest a substantial risk to develop CALD even at higher ages.27–29 The majority of male X-ALD patients develops primary adrenocortical insufficiency (Addison’s disease) which can be the first and often sole symptom in young male X-ALD patients.30 These patients will develop AMN later in life and are at risk of developing CALD.
No relevant genotype–phenotype correlation exists in X-ALD
Based on several different observations, it is now well established that no generalized correlation exists between certain types of mutations in ABCD1 and the two main clinical phenotypes, ie, AMN and CALD, observed in X-ALD patients. Firstly, even large deletions or frameshift mutations leading to the complete absence of protein can lead to the milder AMN phenotype.31 Secondly, within individual kindreds, phenotypes of the entire clinical spectrum of X-ALD have been described.15,32,33 Thirdly, even in monozygotic twins, different clinical phenotypes have been observed.34 However, it cannot be excluded that specific mutations may lead to ABCD1 protein with some residual activity, which could potentially influence the severity of the phenotype.35,36 Further, it is possible that certain mutations exclusively lead to specific phenotypes or affect the age of onset of first AMN symptoms, but this question has not yet been studied in detail. The main reasons for that are the low number of patients with identical mutations as well as the current lack of knowledge concerning the influence of environmental factors because this might confound characteristic phenotypes of particular mutations.
Diagnosis of X-ALD
Typically, when a diagnosis of X-ALD is suspected based on clinical presentation or magnetic resonance imaging abnormalities, biochemical testing for elevated plasma VLCFA levels is performed. VLCFAs are elevated in all male X-ALD patients regardless of age, disease duration, metabolic status, or clinical symptoms. The most frequently used diagnostic parameter is the concentration of total C26:0 (saturated fatty acid with 26 carbon atoms) after hydrolysis.37 Alternatively, the ratio of C26:0/C22:0 or C24:0/C22:0 can be used, because C22:0 remains unchanged or is even slightly reduced in plasma samples of X-ALD patients.38,39
While the measurement of total C26:0 is highly specific in male patients, measurement of VLCFAs can only detect about 85% of heterozygous female carriers.13 Thus, additional mutation analysis of the ABCD1 gene is necessary for proper identification of heterozygous X-ALD females.
One possible diagnostic pitfall is the fact that plasma VLCFA levels strongly depend on dietary intake of VLCFAs, for example, by the consumption of peanuts.40 To circumvent this problem, cultured skin fibroblasts were previously used to confirm the diagnosis. Nowadays, other patient-derived cells, like blood leucocytes, provide a less invasive alternative for confirmation of the diagnosis.41 Furthermore, VLCFAs are not only increased in X-ALD patients, but also in other peroxisomal disorders such as peroxisomal biogenesis defects of the Zellweger syndrome spectrum or single enzyme defects of peroxisomal beta-oxidation (acyl-CoA oxidase deficiency and D-bifunctional protein deficiency). Therefore, mild, atypical, or late-onset variants of these diseases must be considered in the differential diagnosis of VLCFA accumulation.42–45 Although other peroxisomal disorders can usually be excluded by measuring phytanic acid and plasmalogen levels,46 confirmation of the X-ALD diagnosis by ABCD1 mutation analysis is advisable, especially in patients with atypical symptoms or when HSCT is considered.
Mutation detection and ABCD1 pseudogenes
Beside the identification of female carriers and confirmation of the diagnosis in male patients, analysis of mutations in the ABCD1 gene is also essential to characterize X-ALD kindreds and for prenatal diagnosis.
Mutation detection is typically done by analysis of the genomic area of the ABCD1 gene, which contains ten exons and is located at chromosome position Xq28, spanning a genomic region of around 20 kb.1 However, the sequence-based analysis of the ABCD1 gene is complicated by the presence of several nonfunctional pseudogenes on different chromosomes. While various pseudogenes were originally attributed to chromosomes 1, 2, 10, 16, 20, and 22 by polymerase chain reaction analysis of monochromosomal mapping panels and by the use of fluorescence in situ hybridization (FISH), five different pseudogenes are now assigned to chromosomal regions 2p11 (two copies), 10p11, 16p11, and 22q11.31,47 These all consist of exons 7–10 of the ABCD1 gene and are derived from one ancestral duplication, probably to chromosome 2, which was followed by further duplications in a process termed pericentromeric plasticity.47 As these pseudogenes show a high degree of sequence homology to the original ABCD1 gene, special care must be taken in the analysis of genomic DNA. Therefore, primers specific for the ABCD1 gene have been designed for conventional sequence analysis.48
In cases where no mutations can be detected by DNA sequence analysis, methods such as quantitative polymerase chain reaction, multiple ligation-dependent probe amplification (MLPA), or Southern blot analysis have been used for detection of large deletions, duplications, or chromosomal rearrangements.
Next-generation sequencing
The fast development of high-throughput methods, like exome sequencing or whole genome sequencing, provides another opportunity to identify X-ALD, especially in cases presenting with an atypical disease course, or when occurring as comorbidities of unrelated severe conditions, which would previously have been missed.49,50 However, care must be taken in the interpretation of such sequencing results, as depending on the methods used for target enrichment, sequencing, and data analysis, pseudogenes may give rise to false positives. On the other hand, cases might be missed due to too stringent filtering during data analysis.51
One example of difficulties concerning the analysis of data from next-generation sequencing is demonstrated by a report in which the mutation c.1823 G>A in exon 8 of the ABCD1 gene was discussed as a non-deleterious single nucleotide polymorphism variant because it was identified in a patient who was obviously not suffering from X-ALD.52 However, in a previous study, this mutation was clearly shown to cause X-ALD, as it was found in the ABCD1 coding sequence in different patients.53 Therefore, the most plausible explanation for the incorrect detection of the mutation in the former study52 is that the same variant is present in an ABCD1 pseudogene located on chromosome 2 and was by mistake attributed to the real ABCD1 gene.
Prenatal diagnosis
As X-ALD is a monogenic disorder, the risk of affected parents passing on the mutated allele can be clearly predicted and consequently genetic counseling should be offered. Prenatal testing is preferably done by genetic analyses, but biochemical methods are also available for cases where the mutation in the ABCD1 gene has not been established.54 Genetic testing can be performed either by chorionic villus sampling at 11–13 weeks of pregnancy or by amniocentesis at 15–18 weeks’ gestation. Usually, the karyotype of the fetus is determined first, and if male, fetal DNA is analyzed for the previously established familial disease-causing mutation in the ABCD1 gene.55 However, depending on local regulations, different strategies are pursued.56
In the past, the determination of VLCFA levels in cultured amniocytes or cultured chorionic villus cells was the standard method for prenatal diagnosis of X-ALD.57 However, cases have been reported for which normal VLCFA levels were obtained in affected fetuses and thus led to false negative diagnosis.58,59 Therefore, this method should only be considered when genetic analysis is not possible.
Preimplantation genetic diagnosis may be a further option during in vitro fertilization, depending on the particular national regulations. Different strategies have been described for selection of unaffected embryos by analysis of single blastomeres. First, sexing of embryos was used to select female embryos over possibly heavily affected males in a case of established female X-ALD carriership.60 In a different sex selection approach, male embryos derived from a father diagnosed with X-ALD and a healthy mother were selected since these are all unaffected.61
In a more advanced approach, direct detection of the affected allele by multiple displacement amplification of DNA from a single blastomere followed by analysis of microsatellite markers has been used to ensure unaffected offspring.62,63
Finally, a recent report describes the selection of healthy embryos in combination with HLA matching, which gives rise to the possibility of HSCT in affected siblings for whom no matching donor was available before.64,65 However, such an approach depends on applicable regulations and ethical considerations in different countries.
Newborn screening
Many cases of X-ALD are diagnosed early due to extended family screening programs in some but not all countries. Sadly, in the absence of a family history, a situation which is inevitable due to the relatively high rate of ABCD1 mutations occurring de novo (see section on de novo mutations in ABCD1), many patients are detected relatively late and in an advanced stage when therapy is limited. Therefore, the development of newborn screening for X-ALD is a major advancement, as this can ensure timely diagnosis and monitoring of patients. For instance, HSCT is only effective in CALD when performed at an early disease stage, because cerebral inflammation still progresses for 12–18 months after the procedure.8,66 In addition, in patients with subclinical adrenal insufficiency, monitoring and early initiation of hormone replacement therapy can avoid adrenal crises and life-threatening events.30,67
In recent years, substantial progress has been made in the development of suitable methods for high-throughput biochemical screening for X-ALD and other metabolic disorders. Determination of C26:0 lysophosphatidylcholine (C26:0-LPC) from dried blood spots has turned out to be most promising and slightly different methods based on tandem mass spectrometry (MS/MS) were reported by different laboratories.68–71
The feasibility of C26:0-LPC determination has been proven by two large pilot studies based on either liquid chromatography MS/MS72 or flow injection analysis MS/MS.73 Indeed, since the beginning of 2014, X-ALD is included in the New York State newborn screening program,74 and first preliminary estimates suggest a comparable incidence rate for X-ALD as described before. Further newborn screening legislation has been passed in the states of California, Connecticut, and Massachusetts.
In addition to the detection of male X-ALD patients, determination of C26:0-LPC will also identify the majority of female X-ALD carriers as well as patients affected by different peroxisomal biogenesis disorders or single enzyme deficiencies of the peroxisomal beta-oxidation pathway.72,74
Mutations in ABCD1
Due to the X-chromosomal localization of ABCD1 already a single affected allele can lead to disease. Therefore, a tremendous number of different disease-causing mutations have been described in X-ALD. A comprehensive overview of all described mutations can be found in the X-ALD database (http://www.x-ald.nl).75
A large portion of the described mutations lead to an absence of ABCD1 protein. While mutations such as deletions, frameshifts, and nonsense mutations generate truncated proteins, missense mutations often lead to unstable proteins as judged by immunofluorescence microscopy. However, more thorough investigations based on Western blot analysis indicate that many mutated ABCD1 isoforms with single amino acid substitutions are not completely absent, as initially suggested, but are present at strongly reduced levels, which are not detected by standard immunofluorescence microscopy.76 Some variations in ABCD1, such as the “missense mutations” N13T and H97L, do not cause disease but rather represent polymorphisms, which do not affect protein function.53,77
Distribution of mutations
To date, 695 non-recurrent mutations have been described in the X-ALD database, out of which 343 represent missense mutations. Figure 1 shows the distribution of the 209 different amino acids affected by disease-causing missense mutations throughout the ABCD1 protein in comparison with the evolutionary conservation across seven mammalian species.
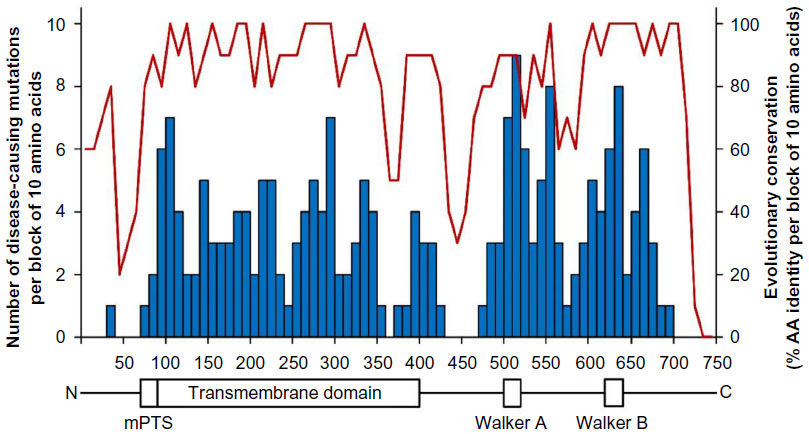 | Figure 1 Distribution of disease-causing amino acid mutations in X-linked adrenoleukodystrophy (X-ALD) in relation to the evolutionary conservation across the ABCD1 protein. |
These missense mutations are rather equally distributed throughout the ABCD1 protein with the exception of three regions that are devoid of disease-causing mutations. The unaffected region in the middle of the protein (AA 430–470) acts as a linker between the transmembrane and the ATP-binding domains of ABCD1, probably allowing a considerable degree of structural flexibility. The very N-terminal part of the protein precedes the PEX19 binding site, which is required for proper peroxisomal targeting of ABCD1.78 Based on the nearly complete absence of mutations within amino acids 1–73, together with the relatively low level of evolutionary conservation, it might be speculated that this region is not absolutely essential for ABCD1 protein function. In a case of defective translation initiation due to a 26-base pair deletion, it was suggested that, despite the presence of an out-of-frame start codon, an alternative internal translation initiation at amino acid position 66 leads to a low level of truncated ABCD166–745 protein. Indeed, all cases of translation initiation mutations in ABCD1 reported to date have presented with the AMN phenotype and none with CALD.35,79,80
Interestingly, no disease-causing mutations have been found after amino acid position 693, although evolutionary conservation remains high up to position 721, suggesting that this region of ABCD1 is not directly involved in substrate transport. This idea is further supported by the finding that amino acids 694–745 are not necessary for ABCD1 function, as truncated ABCD11–693 was also able to restore beta-oxidation of VLCFAs in X-ALD fibroblasts.31 However, two different studies found the very C-terminal region to play a major role in homodimerization and heterodimerization of ABCD1.81,82
De novo mutations in ABCD1
In a considerable number of cases, X-ALD is not inherited but the respective mutations occur spontaneously. Initially, de novo mutation rates of around 5% were described.17,83 Similarly, a comprehensive study based on molecular characterization of 489 families found only 4.1% of index cases to carry de novo mutations in the ABCD1 gene.11 In addition, ten cases of de novo mutations, including three cases of gonosomal or gonadal mosaicism, were described in mothers of index cases in this study. However, a recent population-based Norwegian study reported a higher de novo mutation rate of at least 19%.14 It remains to be established whether these diverging results reflect real differences, are in part caused by the unavoidably small sample size of the Norwegian study, or by possible underestimation in the study by Wang et al due to underdiagnosis.11 The latter could be caused by the fact that spontaneously occurring de novo cases, in particular when presenting with mild phenotypes, might not be recognized as X-ALD patients and therefore were not included in the study set.
However, the initiation of systematic newborn screening for X-ALD will allow systematic identification of at least all male patients and will help to elucidate the rate of de novo mutations. However, it remains to be shown whether newborn screenings can help identify a high number of female index cases, as only about 85% of female carriers can be detected by newborn screening.
Aspects of negative dominance in X-ALD
All four members of the ABCD protein family are half transporters, which have to dimerize in order to form functional units. Some ABCD1 mutations lead to stable and correctly localized proteins but without transporter activity.75 If such a mutated ABCD1 protein dimerizes with a functional monomer, it might exert a dominant-negative effect on its dimerization partner, thus resulting in a nonfunctional transporter.84 Negative dominance could potentially play a role in autologous gene therapy, when introduced functional ABCD1 might form dimers with the endogenous mutated, but stable protein. However, due to high levels of expression of the therapeutic gene, driven by a strong viral promoter, presumably resulting in excess of functional ABCD1, this is probably of minor concern for most cell types.
Similarly, in therapeutic approaches relying on compensatory pharmacological induction of ABCD2, the function of putative ABCD1/ABCD2 heterodimers could be affected by dominant-negative ABCD1. However, given that homodimers of ABCD1 as well as ABCD2 seem to prevail in vivo,82,85 a serious impact of dominant-negative effects on pharmacological gene therapy is unlikely. Owing to X-inactivation, negative dominance should not have any harmful consequences at all for heterozygous carriers.
X-inactivation and clinical symptoms in females
As adrenoleukodystrophy is X-linked, heterozygous X-ALD females were regarded to be more or less unaffected and their disabilities often remained unrecognized and untreated for long times.86 Indeed, the fatal cerebral phenotype of the disease is extremely rare in females and probably only occurs when both alleles are affected.4,87,88
However, the majority of female carriers have elevated plasma VLCFA levels, although to a lesser extent than male patients, and the rate of beta-oxidation of VLCFAs was found to be decreased in fibroblasts.12
During recent more detailed studies, it became apparent that a high number, if not all, of the heterozygous females develop at least some symptoms of AMN and that the severity of symptoms increases with age.12,24,89 It has been speculated for a long time that skewed X-inactivation in females may account for the disease course and severity of symptoms. However, contradictory results concerning skewed X-inactivation have been reported, possibly due to inadequate neurological characterization and consideration of the age of the patients. Additional studies focusing on the specific symptoms of heterozygous females will be needed to finally resolve this question.12,90–95
Modifier genes
The observation that within one family32 or even among monozygotic twins34 one and the same mutation can lead to different X-ALD phenotypes suggests that, in addition to the primary mutation in the ABCD1 gene, other environmental or genetic factors contribute to the phenotype. Indeed, segregation analysis based on 3,862 individuals from 89 kindreds suggested the presence of an autosomal modifier locus.96
Numerous candidate genes were analyzed either by association and/or segregation studies or by analyses of gene expression levels (Table 2). However, a problem faced by most of these studies is the low number of available subjects.
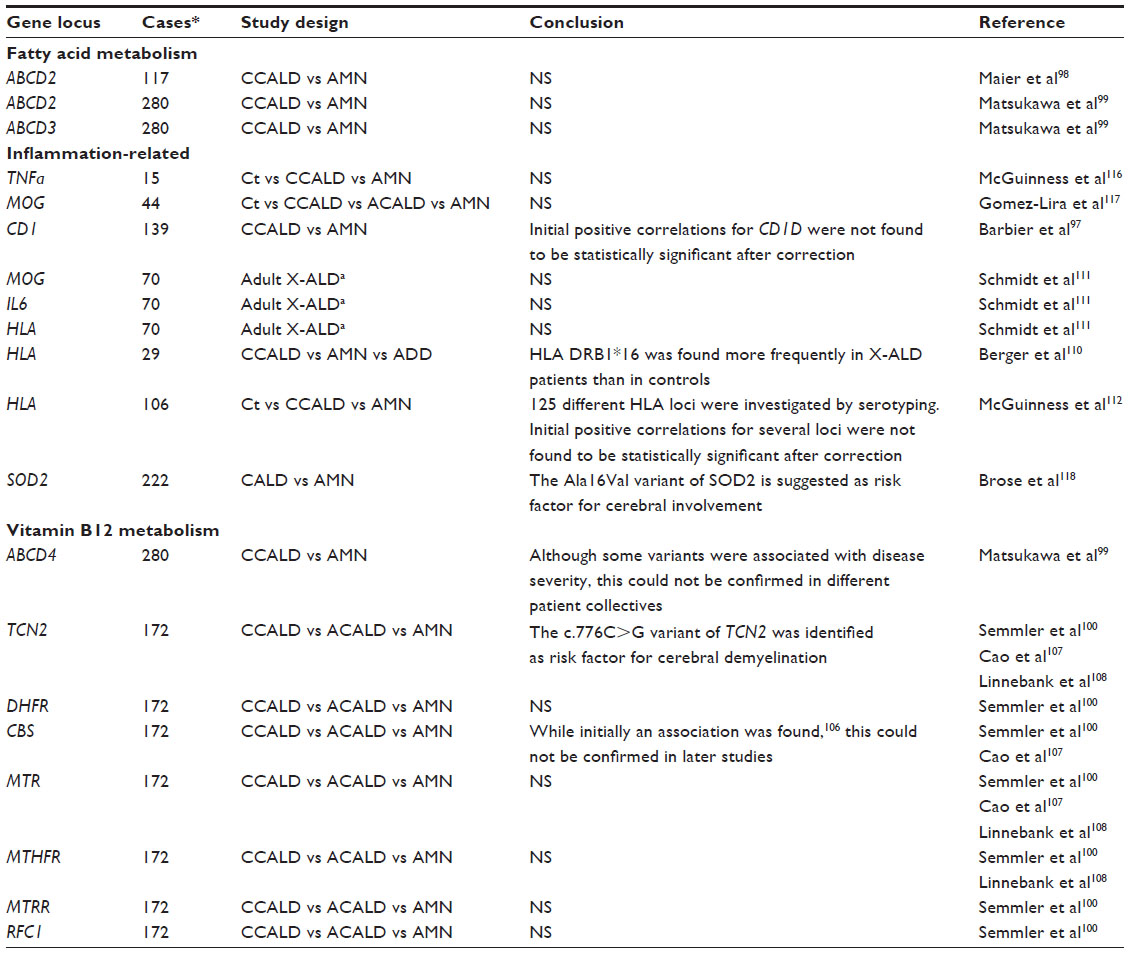 | Table 2 Summary of genes investigated as possible modifiers for X-linked adrenoleukodystrophy |
A second methodological problem in some of the published studies concerned the definition of non-inflammatory subjects. An excellent collective with clinically well defined samples originates from the group led by Patrick Aubourg. In these studies, only male AMN patients older than 45 years of age with a demonstrated absence of brain inflammation by magnetic resonance imaging were included in the non-inflammatory sample set.97–100 As the likelihood for initiation of brain inflammation strongly diminishes with age, X-ALD patients without cerebral involvement at 45 years were considered as cases of “pure AMN”, although rare instances of conversion to CALD still cannot be excluded (see section on phenotypes and their relative frequencies).
Upon overexpression, ABCD2101–104 and, to some extent, ABCD35,102,105 can compensate for the lack of ABCD1 in vivo and in vitro, and therefore represent excellent candidates for a modulation of the phenotype. However, two independent association studies comparing ABCD2 and ABCD3 polymorphisms with different clinical phenotypes failed to determine significant correlations.98,99
Various genes related to vitamin B12 metabolism were investigated for a role as modifiers of the phenotype in X-ALD. While initially a protective role of the CBS gene was suggested,106 this finding could not be confirmed by a more comprehensive study in a different patient collective.100 However, a polymorphism within the TCN2 gene was found to be significantly associated with cerebral demyelination in X-ALD.100,107,108
There are some similarities in the pathology between multiple sclerosis and the cerebral inflammatory demyelination in X-ALD. Some triggers of inflammation may be shared by these two diseases. Certain HLA-DRB1 haplotypes within the major histocompatibility complex represent a confirmed genetic risk factor for multiple sclerosis,109 and were therefore speculated to be good candidates for modification of the X-ALD phenotype. However, previous studies investigating this association were not able to demonstrate significant associations of certain HLA type II haplotypes with cerebral involvement.110–112 Additional studies would be required to resolve this issue. Furthermore, genes involved in fatty acid elongation113 or omega-oxidation of fatty acids114 represent reasonable modifier gene candidates, but unfortunately association studies concerning these genes have not yet been published. Besides direct association studies, expression levels of possible modifier genes have been investigated. While for ABCD2, ABCD3, and ACSVL1 no significant differences in mRNA expression levels were found in brain tissues when comparing CALD and AMN, an altered expression of ACSBG1 and ABCD4 mRNA was observed in normal appearing white matter of CALD patients.115
An obvious, although often unavoidable, problem in carrying out association studies in rare diseases like X-ALD, is the inevitably low number of analyzed subjects. As studies with relatively low sample numbers can only detect genetic associations with a relatively large effect on the disease course, genes with a smaller influence typically remain below the significance threshold level. Therefore, negative results should not necessarily be regarded as exclusion criteria for a possible modifier gene but rather as an indication that it is not a major determinant.
So far, most association studies have focused on the presence or absence of cerebral inflammatory demyelination. However, additional clinical variability, like the age of onset of spinal cord degeneration, may also be influenced by the genetic background of the patients.
In summary, it appears that not one single major factor is responsible for the different clinical phenotypes in X-ALD. More likely, several different genetic as well as environmental factors contribute to some extent to the phenotypic variability in X-ALD.
Substrate specificity of the related peroxisomal ABC transporters
In addition to ABCD1, its two closest homologs, ABCD2 (ALDRP) and ABCD3 (PMP70), are also located in the peroxisomal membrane. It has been reported that, upon overexpression, both of these homologs are in principle able to compensate for dysfunctional ABCD1.5,101–103
However, at the endogenous basal levels, ABCD2 and ABCD3 obviously fail to compensate for ABCD1 deficiency, as they are not able to prevent the clinical manifestations in X-ALD patients. Regarding ABCD2, the main reason for this seems to be the opposite expression pattern of ABCD1 and ABCD2 in many cell types, meaning that in cell types with high levels of ABCD1 expression, ABCD2 is often present at low abundance and vice versa.119–121 A further explanation for the lack of compensation in vivo may be different affinities toward VLCFAs. Different substrate preferences were suggested for ABCD1-3: straight-chain VLCFAs for ABCD1, unsaturated VLCFAs for ABCD2, and branched-chain fatty acids, dicarboxylic acids, and bile acid precursors for ABCD3.122–124 Furthermore, the ABCD family of transporters needs to form dimers in order to be functional. It is still debated whether ABCD1 occurs only in homodimeric units or whether heterodimers with ABCD2 and ABCD3 are also formed in vivo.81,82,85,125 As chimeric heterodimers of ABCD1 and ABCD2 were shown in vitro to be functional, this would raise the possibility of different substrate preferences for such putative heterodimers.126
Conclusion and future outlook
The implementation of newborn screening for X-ALD will ensure that boys affected by X-ALD will not remain undiagnosed, and thus their chance to receive HSCT at an appropriate time will increase. Possibly improved knowledge about different genetic modifiers will in future allow the identification of groups of patients with an increased risk for developing the cerebral form of X-ALD, which could then be monitored more closely. The increased integration of next-generation sequencing into routine clinical practice will enable the detection of additional modifier genes and evaluation of previously suggested modifier genes. To this end, it is important to establish worldwide collaborations for X-ALD research in order to organize systematic collection of data sets for future comprehensive analyses. During the last few years, large consortia, which strongly involve patient organizations, were formed in Europe (European AMN Board) and the USA (ALD Connect). These consortia will allow the establishment of large sample sets for genetic studies and provide high-quality platforms for more thorough metabolic and genetic investigations.
Although X-ALD is a monogenic disorder, the individual genetic background, involving many different genes, may be responsible for the age of onset of AMN symptoms in male as well as in female patients. For the inflammatory demyelination in children and adults, next to genetic modifiers, different environmental factors are likely to be involved as triggers, contributing to the overall complexity of X-ALD. For example, it has been reported that severe head trauma can elicit the rapidly progressive brain inflammation in adult patients. Thus, it is possible that independently of the genetic background, every male X-ALD patient might be at risk of developing the inflammatory form of the disease in response to environmental factors, but the likelihood for this to occur may depend on specific genetic modifiers.
There is some evidence that the amount of VLCFAs may be directly related to the risk of developing inflammatory demyelination.115 Modern lipidomic methods now enable unbiased screening for predictive lipid species and provide an additional possibility for the development of new markers allowing improved prediction of clinical progression.
Detailed knowledge of the involvement of modifying genes, lipids, and environmental factors will not only help us to understand the detailed molecular and pathogenic mechanisms underlying the broad spectrum of clinical heterogeneity in X-ALD, but will also help to develop novel therapeutic strategies and enable a more refined evaluation of therapeutic efficacy in clinical trials for X-ALD.
Acknowledgments
We thank Sonja Forss-Petter and Fabian Dorninger for their comments and critical reading of the manuscript. This work was supported by the Austrian Science Fund (FWF) project P26112-B19.
Disclosure
The authors report no conflicts of interest in this work.
References
Mosser J, Douar AM, Sarde CO, et al. Putative X-linked adrenoleukodystrophy gene shares unexpected homology with ABC transporters. Nature. 1993;361(6414):726–730. | |
Engelen M, Kemp S, de Visser M, et al. X-linked adrenoleukodystrophy (X-ALD): clinical presentation and guidelines for diagnosis, follow-up and management. Orphanet J Rare Dis. 2012;7:51. | |
Moser HW, Smith KD, Watkins PA, Powers J, Moser AB. X-linked adrenoleukodystrophy. In: Scriver R, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. 8th ed. New York, NY, USA: McGraw-Hill Book Co; 2001. | |
Berger J, Forss-Petter S, Eichler FS. Pathophysiology of X-linked adrenoleukodystrophy. Biochimie. 2014;98:135–142. | |
Wiesinger C, Kunze M, Regelsberger G, Forss-Petter S, Berger J. Impaired very long-chain acyl-CoA beta-oxidation in human X-linked adrenoleukodystrophy fibroblasts is a direct consequence of ABCD1 transporter dysfunction. J Biol Chem. 2013;288(26):19269–19279. | |
van Roermund CW, Visser WF, Ijlst L, et al. The human peroxisomal ABC half transporter ALDP functions as a homodimer and accepts acyl-CoA esters. FASEB J. 2008;22(12):4201–4208. | |
Morita M, Imanaka T. Peroxisomal ABC transporters: structure, function and role in disease. Biochim Biophys Acta. 2012;1822(9):1387–1396. | |
Peters C, Charnas LR, Tan Y, et al. Cerebral X-linked adrenoleukodystrophy: the international hematopoietic cell transplantation experience from 1982 to 1999. Blood. 2004;104(3):881–888. | |
Cartier N, Hacein-Bey-Abina S, Bartholomae CC, et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science. 2009;326(5954):818–823. | |
van Geel BM, Poll-The BT, Verrips A, Boelens JJ, Kemp S, Engelen M. Hematopoietic cell transplantation does not prevent myelopathy in X-linked adrenoleukodystrophy: a retrospective study. J Inherit Metab Dis. 2015;38(2):359–361. | |
Wang Y, Busin R, Reeves C, et al. X-linked adrenoleukodystrophy: ABCD1 de novo mutations and mosaicism. Mol Genet Metab. 2011; 104(1–2):160–166. | |
Engelen M, Barbier M, Dijkstra IM, et al. X-linked adrenoleukodystrophy in women: a cross-sectional cohort study. Brain. 2014;137 Pt 3:693–706. | |
Moser AB, Kreiter N, Bezman L, et al. Plasma very long chain fatty acids in 3,000 peroxisome disease patients and 29,000 controls. Ann Neurol. 1999;45(1):100–110. | |
Horn MA, Retterstol L, Abdelnoor M, Skjeldal OH, Tallaksen CM. Adrenoleukodystrophy in Norway: high rate of de novo mutations and age-dependent penetrance. Pediatr Neurol. 2013;48(3):212–219. | |
Jardim LB, da Silva AC, Blank D, et al. X-linked adrenoleukodystrophy: clinical course and minimal incidence in South Brazil. Brain Dev. 2010;32(3):180–190. | |
Takemoto Y, Suzuki Y, Tamakoshi A, et al. Epidemiology of X-linked adrenoleukodystrophy in Japan. J Hum Genet. 2002;47(11):590–593. | |
Bezman L, Moser AB, Raymond GV, et al. Adrenoleukodystrophy: incidence, new mutation rate, and results of extended family screening. Ann Neurol. 2001;49(4):512–517. | |
Di Biase A, Salvati S, Avellino C, et al. X-linked adrenoleukodystrophy: first report of the Italian Study Group. Ital J Neurol Sci. 1998;19(5):315–319. | |
Kirk EP, Fletcher JM, Sharp P, Carey B, Poulos A. X-linked adrenoleukodystrophy: the Australasian experience. Am J Med Genet. 1998;76(5):420–423. | |
Heim P, Claussen M, Hoffmann B, et al. Leukodystrophy incidence in Germany. Am J Med Genet. 1997;71(4):475–478. | |
van Geel BM, Assies J, Weverling GJ, Barth PG. Predominance of the adrenomyeloneuropathy phenotype of X-linked adrenoleukodystrophy in The Netherlands: a survey of 30 kindreds. Neurology. 1994;44(12):2343–2346. | |
Sereni C, Paturneau-Jouas M, Aubourg P, Baumann N, Feingold J. Adrenoleukodystrophy in France: an epidemiological study. Neuroepidemiology. 1993;12(4):229–233. | |
Engelen M, Kemp S, Poll-The BT. X-linked adrenoleukodystrophy: pathogenesis and treatment. Curr Neurol Neurosci Rep. 2014; 14(10):486. | |
Horn MA, Retterstol L, Abdelnoor M, Skjeldal OH, Tallaksen CM. Age-dependent penetrance among females with X-linked adrenoleukodystrophy. Brain. 2015;138 Pt 2:e325. | |
Kemp S, Berger J, Aubourg P. X-linked adrenoleukodystrophy: clinical, metabolic, genetic and pathophysiological aspects. Biochim Biophys Acta. 2012;1822(9):1465–1474. | |
Berger J, Gartner J. X-linked adrenoleukodystrophy: clinical, biochemical and pathogenetic aspects. Biochim Biophys Acta. 2006;1763(12):1721–1732. | |
de Beer M, Engelen M, van Geel BM. Frequent occurrence of cerebral demyelination in adrenomyeloneuropathy. Neurology. 2014;83(24):2227–2231. | |
Costello DJ, Eichler FS, Grant PE, Auluck PK. Case records of the Massachusetts General Hospital. Case 1-2009. A 57-year-old man with progressive cognitive decline. N Engl J Med. 2009;360(2):171–181. | |
van Geel BM, Bezman L, Loes DJ, Moser HW, Raymond GV. Evolution of phenotypes in adult male patients with X-linked adrenoleukodystrophy. Ann Neurol. 2001;49(2):186–194. | |
Dubey P, Raymond GV, Moser AB, Kharkar S, Bezman L, Moser HW. Adrenal insufficiency in asymptomatic adrenoleukodystrophy patients identified by very long-chain fatty acid screening. J Pediatr. 2005; 146(4):528–532. | |
Smith KD, Kemp S, Braiterman LT, et al. X-linked adrenoleukodystrophy: genes, mutations, and phenotypes. Neurochem Res. 1999;24(4):521–535. | |
Berger J, Molzer B, Fae I, Bernheimer H. X-linked adrenoleukodystrophy (ALD): a novel mutation of the ALD gene in 6 members of a family presenting with 5 different phenotypes. Biochem Biophys Res Commun. 1994;205(3):1638–1643. | |
Guimaraes CP, Lemos M, Sa-Miranda C, Azevedo JE. Molecular characterization of 21 X-ALD Portuguese families: identification of eight novel mutations in the ABCD1 gene. Mol Genet Metab. 2002;76(1):62–67. | |
Korenke GC, Fuchs S, Krasemann E, et al. Cerebral adrenoleukodystrophy (ALD) in only one of monozygotic twins with an identical ALD genotype. Ann Neurol. 1996;40(2):254–257. | |
O’Neill GN, Aoki M, Brown RH Jr. ABCD1 translation-initiator mutation demonstrates genotype-phenotype correlation for AMN. Neurology. 2001;57(11):1956–1962. | |
Guimaraes CP, Lemos M, Menezes I, Coelho T, Sa-Miranda C, Azevedo JE. Characterisation of two mutations in the ABCD1 gene leading to low levels of normal ALDP. Hum Genet. 2001;109(6):616–622. | |
Valianpour F, Selhorst JJ, van Lint LE, van Gennip AH, Wanders RJ, Kemp S. Analysis of very long-chain fatty acids using electrospray ionization mass spectrometry. Mol Genet Metab. 2003;79(3):189–196. | |
Moser HW, Moser AB, Frayer KK, et al. Adrenoleukodystrophy: increased plasma content of saturated very long chain fatty acids. Neurology. 1981;31(10):1241–1249. | |
Schutgens RB, Bouman IW, Nijenhuis AA, Wanders RJ, Frumau ME. Profiles of very-long-chain fatty acids in plasma, fibroblasts, and blood cells in Zellweger syndrome, X-linked adrenoleukodystrophy, and rhizomelic chondrodysplasia punctata. Clin Chem. 1993;39(8):1632–1637. | |
Lam C, Wong D, Cederbaum S, Lim B, Qu Y. Peanut consumption increases levels of plasma very long chain fatty acids in humans. Mol Genet Metab. 2012;107(3):620–622. | |
Unterberger U, Regelsberger G, Sundt R, Bernheimer H, Voigtlander T. Diagnosis of X-linked adrenoleukodystrophy in blood leukocytes. Clin Biochem. 2007;40(13–14):1037–1044. | |
Sevin C, Ferdinandusse S, Waterham HR, Wanders RJ, Aubourg P. Autosomal recessive cerebellar ataxia caused by mutations in the PEX2 gene. Orphanet J Rare Dis. 2011;6:8. | |
Regal L, Ebberink MS, Goemans N, et al. Mutations in PEX10 are a cause of autosomal recessive ataxia. Ann Neurol. 2010;68(2):259–263. | |
Tran C, Hewson S, Steinberg SJ, Mercimek-Mahmutoglu S. Late-onset Zellweger spectrum disorder caused by PEX6 mutations mimicking X-linked adrenoleukodystrophy. Pediatr Neurol. 2014;51(2):262–265. | |
Suzuki Y, Shimozawa N, Yajima S, et al. Novel subtype of peroxisomal acyl-CoA oxidase deficiency and bifunctional enzyme deficiency with detectable enzyme protein: identification by means of complementation analysis. Am J Hum Genet. 1994;54(1):36–43. | |
Takemoto Y, Suzuki Y, Horibe R, Shimozawa N, Wanders RJ, Kondo N. Gas chromatography/mass spectrometry analysis of very long chain fatty acids, docosahexaenoic acid, phytanic acid and plasmalogen for the screening of peroxisomal disorders. Brain Dev. 2003;25(7):481–487. | |
Eichler EE, Budarf ML, Rocchi M, et al. Interchromosomal duplications of the adrenoleukodystrophy locus: a phenomenon of pericentromeric plasticity. Hum Mol Genet. 1997;6(7):991–1002. | |
Boehm CD, Cutting GR, Lachtermacher MB, Moser HW, Chong SS. Accurate DNA-based diagnostic and carrier testing for X-linked adrenoleukodystrophy. Mol Genet Metab. 1999;66(2):128–136. | |
Zhan ZX, Liao XX, Du J, et al. Exome sequencing released a case of X-linked adrenoleukodystrophy mimicking recessive hereditary spastic paraplegia. Eur J Med Genet. 2013;56(7):375–378. | |
Zhang Y, Liu Y, Li Y, et al. Exome sequencing identifies mutations in ABCD1 and DACH2 in two brothers with a distinct phenotype. BMC Med Genet. 2014;15(1):105. | |
Claes KB, De Leeneer K. Dealing with pseudogenes in molecular diagnostics in the next-generation sequencing era. Methods Mol Biol. 2014;1167:303–315. | |
Lupski JR, Reid JG, Gonzaga-Jauregui C, et al. Whole-genome sequencing in a patient with Charcot-Marie-Tooth neuropathy. N Engl J Med. 2010;362(13):1181–1191. | |
Dvorakova L, Storkanova G, Unterrainer G, et al. Eight novel ABCD1 gene mutations and three polymorphisms in patients with X-linked adrenoleukodystrophy: the first polymorphism causing an amino acid exchange. Hum Mutat. 2001;18(1):52–60. | |
Wanders RJ, Mooyer PW, Dekker C, Vreken P. X-linked adrenoleukodystrophy: improved prenatal diagnosis using both biochemical and immunological methods. J Inherit Metab Dis. 1998; 21(3):285–287. | |
Maier EM, Roscher AA, Kammerer S, Mehnert K, Conzelmann E, Holzinger A. Prenatal diagnosis of X-linked adrenoleukodystrophy combining biochemical, immunocytochemical and DNA analyses. Prenat Diagn. 1999;19(4):364–368. | |
Lan F, Wang Z, Ke L, et al. A rapid and sensitive protocol for prenatal molecular diagnosis of X-linked adrenoleukodystrophy. Clin Chim Acta. 2010;411(23–24):1992–1997. | |
Moser AB, Moser HW. The prenatal diagnosis of X-linked adrenoleukodystrophy. Prenat Diagn. 1999;19(1):46–48. | |
Carey WF, Poulos A, Sharp P, et al. Pitfalls in the prenatal diagnosis of peroxisomal beta-oxidation defects by chorionic villus sampling. Prenat Diagn. 1994;14(9):813–819. | |
Gray RG, Green A, Cole T, et al. A misdiagnosis of X-linked adrenoleukodystrophy in cultured chorionic villus cells by the measurement of very long chain fatty acids. Prenat Diagn. 1995;15(5):486–490. | |
Handyside AH, Kontogianni EH, Hardy K, Winston RM. Pregnancies from biopsied human preimplantation embryos sexed by Y-specific DNA amplification. Nature. 1990;344(6268):768–770. | |
Iglesias M, Ceballos P, Gimenez C, Garcia-Nebreda MI, Dominguez R, Garcia-Enguidanos A. Pregnancy outcome after preimplantation genetic diagnosis in an affected couple with X-linked adrenoleukodystrophy. Fertil Steril. 2008;90(5):2010. e2011–e2013. | |
Lledo B, Bernabeu R, Ten J, Galan FM, Cioffi L. Preimplantation genetic diagnosis of X-linked adrenoleukodystrophy with gender determination using multiple displacement amplification. Fertil Steril. 2007;88(5):1327–1333. | |
Gigarel N, Frydman N, Burlet P, et al. Single cell co-amplification of polymorphic markers for the indirect preimplantation genetic diagnosis of hemophilia A, X-linked adrenoleukodystrophy, X-linked hydrocephalus and incontinentia pigmenti loci on Xq28. Hum Genet. 2004;114(3):298–305. | |
Rechitsky S, Kuliev A, Tur-Kaspa I, Morris R, Verlinsky Y. Preimplantation genetic diagnosis with HLA matching. Reprod Biomed Online. 2004;9(2):210–221. | |
Kahraman S, Beyazyurek C, Yesilipek MA, et al. Successful haematopoietic stem cell transplantation in 44 children from healthy siblings conceived after preimplantation HLA matching. Reprod Biomed Online. 2014;29(3):340–351. | |
Cartier N, Aubourg P. Hematopoietic stem cell transplantation and hematopoietic stem cell gene therapy in X-linked adrenoleukodystrophy. Brain Pathol. 2010;20(4):857–862. | |
Polgreen LE, Chahla S, Miller W, et al. Early diagnosis of cerebral X-linked adrenoleukodystrophy in boys with Addison’s disease improves survival and neurological outcomes. Eur J Pediatr. 2011; 170(8):1049–1054. | |
Matern D, Oglesbee D, Tortorelli S. Newborn screening for lysosomal storage disorders and other neuronopathic conditions. Dev Disabil Res Rev. 2013;17(3):247–253. | |
Hubbard WC, Moser AB, Liu AC, et al. Newborn screening for X-linked adrenoleukodystrophy (X-ALD): validation of a combined liquid chromatography-tandem mass spectrometric (LC-MS/MS) method. Mol Genet Metab. 2009;97(3):212–220. | |
Sandlers Y, Moser AB, Hubbard WC, Kratz LE, Jones RO, Raymond GV. Combined extraction of acyl carnitines and 26:0 lysophosphatidylcholine from dried blood spots: prospective newborn screening for X-linked adrenoleukodystrophy. Mol Genet Metab. 2012;105(3):416–420. | |
Haynes CA, De Jesus VR. Improved analysis of C26:0-lysophosphatidylcholine in dried-blood spots via negative ion mode HPLC-ESI-MS/MS for X-linked adrenoleukodystrophy newborn screening. Clin Chim Acta. 2012;413(15–16):1217–1221. | |
Theda C, Gibbons K, Defor TE, et al. Newborn screening for X-linked adrenoleukodystrophy: further evidence high throughput screening is feasible. Mol Genet Metab. 2014;111(1):55–57. | |
Turgeon CT, Moser AB, Morkrid L, et al. Streamlined determination of lysophosphatidylcholines in dried blood spots for newborn screening of X-linked adrenoleukodystrophy. Mol Genet Metab. 2015; 114(1):46–50. | |
Vogel BH, Bradley SE, Adams DJ, et al. Newborn screening for X-linked adrenoleukodystrophy in New York State: Diagnostic protocol, surveillance protocol and treatment guidelines. Mol Genet Metab. February 12, 2015. [Epub ahead of print.] | |
Kemp S, Pujol A, Waterham HR, et al. ABCD1 mutations and the X-linked adrenoleukodystrophy mutation database: role in diagnosis and clinical correlations. Hum Mutat. 2001;18(6):499–515. | |
Zhang X, De Marcos Lousa C, Schutte-Lensink N, et al. Conservation of targeting but divergence in function and quality control of peroxisomal ABC transporters: an analysis using cross-kingdom expression. Biochem J. 2011;436(3):547–557. | |
Morita M, Kobayashi J, Yamazaki K, et al. A novel double mutation in the ABCD1 gene in a patient with X-linked adrenoleukodystrophy: analysis of the stability and function of the mutant ABCD1 protein. JIMD Rep. 2013;10:95–102. | |
Halbach A, Lorenzen S, Landgraf C, Volkmer-Engert R, Erdmann R, Rottensteiner H. Function of the PEX19-binding site of human adrenoleukodystrophy protein as targeting motif in man and yeast. PMP targeting is evolutionarily conserved. J Biol Chem. 2005;280(22):21176–21182. | |
Engelen M, van der Kooi AJ, Kemp S, et al. X-linked adrenomyeloneuropathy due to a novel missense mutation in the ABCD1 start codon presenting as demyelinating neuropathy. J Peripher Nerv Syst. 2011;16(4):353–355. | |
Vachalova I, Chandoga J, Petrovic R, Copikova-Cudrakova D, Sykora M, Traubner P. Adrenoleukodystrophy – a new mutation identified. Bratisl Lek Listy. 2007;108(10–11):462–466. | |
Liu LX, Janvier K, Berteaux-Lecellier V, Cartier N, Benarous R, Aubourg P. Homo- and heterodimerization of peroxisomal ATP-binding cassette half-transporters. J Biol Chem. 1999;274(46):32738–32743. | |
Hillebrand M, Verrier SE, Ohlenbusch A, et al. Live cell FRET microscopy: homo- and heterodimerization of two human peroxisomal ABC transporters, the adrenoleukodystrophy protein (ALDP, ABCD1) and PMP70 (ABCD3). J Biol Chem. 2007;282(37):26997–27005. | |
Coll MJ, Palau N, Camps C, Ruiz M, Pampols T, Giros M. X-linked adrenoleukodystrophy in Spain. Identification of 26 novel mutations in the ABCD1 gene in 80 patients. Improvement of genetic counseling in 162 relative females. Clin Genet. 2005;67(5):418–424. | |
Unterrainer G, Molzer B, Forss-Petter S, Berger J. Co-expression of mutated and normal adrenoleukodystrophy protein reduces protein function: implications for gene therapy of X-linked adrenoleukodystrophy. Hum Mol Genet. 2000;9(18):2609–2616. | |
Guimaraes CP, Domingues P, Aubourg P, et al. Mouse liver PMP70 and ALDP: homomeric interactions prevail in vivo. Biochim Biophys Acta. 2004;1689(3):235–243. | |
Jangouk P, Zackowski KM, Naidu S, Raymond GV. Adrenoleukodystrophy in female heterozygotes: underrecognized and undertreated. Mol Genet Metab. 2012;105(2):180–185. | |
Hershkovitz E, Narkis G, Shorer Z, et al. Cerebral X-linked adrenoleukodystrophy in a girl with Xq27-Ter deletion. Ann Neurol. 2002; 52(2):234–237. | |
Chen X, Chen Z, Huang D, Liu X, Gui Q, Yu S. Adult cerebral adrenoleukodystrophy and Addison’s disease in a female carrier. Gene. 2014;544(2):248–251. | |
Habekost CT, Schestatsky P, Torres VF, et al. Neurological impairment among heterozygote women for X-linked adrenoleukodystrophy: a case control study on a clinical, neurophysiological and biochemical characteristics. Orphanet J Rare Dis. 2014;9(1):6. | |
Wang Z, Yan A, Lin Y, Xie H, Zhou C, Lan F. Familial skewed X chromosome inactivation in adrenoleukodystrophy manifesting heterozygotes from a Chinese pedigree. PLoS One. 2013;8(3):e57977. | |
Salsano E, Tabano S, Sirchia SM, et al. Preferential expression of mutant ABCD1 allele is common in adrenoleukodystrophy female carriers but unrelated to clinical symptoms. Orphanet J Rare Dis. 2012;7:10. | |
Maier EM, Kammerer S, Muntau AC, Wichers M, Braun A, Roscher AA. Symptoms in carriers of adrenoleukodystrophy relate to skewed X inactivation. Ann Neurol. 2002;52(5):683–688. | |
Watkiss E, Webb T, Bundey S. Is skewed X inactivation responsible for symptoms in female carriers for adrenoleucodystrophy? J Med Genet. 1993;30(8):651–654. | |
Migeon BR, Moser HW, Moser AB, Axelman J, Sillence D, Norum RA. Adrenoleukodystrophy: evidence for X linkage, inactivation, and selection favoring the mutant allele in heterozygous cells. Proc Natl Acad Sci U S A. 1981;78(8):5066–5070. | |
Pereira Fdos S, Matte U, Habekost CT, et al. Mutations, clinical findings and survival estimates in South American patients with X-linked adrenoleukodystrophy. PLoS One. 2012;7(3):e34195. | |
Moser HW, Moser AB, Smith KD, et al. Adrenoleukodystrophy: phenotypic variability and implications for therapy. J Inherit Metab Dis. 1992;15(4):645–664. | |
Barbier M, Sabbagh A, Kasper E, et al. CD1 gene polymorphisms and phenotypic variability in X-linked adrenoleukodystrophy. PLoS One. 2012;7(1):e29872. | |
Maier EM, Mayerhofer PU, Asheuer M, et al. X-linked adrenoleukodystrophy phenotype is independent of ABCD2 genotype. Biochem Biophys Res Commun. 2008;377(1):176–180. | |
Matsukawa T, Asheuer M, Takahashi Y, et al. Identification of novel SNPs of ABCD1, ABCD2, ABCD3, and ABCD4 genes in patients with X-linked adrenoleukodystrophy (ALD) based on comprehensive resequencing and association studies with ALD phenotypes. Neurogenetics. 2011;12(1):41–50. | |
Semmler A, Bao X, Cao G, et al. Genetic variants of methionine metabolism and X-ALD phenotype generation: results of a new study sample. J Neurol. 2009;256(8):1277–1280. | |
Netik A, Forss-Petter S, Holzinger A, Molzer B, Unterrainer G, Berger J. Adrenoleukodystrophy-related protein can compensate functionally for adrenoleukodystrophy protein deficiency (X-ALD): implications for therapy. Hum Mol Genet. 1999;8(5):907–913. | |
Kemp S, Wei HM, Lu JF, et al. Gene redundancy and pharmacological gene therapy: implications for X-linked adrenoleukodystrophy. Nat Med. 1998;4(11):1261–1268. | |
Pujol A, Ferrer I, Camps C, et al. Functional overlap between ABCD1 (ALD) and ABCD2 (ALDR) transporters: a therapeutic target for X-adrenoleukodystrophy. Hum Mol Genet. 2004;13(23):2997–3006. | |
Muneer Z, Wiesinger C, Voigtlander T, Werner HB, Berger J, Forss-Petter S. ABCD2 is a strong modifier of the metabolic impairments in peritoneal macrophages of ABCD1-deficient mice. PLoS One. 2014;9(9):e108655. | |
Braiterman LT, Zheng S, Watkins PA, et al. Suppression of peroxisomal membrane protein defects by peroxisomal ATP binding cassette (ABC) proteins. Hum Mol Genet. 1998;7(2):239–247. | |
Linnebank M, Semmler A, Kleijer WJ, et al. The cystathionine beta-synthase variant c.844_845ins68 protects against CNS demyelination in X-linked adrenoleukodystrophy. Hum Mutat. 2006;27(10):1063–1064. | |
Cao GN, Bao XH, Xiong H, Wu Y, Wu XR. [Association of genetic polymorphisms in methionine metabolism genes with X-linked adrenoleukodystrophy]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2011;28(3):279–282. Chinese. | |
Linnebank M, Kemp S, Wanders RJ, et al. Methionine metabolism and phenotypic variability in X-linked adrenoleukodystrophy. Neurology. 2006;66(3):442–443. | |
Sawcer S, Hellenthal G, Pirinen M, et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature. 2011;476(7359):214–219. | |
Berger J, Bernheimer H, Fae I, et al. Association of X-linked adrenoleukodystrophy with HLA DRB1 alleles. Biochem Biophys Res Commun. 1995;216(2):447–451. | |
Schmidt S, Marrosu GM, Kolsch H, et al. Genetic variations and humoral immune responses to myelin oligodendroglia glycoprotein in adult phenotypes of X-linked adrenoleukodystrophy. J Neuroimmunol. 2003;135(1–2):148–153. | |
McGuinness MC, Powers JM, Bias WB, et al. Human leukocyte antigens and cytokine expression in cerebral inflammatory demyelinative lesions of X-linked adrenoleukodystrophy and multiple sclerosis. J Neuroimmunol. 1997;75(1–2):174–182. | |
Ofman R, Dijkstra IM, van Roermund CW, et al. The role of ELOVL1 in very long-chain fatty acid homeostasis and X-linked adrenoleukodystrophy. EMBO Mol Med. 2010;2(3):90–97. | |
Sanders RJ, Ofman R, Duran M, Kemp S, Wanders RJ. Omega-oxidation of very long-chain fatty acids in human liver microsomes. Implications for X-linked adrenoleukodystrophy. J Biol Chem. 2006; 281(19):13180–13187. | |
Asheuer M, Bieche I, Laurendeau I, et al. Decreased expression of ABCD4 and BG1 genes early in the pathogenesis of X-linked adrenoleukodystrophy. Hum Mol Genet. 2005;14(10):1293–1303. | |
McGuinness MC, Griffin DE, Raymond GV, Washington CA, Moser HW, Smith KD. Tumor necrosis factor-alpha and X-linked adrenoleukodystrophy. J Neuroimmunol. 1995;61(2):161–169. | |
Gomez-Lira M, Marzari MG, Uziel G, Pignatti P, Rizzuto N, Salviati A. Myelin oligodendrocyte glycoprotein (MOG) polymorphisms and adrenoleukodystrophy. J Neuroimmunol. 2000;111(1–2):245–247. | |
Brose RD, Avramopoulos D, Smith KD. SOD2 as a potential modifier of X-linked adrenoleukodystrophy clinical phenotypes. J Neurol. 2012;259(7):1440–1447. | |
Troffer-Charlier N, Doerflinger N, Metzger E, Fouquet F, Mandel JL, Aubourg P. Mirror expression of adrenoleukodystrophy and adrenoleukodystrophy related genes in mouse tissues and human cell lines. Eur J Cell Biol. 1998;75(3):254–264. | |
Berger J, Albet S, Bentejac M, et al. The four murine peroxisomal ABC-transporter genes differ in constitutive, inducible and developmental expression. Eur J Biochem. 1999;265(2):719–727. | |
Weber FD, Wiesinger C, Forss-Petter S, et al. X-linked adrenoleukodystrophy: very long-chain fatty acid metabolism is severely impaired in monocytes but not in lymphocytes. Hum Mol Genet. 2014; 23(10):2542–2550. | |
Genin EC, Geillon F, Gondcaille C, et al. Substrate specificity overlap and interaction between adrenoleukodystrophy protein (ALDP/ABCD1) and adrenoleukodystrophy-related protein (ALDRP/ABCD2). J Biol Chem. 2011;286(10):8075–8084. | |
van Roermund CW, Ijlst L, Wagemans T, Wanders RJ, Waterham HR. A role for the human peroxisomal half-transporter ABCD3 in the oxidation of dicarboxylic acids. Biochim Biophys Acta. 2014;1841(4):563–568. | |
Ferdinandusse S, Jimenez-Sanchez G, Koster J, et al. A novel bile acid biosynthesis defect due to a deficiency of peroxisomal ABCD3. Hum Mol Genet. 2015;24(2):361–370. | |
Tanaka AR, Tanabe K, Morita M, et al. ATP binding/hydrolysis by and phosphorylation of peroxisomal ATP-binding cassette proteins PMP70 (ABCD3) and adrenoleukodystrophy protein (ABCD1). J Biol Chem. 2002;277(42):40142–40147. | |
Geillon F, Gondcaille C, Charbonnier S, et al. Structure-function analysis of peroxisomal ATP-binding cassette transporters using chimeric dimers. J Biol Chem. 2014;289(35):24511–24520. | |
Analysis Tool Web Services from the EMBL-EBI. (2013) McWilliam H, Li W, Uludag M, Squizzato S, Park YM, Buso N, Cowley AP, Lopez R. Nucleic acids research 2013 Jul;41(Web Server issue):W597–600. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.