")
Back to Journals » Drug Design, Development and Therapy » Volume 8
Tetrandrine protects mouse retinal ganglion cells from ischemic injury
Authors Li W, Yang C, Lu J, Huang P, Barnstable C, Zhang C, Zhang S
Received 4 October 2013
Accepted for publication 12 December 2013
Published 21 March 2014 Volume 2014:8 Pages 327—339
DOI https://doi.org/10.2147/DDDT.S55407
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Weiyi Li,1,2 Chen Yang,2 Jing Lu,2 Ping Huang,1 Colin J Barnstable,2 Chun Zhang,1 Samuel S Zhang2,3
1Department of Ophthalmology, Peking University Third Hospital, Peking University Eye Center, Beijing, People's Republic of China; 2Department of Neural and Behavioral Sciences, Penn State University, Hershey, PA, USA; 3Singapore Eye Research Institute, Singapore National Eye Centre, Singapore
Abstract: This study aimed to determine the protective effects of tetrandrine (Tet) on murine ischemia-injured retinal ganglion cells (RGCs). For this, we used serum deprivation cell model, glutamate and hydrogen peroxide (H2O2)-induced RGC-5 cell death models, and staurosporine-differentiated neuron-like RGC-5 in vitro. We also investigated cell survival of purified primary-cultured RGCs treated with Tet. An in vivo retinal ischemia/reperfusion model was used to examine RGC survival after Tet administration 1 day before ischemia. We found that Tet affected RGC-5 survival in a dose- and time-dependent manner. Compared to dimethyl sulfoxide treatment, Tet increased the numbers of RGC-5 cells by 30% at 72 hours. After 48 hours, Tet protected staurosporine-induced RGC-5 cells from serum deprivation-induced cell death and significantly increased the relative number of cells cultured with 1 mM H2O2 (P<0.01). Several concentrations of Tet significantly prevented 25-mM-glutamate-induced cell death in a dose-dependent manner. Tet also increased primary RGC survival after 72 and 96 hours. Tet administration (10 µM, 2 µL) 1 day before retinal ischemia showed RGC layer loss (greater survival), which was less than those in groups with phosphate-buffered saline intravitreal injection plus ischemia in the central (P=0.005, n=6), middle (P=0.018, n=6), and peripheral (P=0.017, n=6) parts of the retina. Thus, Tet conferred protective effects on serum deprivation models of staurosporine-differentiated neuron-like RGC-5 cells and primary cultured murine RGCs. Furthermore, Tet showed greater in vivo protective effects on RGCs 1 day after ischemia. Tet and ciliary neurotrophic factor maintained the mitochondrial transmembrane potential (ΔΨm) of primary cultured RGCs and inhibited the expression of activated caspase-3 and bcl-2 in ischemia/reperfusion-insult retinas.
Keywords: RGC-5, serum deprivation, glaucoma, mitochondrial membrane potential, apoptosis
Introduction
Tetrandrine (Tet) is a bisbenzylisoquinoline alkaloid extracted from the root of the Chinese herb creeper Stephania tetrandra.1,2 It is a calcium channel blocker3,4 that inhibits lipid peroxidation5 and the generation of reactive oxygen species (ROS)6 and suppresses the production of cytokines and inflammatory mediators in the brain after ischemia reperfusion injury,6,7 anoxia,8 or Alzheimer’s disease.9 Tet ameliorates amyloid-β(1-42)-induced spatial learning and memory impairment, and the beneficial effect of tetrandrine treatment may be linked to the inhibition of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) activity and downregulation of interleukin-1β and tumor necrosis factor-α.9 Furthermore, it has been found that Tet can increase neuroprotective brain-derived neurotrophic factor (BDNF) protein levels in the rat hippocampus and regulate the central monoaminergic neurotransmitter system.10
Cytosolic ionized calcium often increases within seconds or minutes after an injury or other cell damage, long before cell death, and the increased calcium concentration activates various signaling processes that include phospholipases, endonucleases, proteases, and protein kinases,11–13 followed by both apoptotic and necrotic cell death of neurons,14,15 including retinal neurons.16,17 For example, excitotoxic activation of calpain, a calcium-dependent protease, may facilitate necrosis and apoptosis after cerebral hypoxia-induced ischemia.17,18 There have been no studies to indicate a neuroprotective action of Tet on retinal ganglion cells (RGCs), although neuroprotective effects on neurons of the brain have been reported.9,10 To determine the neuroprotective effects of Tet on RGCs, we utilized a series of in vitro models of cell death, namely serum deprivation as well as glutamate and hydrogen peroxide (H2O2) treatment of RGC-5 cells, staurosporine (SSP)-induced RGC-5 cells, and purified RGCs in culture. We also used an in vivo model of transient retinal ischemia to determine whether Tet would protect RGCs from retinal ischemia reperfusion injury.
Materials and methods
Animals
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health.19 The protocol was approved by the IACUC of the Penn State University College of Medicine (Permit Number, 2006-080) and the Peking University Third Hospital (Permit Number, 11161). All surgeries were performed under sodium pentobarbital anesthesia and all efforts were made to minimize suffering. All animals were housed in temperature-controlled rooms with a 12-hour light/dark cycle and free access to food and water.
Reagents
Tet (C38H42O6 N2, molecular weight 622.730; Figure 1A; Sigma-Aldrich, St Louis, MO, USA) was dissolved in dimethyl sulfoxide (DMSO), and 10% 1 normal hydrogen chloride was added. The solution was diluted with phosphate-buffered saline (PBS) to a concentration of 1 mM, adjusted to pH 7.35, and sterilized by passage through a 0.22 μm filter. Gentamicin, Dulbecco’s PBS (D-PBS), progesterone, B27, sodium pyruvate, glutamine, and ciliary neurotrophic factor (CNTF) were obtained from Life Technologies (Carlsbad, CA, USA); fetal bovine serum (FBS) from Atlanta Biologicals, Inc. (Flowery Branch, GA, USA); SSP from Alexis Biochemicals USA (San Diego, CA, USA); WST-8 assay kit (CCK-8) from Dojindo Laboratories (Kumamoto, Japan); BDNF from PeproTech (Rocky Hill, NJ, USA); ketamine from Mylan GmbH (Zurich, Switzerland); xylazine from Celgene Corporation (Summit, NJ, USA); atropine ointment from Bausch and Lomb (Rochester, NY, USA); and vetropolycin ointment from PharmaDerm (Melville, NY, USA).
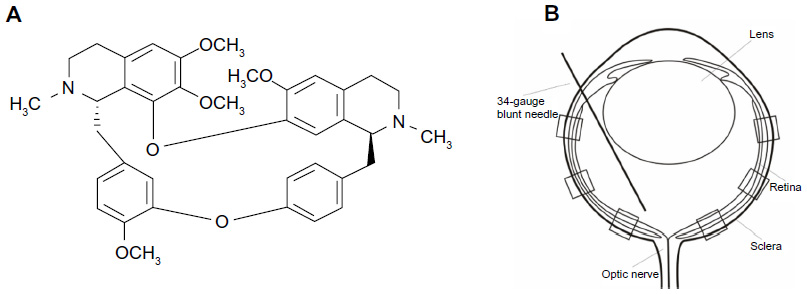 | Figure 1 Structural formula of Tet and a schematic of intravitreal treatment and partitions of the retina for RGC counting. |
The antibody integrin αM (2LPM19c) (MAC-1, CD11b) was purchased from Santa Cruz Biotechnology, Inc. (Dallas, Texas, USA), affinity-purified goat anti-mouse immunoglobulin G (IgG) (H+L) and rat anti-mouse IgG (H+L) antibody from Jackson ImmunoResearch Laboratories, Inc. (West Grove, PA, USA), and mouse anti-rat CD90.2 antibody from BD Biosciences (San Jose, CA, USA). Antibodies to Bcl-2 and cleaved caspase-3 (Asp175) were purchased from Beyotime (Haimen, People’s Republic of China) and JC-10 from AAT Bioquest, Inc. (Sunnyvale, CA, USA).
Unless noted, all other reagents were obtained from Sigma-Aldrich.
RGC-5 cell line
Cultures of the rat retinal ganglion cell line (RGC-5) (ATCC; Manassas, VA, USA) were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 4.5 mg/L glucose and 2 mM l-glutamine, supplemented with 10% FBS, and 10 μg/mL gentamicin in a humidified atmosphere of 95% air and 5% CO2 at 37°C. For the experiments, the RGC-5 cells were plated on 96-well culture plates in DMEM with or without 10% FBS at a density of 5 × 104 cells/mL.
SSP-induced RGC-5 line
A protocol developed by Yang et al,20 with modification, was used to induce RGC-5 cells, which led to their differentiation into highly branched, nonmitotic neuron-like cells. RGC-5 cells were cultured in DMEM with 10% FBS and plated onto 96-well plates, coated with a poly-d-lysine and laminin, at a density of 1 × 105 cells/mL. Cells were allowed to adhere to the substrate for 1 hour at 37°C and were then treated with the general kinase inhibitor SSP (1 μM) for 1 hour. Following differentiation, the medium was changed to a Neurobasal® medium (Life Technologies, Carlsbad, CA, USA) with Tet and the number of cells were counted 24 hours and 48 hours after treatment.
Glutamate- and H2O2-mediated cell death
l-glutamic acid (glutamate) and H2O2 were used to induce cell death in RGC-5 cells. Following treatment with various concentrations of Tet for 6 hours, RGC-5 cells were exposed to glutamate (25 mM) or H2O2 (1 μM) for 12 hours to 72 hours depending on the experiment. Cell numbers were determined with a WST-8 assay (CCK-8) described below. The number of living cells in each well was expressed as a value relative to the control value.
WST-8 assay
The relative numbers of the living RGC-5 and SSP-induced RGC-5 cells were determined with a WST-8 assay kit. WST-8 (2-[2-methoxy-4-nitrophenyl]-3-[4-nitrophenyl]-5-[2,4-disulfophenyl]-2H-tetrazolium, monosodium salt) produces a water-soluble formazan dye on bioreduction in the presence of an electron carrier, 1-methoxy-5-methylphenazinium methyl sulfate. After exposure to Tet under the different conditions, the medium was replaced by a 10% WST-8 solution in DMEM. The number of living cells in each well was determined by measuring the optical density at 460 nm with a microplate reader (680XR; Bio-Rad Laboratories, Inc., Hercules, CA, USA) and was expressed as a value relative to the control value.
Purified primary culture of murine RGCs
The protocols used to purify RGCs from mice, rats, and human neonatal retina21–25 were adapted for this study. A two-step immunopanning procedure was performed.
Retinas from 24 0–2-day-old C57Bl/6 pups of either sex were dissected on ice and incubated at 37°C for 30 minutes in 33 U/mL papain, 70 U/mL collagenase, 0.004% deoxyribonuclease, and 0.2 mg/mL l-cysteine in D-PBS with Ca2+ and Mg2+ containing 0.2 mg/mL bovine serum albumin (BSA). To yield a suspension of single cells, the tube was gently agitated ten times until the suspension was cloudy. Then, 2 mL 1% trypsin inhibitor BSA solution was added to the tube and the cells were collected by centrifugation at 80 × gravity for 10 minutes. The cell pellet was resuspended in 2 mL 0.02% BSA in D-PBS containing integrin αM (Mac-1) antibody.
Sterile petri dishes (100 and 60 mm in diameter; BD Biosciences) were prepared for immunopanning. The 100 mm petri dishes were coated with 10 μg/mL affinity-purified goat anti-mouse IgG (H+L) antibody, and the 60 mm petri dishes were coated with 10 μg/mL affinity-purified rat anti-mouse IgG (H+L) antibody in 50 mM Tris (pH 9.5) at 4°C overnight. After washing, the surface of the 100 mm petri dishes was blocked with 0.2 mg/mL BSA in D-PBS for 1 hour. The 60 mm petri dishes were incubated with mouse anti-rat CD90.2 antibody for 1 hour at room temperature. The retinal cells in 0.02% BSA were placed in a 100 mm petri dish precoated with affinity-purified goat anti-mouse IgG (H+L) antibody for 20 minutes at 37°C. Nonadherent cells were transferred to the second 100 mm petri dish for 20 minutes at 37°C. The nonadherent cells were passed through a mesh filter (40 μm pore size; BD Biosciences) to obtain single cells, which were then transferred to the 60 mm dish precoated with rat anti-mouse CD90.2 antibody and incubated for 30 minutes at 37°C. After removal of nonadherent cells (collected for non-RGC testing), the dish was washed ten times with D-PBS. Adherent cells were mechanically scraped off in Neurobasal® medium containing 1% BSA, selenium (6.7 ng/mL), transferrin (5.5 μg/mL), putrescine (60 μM), 3,5,3-triiodothyronine (T3, 100 nM), progesterone (20 nM), B27 (1%), sodium pyruvate (1 mM), glutamine (2 mM), BDNF (50 ng/mL), CNTF (10 ng/mL), forskolin (5 μM), and gentamicin (10 μg/mL). The cells were plated at a low density of approximately 1,000 cells/cm2 on a surface precoated with poly-d-lysine and laminin. Cultures were maintained at 37°C in a humidified atmosphere containing 5% CO2 and 95% air.
Cell counting of primary cultured RGCs
Purified RGCs were cultured in 96-well plates. Five 200 × 250 μm2 microscopic fields (central, superior, inferior, left, and right) of each well were chosen to count the RGCs. The corresponding regions of the RGCs with healthy somas that had axons or dendrites at least two times longer than their cell body were counted.
Mitochondrial membrane potential in primary cultured RGCs (ΔΨm) by JC-10 fluorescence and flow cytometry
The ΔΨm of primary cultured RGCs was determined using the fluorescent probe JC-10. Briefly, primary cultured RGCs were exposed to CNTF (12.5 ng/mL), Tet (10 nM), or the mitochondrial uncoupler carbonyl cyanide-4- (trifluoromethoxy)phenylhydrazone (FCCP) (5 μM) for 1 hour and then incubated in culture medium containing 3 μM JC-10 for 1 hour at room temperature. The cells were washed with PBS and analyzed by flow cytometry. Photomultiplier settings were adjusted to detect JC-10 monomer and aggregate fluorescence on the FL1 (525 nm) and FL2 (595 nm) detectors. The fluorescence ratio at these wavelengths was used to monitor changes in mitochondrial membrane potential.26
Intravitreal injection of tetrandrine
The protocol of performing an intravitreal injection has been previously described (Figure 1B).27 For the intravitreal injection of Tet into a transient retina ischemic model, 20 BABL/c mice (24 eyes) of either sex (4-week-old mice, 18–24 g) were used. Animals were anesthetized with a cocktail of intraperitoneal ketamine (75 mg/kg) and xylazine (5 mg/kg). Atropine ointment was applied to achieve mydriasis. Twenty-four hours before ischemic insult, the mice received intravitreal injections of Tet in the experimental group or of PBS in the control group. All injections were performed under visual control using an ophthalmic microscope. Mice in both experimental and control groups received 2 μL intravitreal injections. Intravitreal injections were performed with a 34-gauge blunt needle attached to a 5 μL syringe (Hamilton Company; Reno, NV, USA) after 10–0 needle puncture through the sclera, approximately 1 mm behind the limbus. The needle remained in the vitreous cavity for 1 minute after the injection and was then gently withdrawn. Vetropolycin ointment was used on the injected eyes after the operation. The injected animals were allowed to recover for 24 hours before being subjected to ischemic insults.
Transient retina ischemic model
The transient retina ischemic model was induced according to a previously reported protocol,27–29 with a modification. Transient retinal ischemia was induced in the right eyes, and the contralateral eyes served as nonischemic control. Briefly, animals were anesthetized with an intraperitoneal injection of ketamine (75 mg/kg) and xylazine hydrochloride (5 mg/kg). Atropine ointment was applied to achieve mydriasis. The anterior chamber of the right eye was then cannulated with a 30-gauge needle connected to a sterile saline-filled bottle. Intraocular pressure (IOP) was maintained at 110 mmHg for 60 minutes by lifting the bottle to a height of 1,496 mm above the eyes. Vetropolycin ophthalmic ointment was applied to treated eyes after the operation.
Immunofluorescence staining of retinas
For Bcl-2, cleaved caspase-3 (Asp175), and 4′,6-diamidino-2-phenylindole (DAPI) staining 1 and 3 days after ischemia, the eyes were quickly enucleated and dissected, and the posterior eyecups were placed in a chilled fixative (4% paraformaldehyde in 0.1 M phosphate buffer, pH 7.4) for 6 hours. After washing three times, the fixed retinas were transferred into 30% sucrose containing 0.1 M PBS overnight at 4°C and then embedded with Tissue-Tek® O.C.T Compound (Sakura Finetek; Tokyo, Japan) at optimal cutting temperature. Cryostat sections of the retina (12 μm, sagittal) were mounted onto poly-l-lysine-coated slides (VWR International, Radnor, PA, USA). The specimens were blocked in 2% horse serum and 2% BSA in Triton 100 (Sigma-Aldrich) for 60 minutes. The Bcl-2 and cleaved caspase-3 (Asp175) antibodies (1:500) were incubated overnight at 4°C. After three 5-minute rinses in PBS, Dylight488 (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA) secondary antibody (1:1,000) and DAPI (1:50,000) were added and incubated at room temperature in the dark. After three 5-minute rinses in PBS, the slides were covered with Vectashield Mounting Medium (Sigma-Aldrich). Fluorescence signals were visualized by laser-scanning confocal microscopy (Olympus Corporation, Tokyo, Japan).
Cell counting of RGCs in retinal cross sections
RGCs were counted using published procedures30 with little modification. Briefly, each retinal section was divided into central, middle, and peripheral regions (1, 2, and 3 mm from the optic disc, respectively). Microscopic fields measuring 210 × 210 μm2 were chosen to count DAPI-labeled RGCs. Six microscopic fields were used in each retina for counting (Figure 1B). Corresponding regions from each retina of the experimental and control groups were used for counting.
Statistical analysis
Statistical analyses of the means for more than two groups were performed using one-way analysis of variance. The Student’s t-test was applied for samples with an insignificant difference in variance. The null hypothesis was rejected at the 0.05 level. All statistical computations were carried out using SPSS 13.0 (IBM Corporation, Armonk, NY, USA). The relative cell numbers are expressed as the mean ± standard deviation.
Results
Protective influence of Tet on RGC-5 and SSP-induced neuron-like cell death resulting from serum deprivation
Serum deprivation models were used to evaluate the protective effects of Tet on RGC-5 cell survival. The relative cell numbers from control, 0.01% DMSO, and Tet-treated groups of RGC-5 cells are shown in Figure 2. All cell numbers were measured at 24 hours, 72 hours, and 96 hours after treatment in both serum deprived (Figure 2A) and 10% FBS growth conditions (Figure 2B).
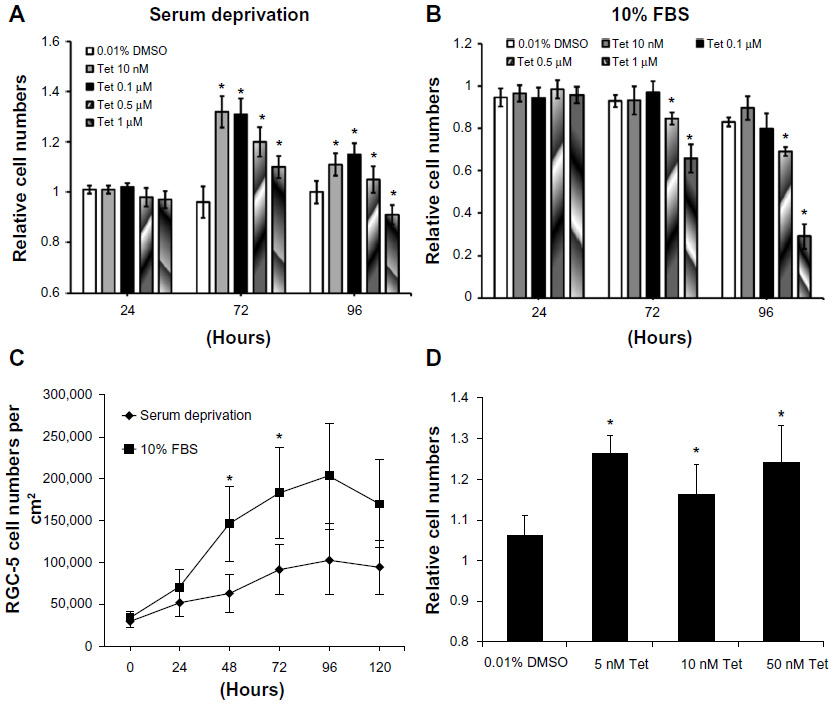 | Figure 2 Effects of Tet on the survival of RGC-5 cells and SSP-induced neuron-like cells under serum deprivation conditions. |
Under conditions of serum deprivation, as shown in Figure 2A, the RGC-5 cell numbers increased 30% at 72 hours using concentrations of Tet from 10 nM (1.32±0.060; P=1x10–11) to 0.1 μM (1.31±0.080; P=1×10–11) as compared to the cells treated with DMSO alone. At 96 hours after Tet treatment, the increase was less pronounced but still significant (10 nM, 1.11±0.041; P=0.001; 0.1 μM, 1.15±0.077; P=3.8×10–5). However, higher concentrations of Tet, 0.5 μM (1.20±0.059, P=1.96×10–7) and 1 μM (1.10±0.043, P=3.95×10–4), did not increase the cell numbers compared to DMSO alone at 72 hours, and 1 μM Tet treatment resulted in a reduction in cell numbers at 96 hours (0.91±0.044, P=0.004). For cells grown in 10% FBS, lower concentrations of Tet caused no significant change in cell numbers, and both 0.5 μM and 1 μM Tet resulted in a significant decrease in relative numbers of RGC-5 cells (for 1 μM, P=3.16×10–5) compared to DMSO alone (Figure 2B). The absolute numbers of RGC-5 cells in 10% FBS (Figure 2C) were significantly higher than those under the serum deprivation condition at 48 hours (P=1.5×10–9) and 72 hours (P=1.9×10–7). These results suggest that Tet has little effect on cells that are growing rapidly in 10% FBS, but can protect cells under stressed conditions.
To eliminate confounding factors related to cell division, we examined Tet function in postmitotic RGC-5-derived neuron-like cells. SSP (1 μM) optimally induced and maintained neuron-like morphology in RGC-5 cells from 2 hours to 72 hours (data not shown). We measured the protection of SSP-induced neuron-like cells at 48 hours after treatment with 5 nM (P=9.67×10–5), 10 nM (P=9.67×10–5), and 50 nM (P=2.58x10–3) Tet (Figure 2D). The numbers of SSP-induced neuron-like cells increased approximately 16% to 26% after Tet pretreatment, as compared to DMSO alone (Figure 2D). This suggests that Tet promotes survival of postmitotic cells, which more closely represent mature neurons.
Tet protected cells from oxidative stress and glutamate-induced neurotoxicity
To determine whether Tet protects cells from oxidative stress and glutamate neurotoxicity, insults closely related to glaucoma, we examined cell survival in response to H2O2 and glutamate stresses using a WST-8 cell proliferation assay (Figure 3). As shown in Figure 3A, 100 nM Tet, but not 1 nM or 10 nM (data not shown), was able to significantly retard the reduction in cell numbers caused by 1 mM H2O2 (P=4.2x10–4), although they did not return to control levels.
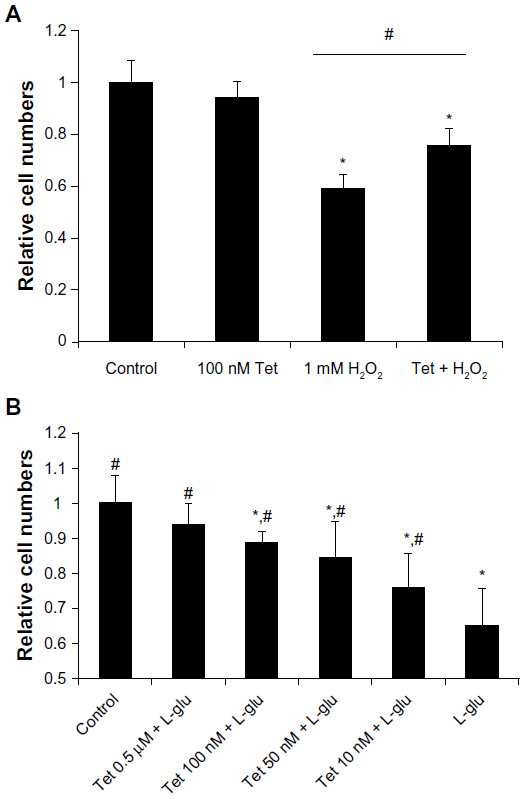 | Figure 3 Tet protected RGC-5 cells from H2O2 injury and l-glutamate excitotoxicity. |
Similarly, a range of Tet concentrations from 10 nM to 0.5 μM significantly prevented the cell death induced by 25 mM glutamate-induced neurotoxicity, in a dose-dependent manner (Figure 3B), with cell numbers almost reaching control levels after Tet treatment with the highest concentration (0.5 μM). Together, these results indicate that Tet can promote cell survival in the context of oxidative stress or glutamate-induced neurotoxicity.
Tet slows down the loss of primary cultured RGCs in vitro
To confirm that Tet can exert protective effects on primary cultures, we examined the effects of Tet on the survival of murine primary RGCs. The immunopanning method,21–25 with modifications, was used to isolate RGCs from neonatal mouse retinas. Primary RGCs were grown at a low density (approximately 1,000 cells/cm2) for 4 days and were characterized with a neuron-specific antibody (Figure 4A). The cells extended long processes from variably sized cell bodies (Figure 4B), and exhibited a gradual loss of viability with a more stable period of approximately 40%–60% survival between 3 days and 9 days after isolation (Figure 4C). The primary RGCs from this period were used to examine Tet function.
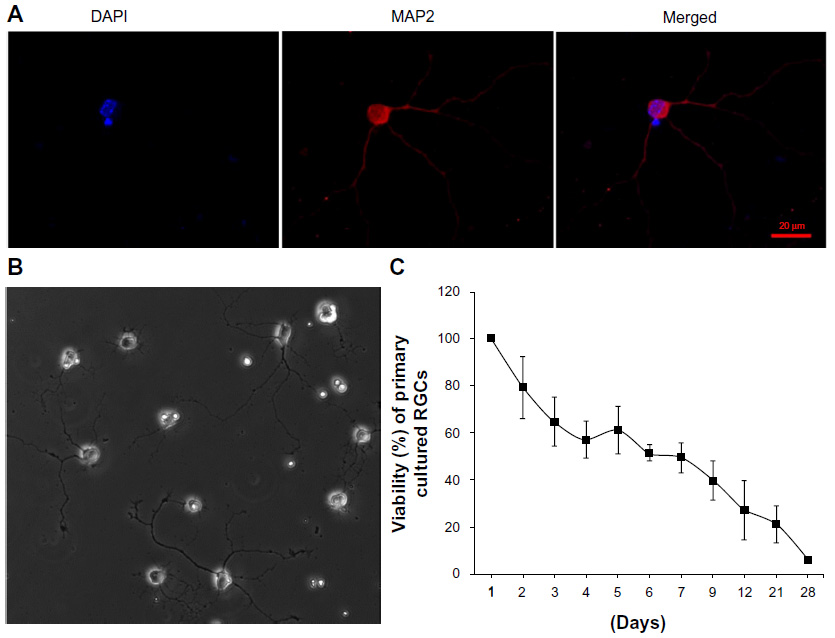 | Figure 4 The identification, morphology, and survival curve of primary cultured RGCs. |
Two concentrations of Tet, 1 nM and 10 nM, were investigated with regard to their relative capacity to protect RGCs from death after isolation, as compared to DMSO alone (Figure 5). Where the cell count immediately after isolation was deemed to be 100%, 72 hours after Tet treatment, significantly increased RGC survival was observed in both the 1 nM (35.7%±10.4%, P=0.009) and 10 nM (56%±10.5%, P=3.8×10–4) Tet groups as compared to DMSO alone. At 96 hours after Tet treatment, a significant increase in RGC survival rate was still detected in both the 1 nM (36.6%±15.5%, P=0.011) and 10 nM (40.6%±8.5%, P=0.006) Tet groups. There were no differences between 1 nM Tet, 10 nM Tet, and DMSO groups at 120 hours after treatment (P>0.05). These results confirmed that Tet increased the survival rates of primary cultured RGCs, but Tet could not extend the survival times for primary cultured RGCs under the culture conditions tested.
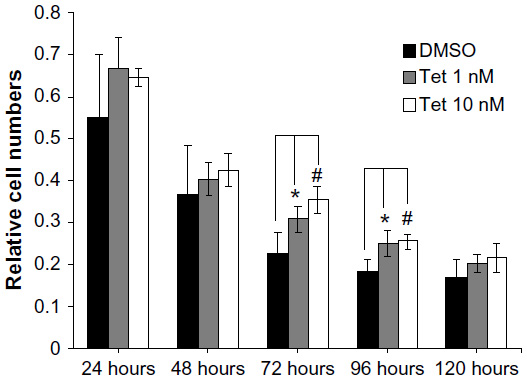 | Figure 5 Time-dependent effect of Tet on the viability of primary cultured RGCs. |
Tet preserves ischemic RGCs in vivo
To evaluate the neuroprotective effects of Tet in vivo, we measured cell numbers in the RGC layers (ganglion cell layers [GCL]) of the retina following transient retinal ischemia/reperfusion (I/R) injury with or without Tet pretreatment. Tet was injected intravitreally (2 μL of 10 nM), while the same volume of 1 × PBS and CNTF (12.5 ng/mL) served as negative and positive controls. Six groups: Tet–I/R (Figure 6A–C), CNTF–I/R (Figure 6D–F), PBS–I/R (Figure 6G–I), Tet-alone (Figure 6J–L), CNTF-alone (Figure 6M–O), and PBS-alone (Figure 6P–R) – were used to examine the numbers of cells in the GCL 24 hours and 72 hours after I/R insult.
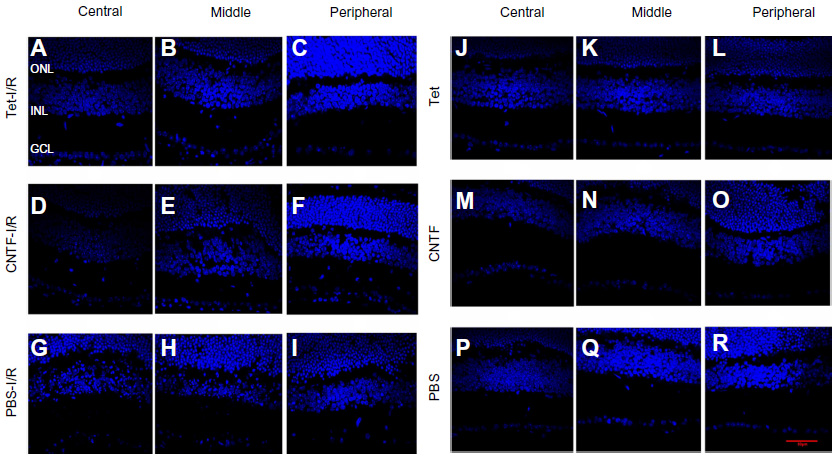 | Figure 6 Tet protects RGCs from ischemic injury in vivo. |
Cell loss was clear in the GCLs 24 hours after I/R stress in the Tet–I/R (Figure 6A–C), CNTF–I/R (Figure 6D–F), and PBS–I/R (Figure 6G–I) groups, whereas no cell loss was evident in the Tet-alone (Figure 6J–L), CNTF-alone (Figure 6M–O), and PBS-alone (Figure 6P–R) groups. No cell loss was seen in the GCL in the Tet-alone (Figure 6J–L) and CNTF-alone (Figure 6M–O) groups in comparison to the PBS–I/R group (Figure 6P–R). This effect was evident across all retinal regions. Cell survival in the central, middle, and peripheral GCL was 61.8%±27.7%, 61.6%±18%, and 44.8%±20.2%, respectively, in Tet–I/R retinas, and 9.6%±7.9%, 13.2%±5.6%, and 16.8%±13.6%, respectively, in PBS–I/R retinas (Figure 7A). Cell survival was significantly better in the Tet–I/R group compared to the PBS–I/R group in the central (P=0.004), middle (P=0.001), and peripheral (P=0.046) GCL. The results were similar in CNTF-I/R retinas.
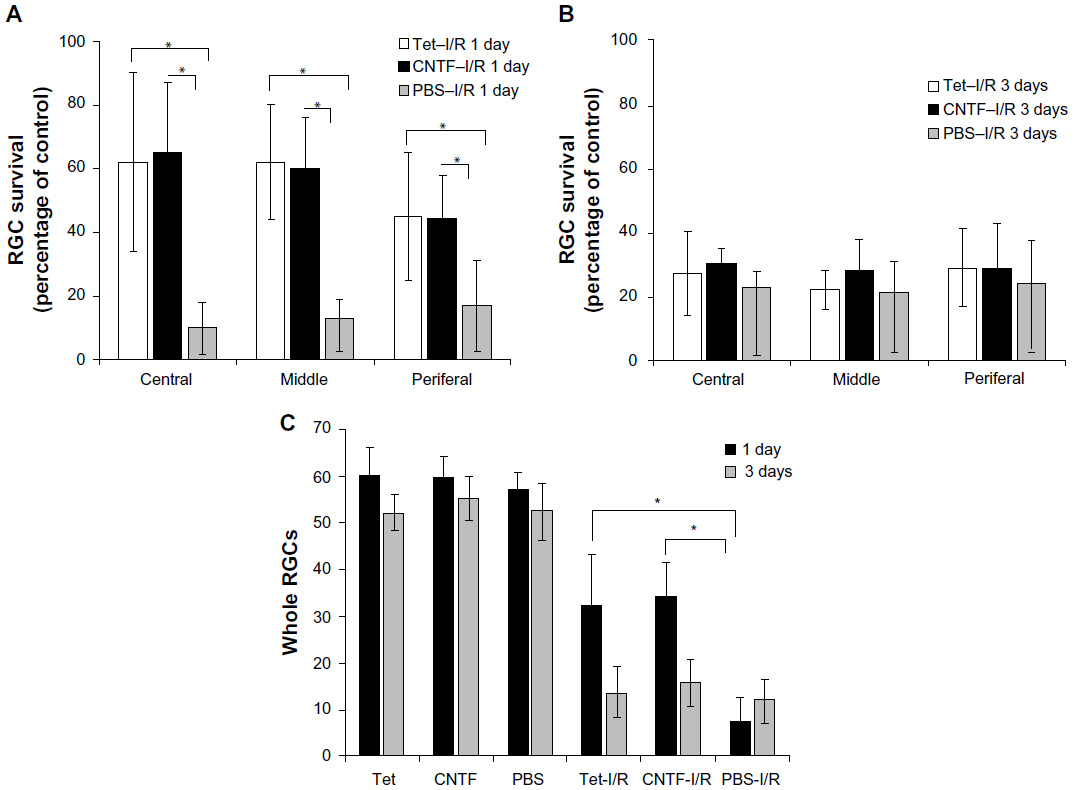 | Figure 7 Statistical analysis of RGC survival following Tet application in vivo. |
We examined the effects of Tet on RGCs 3 days after I/R and observed no significant differences in GCL cell survival in the Tet–I/R and CNTF–I/R groups versus the PBS–I/R group throughout the retinal regions (Figure 7B and C; P>0.05). These results suggest a single dose of Tet protects retinal neurons immediately after I/R injury, but only for a limited time, comparable to the protection provided by CNTF.
Effects of Tet and CNTF on mitochondrial transmembrane potential (ΔΨm)
The staining of primary cultured RGCs with the lipophilic cation JC-10 permits measurement of ΔΨm within intact cells. JC-10 has the unique property of forming J-aggregates locally and spontaneously under high mitochondrial ΔΨm; the aggregates fluoresce red and the monomeric form fluoresces green. Figure 8 shows that about 65% of PBS-, 56% of FCCP-, 86% of Tet-, and 80% of CNTF-treated RGCs (Figure 8A–D) exhibited red and green fluorescence. The percentage of cells with polarized mitochondria in RGCs exposed to PBS, FCCP, Tet, and CNTF for up to 60 minutes were about 22%, 29%, 4%, and 5%, respectively (Figure 8A–D). Figure 8E–H show the survival of RGCs 24 hours after exposure to PBS, FCCP, Tet, and CNTF, respectively, for up to 60 minutes.
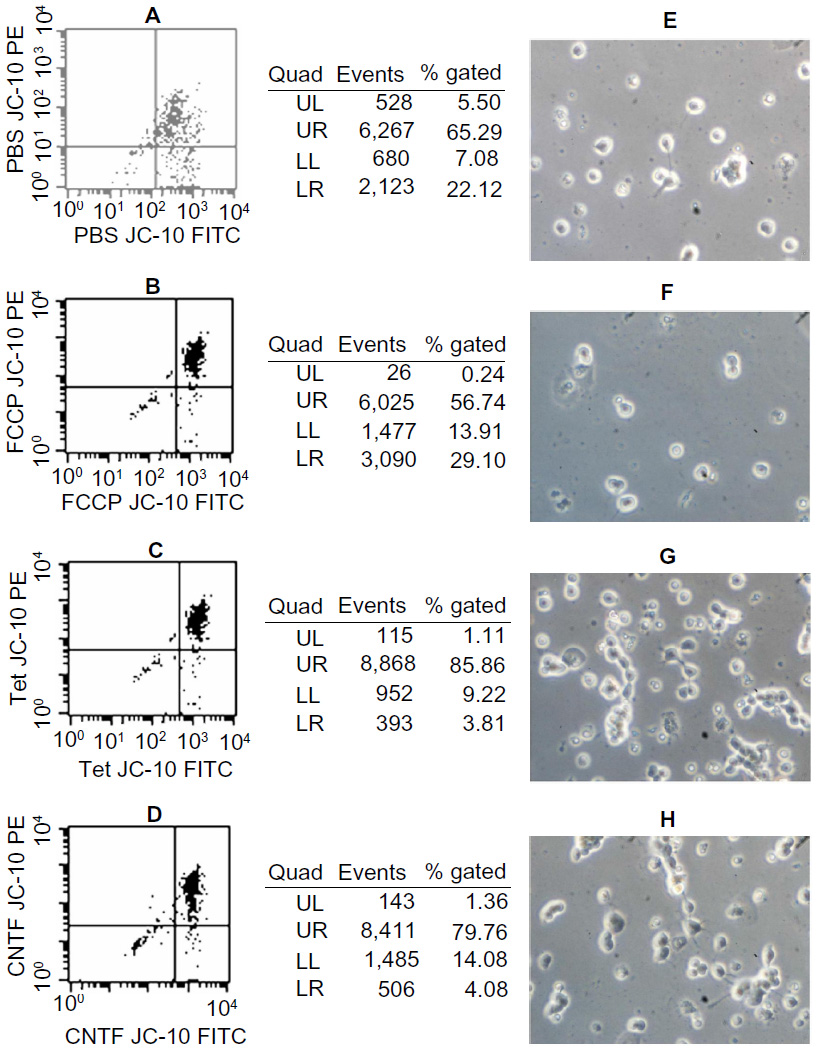 | Figure 8 Effects of Tet and CNTF on primary cultured RGCs ΔΨm. |
Effects of Tet and CNTF on activated caspase-3 and Bcl-2 expression in I/R insult retinas
Bcl-2 and cleaved caspase-3 (Asp175) were detected in the retinas one day after I/R. Expression was lower in the GCL and outer nuclear layer cells of retinas in the Tet–I/R (Figures 9A–C and 10A–C) and CNTF–I/R groups (Figures 9D–F and 10D–F) than in the PBS–I/R groups (Figures 9G–I and 10G–I). They were nearly undetectable in the retinas of PBS-alone (Figures 9J–L and 10J–L), Tet-alone (Figures 9M–O and 10M–O), and CNTF-alone (Figures 9P–R and 10P–R) groups.
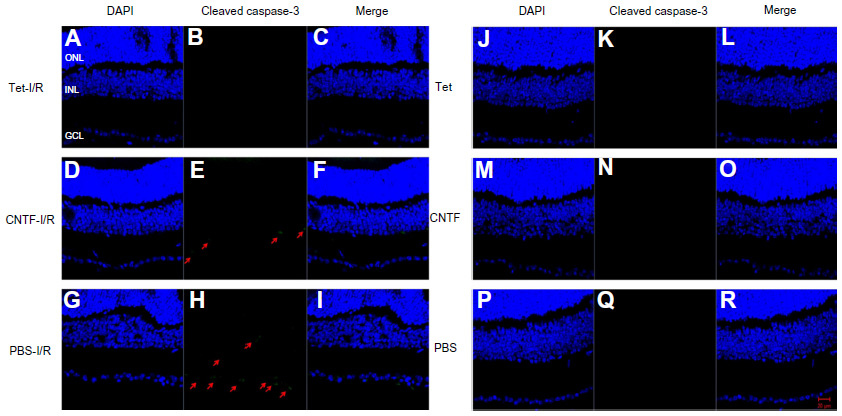 | Figure 9 Tet and CNTF inhibited the expression of cleaved caspase-3 in the retinas 1 day after I/R insult. |
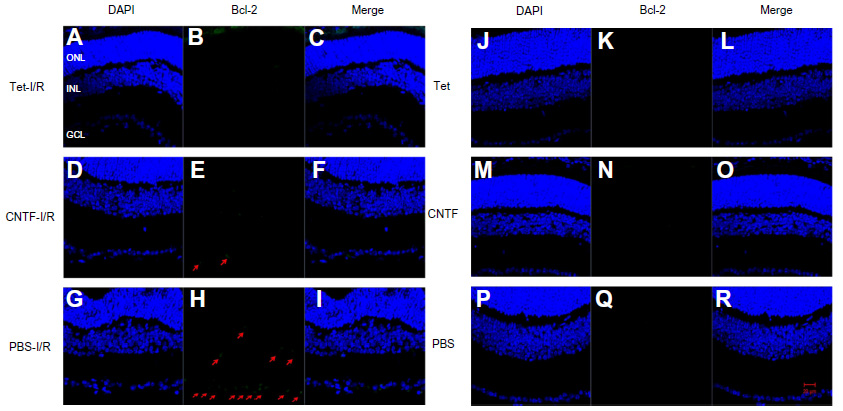 | Figure 10 Tet and CNTF inhibited Bcl-2 expression 1 day after I/R insult. |
Discussion
Our previous experiments suggested that 0.3% Tet exhibits an ocular antihypertensive effect that is comparable to 0.5% timolol in a laser-induced glaucoma model,31 suggesting that Tet may be a potentially useful IOP-lowering agent in the treatment of glaucoma. Herein, we provide evidence that pretreatment with Tet can also promote the survival of cells in a wide variety of conditions. We measured protection of RGC-5 cells in the contexts of serum deprivation (10–500 nM of Tet), glutamate neurotoxicity (10–500 nM of Tet), H2O2 (100 nM of Tet), protection of 1 μM SSP-differentiated RGC-5 cells (5–100 nM of Tet), and purified RGCs (1 nM and 10 nM of Tet) in vitro, as well as RGCs in vivo following retinal ischemic injury (10 μM of Tet). Together, the results showed a direct neuroprotective action of Tet on RGCs, which is consistent with a previous study on neuronal apoptosis in the hippocampus.9
The RGC-5 cell line, derived by transforming postnatal day 1 rat retinal cells with Ψ2EA1 virus,32 was used to evaluate the protective roles of Tet in the context of glaucomatous insults such as ischemic serum deprivation, glutamate, and H2O2, although RGC-5 is not fully characterized as a primary RGC cell line,33 but as a retinal neural cell line.34
A wide range of Tet concentrations (10 nM to 1 μM) promoted both proliferating and SSP-induced neuron-like RGC-5 cell survival following serum deprivation, glutamate excitotoxicity, and H2O2 treatment. In this study, the SSP-differentiated RGC-5 cells developed into highly branched cells that morphologically resembled cultured RGCs, which is consistent with previously reported results.35
H2O2, which is involved in one of the mechanisms of glaucomatous pathogenesis, is a ROS that induces significant toxic oxidative stress in a variety of tissues and causes damage by oxidizing cellular biomolecules, eventually leading to cell death.36 Tet was shown to induce depression of H2O2 production and inhibition of O2 radical generation,37 efficiently react with OH− and scavenge O2– radicals,38–40 and protect from ROS and mitochondrial permeability transition induction,6,41 thus protecting cells from oxidative stress-induced apoptosis. Glutamate excitotoxicity is another important molecular mechanism in RGC degeneration during glaucoma and other retinal diseases with ischemic involvement.42 Tet reportedly prevents glutamate excitatory toxicity in cultured rat cerebellar granule cells,43 and our present study extends this observation to l-glutamate-treated RGC-5 cells. The Tet-induced reduction in intracellular Ca2+ levels, as has been described in cultured neurons of fetal mice,3,44 could limit the toxic Ca2+ overload resulting from prolonged activation of the Ca2+-permeable N-methyl-D-aspartate receptors, which trigger calcium influx, organelle stress, and activation of proapoptotic pathways.42 In addition to protection against ischemic insult, Tet exhibited multiple functions with regard to the promotion of cell survival.41,43
In this study, we also used primary cultured RGCs and a transient retinal ischemia animal model to confirm the neuroprotective function of Tet on RGCs. Tet caused a significant increase in primary RGC survival, in agreement with the effect of CNTF on primary RGC numbers45 and the effect of Tet on neuronal apoptosis in the hippocampus,46 indicating that Tet protects RGCs directly. Furthermore, Tet treatment resulted in a significant increase in RGC survival in vivo, with transient elevation of IOP, 1 day after I/R. The protective effects of Tet could involve multiple pharmacologic actions such as blocking the calcium influx,3,4 inhibiting lipid peroxidation,5 reducing the generation of ROS,6 suppressing the production of cytokines and inflammatory mediators, and reducing neuronal apoptosis.9,47
After induction of high IOP, topical administration of 0.1%–0.3% (1.6–4.8 μM) Tet, twice daily, for 1 week led to a significant decline in IOP in adult Sprague Dawley rats.31 Although this involved a different route of drug application, the concentrations of Tet used in our previous study were equal to or less than the concentration achieved by an intravitreal injection of 2 μL of 10 μM Tet that markedly protected RGCs against ischemic reperfusion.
We also discovered that Tet maintains ΔΨm in primary cultured RGCs and reduces I/R-induced RGC apoptosis by inhibiting expression of cleaved caspase-3 and Bcl-2 one day after I/R. The mechanism of this effect remains unknown.
Although a protective effect of Tet on RGCs was observed 1 day after ischemia, a single injection did not prevent damage to RGCs 3 days after that. The most likely explanation for this is that the metabolism or clearance of Tet caused the concentration to drop below a therapeutic range, although reperfusion might result in greater loss of tissue with secondary injury than that caused by the initial ischemia.17,48 Overall, our results support the suggestion that Tet may be a novel medication that imparts effective neuroprotection.
Acknowledgments
This work was supported by the National Natural Science Foundation of China grant 30371504, 30772376, 81300756, 81041008, and 81170888, China Scholarship Council grant 2011601084, Research Fund for the Doctoral Program of Higher Education of China grant 20100001120100, 20120001110108, and 20130001110099, and NIH grant EY013865.
Disclosure
The authors report no conflicts of interests in this work.
References
Bhakuni DS, Jain S, Singh AN. Biosynthesis of the bisbenzylisoquinoline alkaloid, tetrandrine. Phytochemistry. 1980;19:2347–2350. | |
Ho LJ, Chang DM, Lee TC, Chang ML, Lai JH. Plant alkaloid tetrandrine downregulates protein kinase C-dependent signaling pathway in T cells. Eur J Pharmacol. 1999;367:389–398. | |
Bickmeyer U, Wiegand H. Tetrandrine effects on calcium currents in cultured neurones of foetal mice. Neuroreport. 1993;4:938–940. | |
Jin Q, Kang C, Soh Y, et al. Tetrandrine cytotoxicity and its dual effect on oxidative stress-induced apoptosis through modulating cellular redox states in Neuro 2a mouse neuroblastoma cells. Life Sci. 2002;71:2053–2066. | |
Xu Y, Aluru NR. Carbon nanotube screening effects on the water-ion channels. Appl Phys Lett. 2008;93:43122. | |
Shen DF, Tang QZ, Yan L, et al. Tetrandrine blocks cardiac hypertrophy by disrupting reactive oxygen species-dependent ERK1/2 signalling. Br J Pharmacol. 2010;159:970–981. | |
Shen YC, Chen CF, Sung YJ. Tetrandrine ameliorates ischaemia-reperfusion injury of rat myocardium through inhibition of neutrophil priming and activation. Brit J Pharmacol. 1999;128:1593–1601. | |
Wang ZF, Xue CS, Zhou QX, Wan ZB, Luo QS. Effects of tetrandrine on changes of NMDA receptor channel in cortical neurons of rat induced by anoxia. Zhongguo Yao Li Xue Bao. 1999;20:729–732. | |
He FQ, Qiu BY, Zhang XH, et al. Tetrandrine attenuates spatial memory impairment and hippocampal neuroinflammation via inhibiting NF-kappaB activation in a rat model of Alzheimer’s disease induced by amyloid-beta(1–42). Brain Res. 2011;1384:89–96. | |
Gao S, Cui YL, Yu CQ, Wang QS, Zhang Y. Tetrandrine exerts antidepressant-like effects in animal models: role of brain-derived neurotrophic factor. Behav Brain Res. 2013;238:79–85. | |
Trump BF, Berezesky IK. Calcium-mediated cell injury and cell death. FASEB J. 1995;9:219–228. | |
Nicotera P, Orrenius S. The role of calcium in apoptosis. Cell Calcium. 1998;23:173–180. | |
Sattler R, Tymianski M. Molecular mechanisms of glutamate receptor-mediated excitotoxic neuronal cell death. Mol Neurobiol. 2001;24:107–129. | |
Nicotera P, Leist M. Energy supply and the shape of death in neurons and lymphoid cells. Cell Death Differ. 1997;4:435–442. | |
Chen HS, Pellegrini JW, Aggarwal SK, et al. Open-channel block of N-methyl-D-aspartate (NMDA) responses by memantine: therapeutic advantage against NMDA receptor-mediated neurotoxicity. J Neurosci. 1992;12:4427–4436. | |
Osborne NN, Wood JP, Chidlow G, Casson R, DeSantis L, Schmidt KG. Effectiveness of levobetaxolol and timolol at blunting retinal ischaemia is related to their calcium and sodium blocking activities: relevance to glaucoma. Brain Res Bull. 2004;62:525–528. | |
Osborne NN, Casson RJ, Wood JP, Chidlow G, Graham M, Melena J. Retinal ischemia: mechanisms of damage and potential therapeutic strategies. Prog Retin Eye Res. 2004;23:91–147. | |
Blomgren K, Zhu C, Wang X, et al. Synergistic activation of caspase-3 by m-calpain after neonatal hypoxia-ischemia: a mechanism of “pathological apoptosis”? J Biol Chem. 2001;276:10191–10198. | |
National Research Council (US) Committee for the Update of the Guide for the Care and Use of Laboratory Animals. Guide for the Care and Use of Laboratory Animals. 8th ed. Washington (DC): National Academies Press (US); 2011. | |
Yang H, Lee BK, Kook KH, Jung YS, Ahn J. Protective effect of grape seed extract against oxidative stress-induced cell death in a staurosporine-differentiated retinal ganglion cell line. Curr Eye Res. 2012;37:339–344. | |
Barres BA, Silverstein BE, Corey DP, Chun LL. Immunological, morphological, and electrophysiological variation among retinal ganglion cells purified by panning. Neuron. 1988;1:791–803. | |
Kawasaki A, Otori Y, Barnstable CJ. Muller cell protection of rat retinal ganglion cells from glutamate and nitric oxide neurotoxicity. Invest Ophthalmol Vis Sci. 2000;41:3444–3450. | |
Dun Y, Mysona B, Van Ells T, et al. Expression of the cystine-glutamate exchanger (x(c)(-)) in retinal ganglion cells and regulation by nitric oxide and oxidative stress. Cell Tissue Res. 2006;324:189–202. | |
Zhang XM, Li Liu DT, Chiang SW, et al. Immunopanning purification and long-term culture of human retinal ganglion cells. Mol Vis. 2010;16:2867–2872. | |
Huang P, Zhang C, Wang W, Zhang SM, Barnstable CJ. Purified mouse retinal ganglion cell culture. Zhonghua Yan Ke Za Zhi. 2011;47:129–133. | |
Mathur A, Hong Y, Kemp BK, Barrientos AA, Erusalimsky JD. Evaluation of fluorescent dyes for the detection of mitochondrial membrane potential changes in cultured cardiomyocytes. Cardiovasc Res. 2000;46:126–138. | |
Zhang C, Li H, Liu MG, et al. STAT3 activation protects retinal ganglion cell layer neurons in response to stress. Exp Eye Res. 2008;86:991–997. | |
Li H, Tran VV, Hu Y, Mark Saltzman W, Barnstable CJ, Tombran-Tink J. A PEDF N-terminal peptide protects the retina from ischemic injury when delivered in PLGA nanospheres. Exp Eye Res. 2006;83:824–833. | |
Zhang S, Li W, Wang W, Zhang SS, Huang P, Zhang C. Expression and activation of STAT3 in the astrocytes of optic nerve in a rat model of transient intraocular hypertension. PLoS One. 2013;8:e55683. | |
Doh SH, Kim JH, Lee KM, Park HY, Park CK. Retinal ganglion cell death induced by endoplasmic reticulum stress in a chronic glaucoma model. Brain Res. 2010;1308:158–166. | |
Huang P, Xu Y, Wei R, et al. Efficacy of tetrandrine on lowering intraocular pressure in animal model with ocular hypertension. J Glaucoma. 2011;20:183–188. | |
Krishnamoorthy RR, Agarwal P, Prasanna G, et al. Characterization of a transformed rat retinal ganglion cell line. Brain Res Mol Brain Res. 2001;86:1–12. | |
Wood JP, Chidlow G, Tran T, Crowston JG, Casson RJ. A comparison of differentiation protocols for RGC-5 cells. Invest Ophthalmol Vis Sci. 2010;51:3774–3783. | |
Krishnamoorthy RR, Clark AF, Daudt D, Vishwanatha JK, Yorio T. A forensic path to RGC-5 cell line identification: lessons learned. Invest Ophthalmol Vis Sci. 2013;54:5712–5719. | |
Frassetto LJ, Schlieve CR, Lieven CJ, et al. Kinase-dependent differentiation of a retinal ganglion cell precursor. Invest Ophthalmol Vis Sci. 2006;47:427–438. | |
Gilgun-Sherki Y, Melamed E, Offen D. Oxidative stress induced-neurodegenerative diseases: the need for antioxidants that penetrate the blood brain barrier. Neuropharmacology. 2001;40:959–975. | |
Seow WK, Ferrante A, Li SY, Thong YH. Antiphagocytic and antioxidant properties of plant alkaloid tetrandrine. Int Arch Allergy Appl Immunol. 1988;85:404–409. | |
Shi X, Mao Y, Saffiotti U, et al. Antioxidant activity of tetrandrine and its inhibition of quartz-induced lipid peroxidation. J Toxicol Environ Health. 1995;46:233–248. | |
Cao ZF. Scavenging effect of tetrandrine of active oxygen radicals. Planta Med. 1996;62:413–414. | |
Ye J, Ding M, Zhang X, Rojanasakul Y, Shi X. On the role of hydroxyl radical and the effect of tetrandrine on nuclear factor – kappaB activation by phorbol 12-myristate 13-acetate. Ann Clin Lab Sci. 2000;30:65–71. | |
Fernandes MA, Custodio JB, Santos MS, Moreno AJ, Vicente JA. Tetrandrine concentrations not affecting oxidative phosphorylation protect rat liver mitochondria from oxidative stress. Mitochondrion. 2006;6:176–185. | |
Almasieh M, Wilson AM, Morquette B, Cueva Vargas JL, Di Polo A. The molecular basis of retinal ganglion cell death in glaucoma. Prog Retin Eye Res. 2012;31:152–181. | |
Koh SB, Ban JY, Lee BY, Seong YH. Protective effects of fangchinoline and tetrandrine on hydrogen peroxide-induced oxidative neuronal cell damage in cultured rat cerebellar granule cells. Planta Med. 2003;69:506–512. | |
Wiegand H, Meis S, Gotzsch U. Inhibition by tetrandrine of calcium currents at mouse motor nerve endings. Brain Res. 1990;524:112–118. | |
Lingor P, Tönges L, Pieper N, et al. ROCK inhibition and CNTF interact on intrinsic signalling pathways and differentially regulate survival and regeneration in retinal ganglion cells. Brain. 2008;131:250–263. | |
Chen L, Chen L, Lv Y, et al. Tetrandrine ameliorates cognitive impairment via inhibiting astrocyte-derived S100B activation in a rat model of chronic cerebral hypoperfusion. Neurol Res. 2013;35:614–621. | |
He FQ, Qiu BY, Li TK, et al. Tetrandrine suppresses amyloid-beta-induced inflammatory cytokines by inhibiting NF-kappaB pathway in murine BV2 microglial cells. Int Immunopharmacol. 2011;11:1220–1225. | |
Chen B, Tang L. Protective effects of catalase on retinal ischemia/reperfusion injury in rats. Exp Eye Res. 2011;93:599–606. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.