")
Back to Journals » OncoTargets and Therapy » Volume 7
Targeted therapy for Hodgkin lymphoma and systemic anaplastic large cell lymphoma: focus on brentuximab vedotin
Authors Chen X, Soma L, Fromm J
Received 7 September 2013
Accepted for publication 2 November 2013
Published 19 December 2013 Volume 2014:7 Pages 45—56
DOI https://doi.org/10.2147/OTT.S39107
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Xueyan Chen, Lorinda A Soma, Jonathan R Fromm
Department of Laboratory Medicine, University of Washington Medical Center, Seattle, WA, USA
Abstract: Despite the relative success of chemotherapy for Hodgkin lymphoma (HL) and systemic anaplastic large cell lymphoma (ALCL), novel therapeutic agents are needed for refractory or relapsed patients. Targeted immunotherapy has emerged as a novel treatment option for these patients. Although unconjugated anti-cluster of differentiation (CD)30 antibodies showed minimal antitumor activity in early clinical trials, development of antibody–drug conjugates (ADCs) appears promising. Brentuximab vedotin is an ADC composed of an anti-CD30 antibody linked to a potent microtubule-disrupting agent monomethyl auristatin E (MMAE). It has the ability to target CD30-positive tumor cells and, once bound to CD30, brentuximab vedotin is internalized and MMAE is released to induce cell cycle arrest and apoptosis. In two phase II trials, objective response was reported in 75% and 86% of patients with refractory or relapsed HL and systemic ALCL, respectively, with an acceptable toxicity profile. Based on these studies, the US Food and Drug Administration (FDA) granted accelerated approval of brentuximab vedotin in August 2011 for the treatment of refractory and relapsed HL and ALCL. We review the key characteristics of brentuximab vedotin, clinical data supporting its therapeutic efficacy, and current ongoing trials to explore its utility in other CD30-positive malignancies.
Keywords: classical Hodgkin lymphoma, systemic anaplastic large cell lymphoma, CD30, brentuximab vedotin, SGN-35
Introduction
Antibody-based immunotherapy has become a pivotal component of cancer therapeutics since rituximab (anti-cluster of differentiation [CD]20), the first monoclonal antibody used in cancer treatment, was approved in the late 1990s. The monoclonal antibodies selectively recognize the antigens on the surface of target cells, induce complement-dependent cytotoxicity (CDC) and antibody-dependent cell-mediated cytotoxicity (ADCC), and interrupt the signaling cascade initiated by the target molecules. However, in most cases, the unconjugated monoclonal antibodies show suboptimal single-agent activity and need to be used in combination with other chemotherapy agents.1 Significant efforts have been made to enhance the activity of monoclonal antibodies through various modifications. One strategy is to attach cytotoxic drugs to tumor antigen-specific antibodies, generating antibody–drug conjugates (ADCs). In theory, ADCs selectively bind and deliver cytotoxic drugs to tumor cells with significantly decreased systemic toxicity.
CD30, a cell membrane protein, is strongly expressed on certain neoplasms but not on the majority of normal cells, making it an ideal target for ADC-based therapy. CD30-positive Hodgkin lymphoma (HL) and systemic anaplastic large cell lymphoma (ALCL) are both aggressive lymphomas and potentially curable with front-line multi-agent chemotherapy regimens.2,3 However, patients who have refractory or relapsed disease after initial therapies carry a poor prognosis. Given the expression profile of CD30, antibody-based therapy through CD30 antigen was attempted in the treatment of refractory or relapsed HL and systemic ALCL. Although unconjugated anti-CD30 antibodies showed minimal clinical activity in HL and ALCL despite the encouraging results in preclinical studies,4,5 the data supported continued study, with the eventual development of the anti-CD30 ADC, brentuximab vedotin, which paved new avenues for treatment of CD30-positive tumors.
Classical Hodgkin lymphoma
HL is a relatively common lymphoid neoplasm, with an estimated 9,000 new cases diagnosed annually in the US.6 It has a bimodal distribution of occurrence and is the most common malignancy in adolescents (15–19 years).7 The neoplastic cells in classical HL, termed Hodgkin and Reed-Sternberg (HRS) cells, are derived from germinal center B cells that have acquired unfavorable immunoglobulin variable chain gene mutations that normally would have undergone apoptosis;8 the mechanisms that prevent HRS cell apoptosis are the subject of current research. HRS cells are unique in that they have lost expression of most B-cell-specific genes and acquired expression of other lineage markers. In the majority of HL, HRS cells account for only a small fraction of the tumor mass, but attract many other cells by secreting chemokines and cytokines, such as thymus- and activation-regulated chemokines (TARC) that attract type-2 helper T cells, to form a heterogeneous, inflammatory-rich background. This microenvironment provides essential survival signals and helps HRS cells escape from cytotoxic T-cell or natural killer (NK)-cell-mediated attacks.9
Despite the fact that the first-line multi-agent chemotherapy (eg, doxorubicin, bleomycin, vinblastine, and dacarbazine [ABVD]) and radiotherapy for HL lead to high remission rates of approximately 80%–90%, a portion of patients with advanced-stage HL (20%–30%) are refractory to therapy or relapse after initial treatment.10 The standard management of these refractory or relapsed patients is second-line salvage therapy followed by autologous stem cell transplant (auto-SCT), which can induce durable response in approximately 50% of selected chemotherapy-sensitive patients.11–13 For patients who relapse after auto-SCT, standard of care has not been established; the patient may be offered palliative chemotherapy, reduced-intensity SCT (allo-SCT), or involvement in a clinical trial. Although allo-SCT remains an option to achieve long-term remission via the graft-versus-lymphoma effect, most studies have implicated frequent disease relapse or progression in more than 50% of patients, and the outcome after allo-SCT failure is exceedingly poor.14–17
Pediatric and adolescent patients with HL have a significantly better survival rate than adults (5-year survival rate, 96% versus 88%, respectively; P<0.001),7 and only a small number of patients will relapse (~10% of early-stage and up to 25% of advanced-stage disease).18 In patients who fail the initial therapy, the response rate to salvage therapy is around 70%, and survival rate after auto-SCT is around 50% in this patient group.18 Despite the relatively high overall survival for HL, the survivors are at risk of long-term complications and treatment-related mortality, especially pediatric, adolescent, and young adult patients. Recent trials in pediatric and adolescent HL have investigated chemotherapy regimens of varying dose intensities and reduction in radiotherapy usage based on risk group stratification.19–23 These approaches appeared to maintain the high response rate while reducing the burden of treatment. Despite these successes, in both pediatric and adult patients with HL, novel therapeutic options with improved efficacy are needed for the treatment of refractory or relapsed disease as well as to decrease therapy-related long-term morbidity and mortality.
Anaplastic large cell lymphoma
Systemic ALCL is an aggressive mature T-cell lymphoma that accounts for 10%–15% and 2%–8% of non-Hodgkin lymphomas in children and adults, respectively.24 The neoplastic cells are characteristically large, so-called hallmark cells, with abundant cytoplasm and eccentric, horseshoe- or kidney-shaped nuclei. A significant proportion of systemic ALCL patients harbor the t(2;5)(p23;q35) translocation, leading to fusion of NPM (nucleophosmin) gene with ALK (anaplastic lymphoma kinase) gene and subsequent expression of constitutively active NPM-ALK tyrosine kinase.25 ALK expression in systemic ALCL varies with age, with ALK-positive ALCL more frequently presenting at a young age, while the peak incidence of ALK-negative ALCL is in adults (54–61 years).26 The majority of ALCL in pediatric patients is ALK-positive.27 Systemic ALK-positive and ALK-negative ALCL cannot be separated based on morphological features alone, but are clinically and genetically distinct.28,29 The most recent World Health Organization (WHO) Classification recognizes ALK-positive ALCL as a distinct disease entity and ALK-negative ALCL as a provisional entity based on expression of ALK by immunohistochemistry or cytogenetic/molecular methods.30 The molecular features defining ALK-negative ALCL are not well understood.28 Systemic ALCL is distinguished from primary cutaneous ALCL, which is a separate disease entity that is localized to the skin. Primary cutaneous ALCL is within the spectrum of CD30-positive cutaneous lymphoproliferative disorders and generally follows an indolent course.31
The first-line therapy for adult patients with systemic ALCL is a multi-agent, anthracycline-containing regimen, in most cases CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone). In a retrospective study with long-term (8 years) follow-up, the overall response rates to first-line therapy were 86% and 68% in patients with ALK-positive and ALK-negative ALCL, respectively, and the 8-year overall survival rates were 82% and 49%, respectively.32 Data from the German High-Grade non-Hodgkin Lymphoma Study Group (DSHNHL), which included 78 ALK-positive and 113 ALK-negative ALCL patients treated with CHOP, showed 3-year overall survival rates of 89.8% and 62.1%, respectively.33 Given that ALK-negative ALCL is more common in older patients, the significance of ALK positivity as an independent prognostic factor remains debated. Several published series suggested age, rather than ALK status, is a prominent prognostic factor in ALCL.28,32,34 In patients with refractory or relapsed ALCL, there is no established standard treatment and therapeutic options are limited, as only a few agents have shown consistent activity. In a phase II study evaluating the efficacy and tolerability of pralatrexate, a novel antifolate methotrexate analog, in refractory or relapsed peripheral T-cell lymphoma, only 6 of 17 patients with ALCL responded to pralatrexate (overall response rate of 35%).35 Crizotinib (PF-02341066), an oral ALK tyrosine kinase inhibitor, has been approved for the treatment of advanced ALK-positive non-small cell lung cancer and is now undergoing clinical trials for other ALK-positive diseases, including ALCL. Preliminary results of a phase I crizotinib dose-escalation study in pediatric patients with relapsed or refractory disease reported that seven of eight patients with ALCL achieved complete remission (88%).36 A few small case series also showed successful treatment in patients with relapsed or refractory ALCL.37,38 Other ALK inhibitors are also currently being evaluated in various stages from preclinical to phase I studies.
The standard treatment for pediatric and adolescent ALCL is still under investigation, and varying chemotherapy strategies have been used in different studies with similar success rates.39–41 The European Intergroup for Childhood Non-Hodgkin Lymphoma reported 5-year overall survival and event-free survival of 81% and 69%, respectively (various treatment schedules were used, which included methotrexate, cyclophosphamide, ifosfamide, doxorubicin, and etoposide).41 Despite the varying protocols, ALCL in pediatric and adolescent patients still has a relapse rate of 25%–30%. The therapies for refractory or relapsed disease are highly variable, ranging from single-agent chemotherapy to allo-SCT, with some success, but no established standard.41 Overall, large clinical trials are needed to establish standard treatment in the setting of refractory or relapsed disease as well as to develop new therapeutic strategies in both pediatric and adult patients.
CD30 antigen in lymphoma
The leukocyte activation marker CD30 is a 120 KDa type I transmembrane cytokine receptor from the tumor necrosis factor receptor (TNFR) superfamily. Ligand binding to CD30 activates the downstream intracellular signal through interaction with multiple members of the TNFR-associated factor (TRAF) family, and is capable of promoting cell survival, proliferation, or death via different signaling pathways.42,43 The biologic activities of CD30 are dependent on the downstream signaling pathways in different cell types as well as on the microenvironment in which the signal is provided. In ALCL cell lines, activation of CD30 leads to induction of apoptosis through degradation or intracellular depletion of TRAF2 (which is involved in the nuclear factor [NF]-κB pathway activation and anti-apoptotic signaling), whereas HL cells are insensitive to CD30-induced apoptosis due to a constitutively activated NF-κB pathway.44
As with other TNFRs, CD30 can generate an 85 kDa soluble form (sCD30) that is detectable in the sera of patients with CD30-positive tumors.45 Given that sCD30 level appears to be a reliable marker for tumor burden, studies have suggested that a high serum level of sCD30 is associated with advanced-stage disease and adverse prognosis in both HL and ALCL.46–49
In non-pathologic conditions, CD30 expression is restricted to activated T cells, B cells, and NK cells, and to Epstein–Barr virus-infected cells, but not to resting T cells, B cells, monocytes, or cells outside of the immune system.50,51 Among hematopoietic malignancies, CD30 is consistently expressed in HL and ALCL, and variably expressed in other B- and T cells and myeloid neoplasms, including lymphomatoid papulosis,52 primary mediastinal B-cell lymphoma, diffuse large B-cell lymphoma, primary effusion lymphoma, adult T-cell leukemia/lymphoma,53 mast cell neoplasm,53 and mycosis fungoides (MF).54 The unique expression pattern of CD30 makes this surface antigen an attractive target for selective drug delivery.
First-generation anti-CD30 monoclonal antibodies: MDX-060 and SGN-30
A variety of anti-CD30 monoclonal antibodies have been tested in clinical trials to treat patients with refractory or relapsed CD30-positive lymphomas.55 MDX-060, also known as 5F11, is a humanized immunoglobulin G1 (IgG1) kappa anti-CD30 monoclonal antibody that has showed in vitro and in vivo antitumor activity against HL and ALCL cell lines, presumably mediated by immune mechanisms, including ADCC and CDC.56,57 In addition, a pronounced additive apoptotic effect had been observed in MDX-060 combined with other chemotherapeutic agents. Given the promising preclinical data, MDX-060 was tested in a phase I/II trial in patients with HL (n=63), ALCL (n=7), and CD30-positive T-cell lymphoma (n=2);4 however, only limited activity was observed in patients treated with MDX-060 as a single agent.4 Of the 72 patients, objective responses were observed in six patients; two patients with ALCL and two with HL achieved complete responses and another two HL patients achieved partial responses.
SGN-30, also known as cAC10, is a chimeric anti-CD30 monoclonal antibody constructed with the variable regions of anti-CD30 murine monoclonal AC10 and the constant regions of human gamma 1 heavy chain and kappa light chain.56 It recognizes a specific CD30 epitope that is unique from other anti-CD30 antibodies. Preclinical SGN-30 studies have demonstrated antitumor activity in HL and ALCL cell lines and in xenograft mouse models.58 Furthermore, HL cells treated with SGN-30 were highly sensitized to multiple agents used in the first-line therapies.59 In a phase II study, 79 patients with either refractory/recurrent HL (n=38) or systemic ALCL (n=41) were treated with SGN-30 to determine the safety and objective response.5 In the HL group, no objective responses were observed and 11 patients had stable disease. Of the 41 patients with ALCL, two achieved a complete response and five achieved a partial response.
Given the low response rate in phase I/II trials of first-generation anti-CD30 antibodies, perhaps secondary to poor antigen binding and/or effector activation, a few modified anti-CD30 antibodies, including MDX-1401 and XmAb2513, were constructed with improved FCγRIIIA receptor affinity to stimulate more potent ADCC.59–61 The modified antibodies showed enhanced ADCC when compared with MDX-060 in CD30-positive cell lines and xenograft mouse models. However, in phase I trials, no objective responses were reported in patients with refractory/relapsed HL treated with MDX-1401,62 and one patient (of 17 with HL) achieved partial response with XbAb2513 treatment.63,64 The overall minimal clinical activity of unconjugated anti-CD30 monoclonal antibodies redirected the research efforts from ADCC to directly attacking target cells to enhance antitumor activity, including the development of ADC. This strategy utilizes the antibody as a delivery system and demonstrates antitumor activity independent of ADCC.
Anti-CD30 antibody–drug conjugates: brentuximab vedotin (SGN-35)
Chemistry and mechanism of action
Brentuximab vedotin is an ADC that comprises an anti-human CD30 chimeric IgG1 antibody cAC10 (produced in Chinese hamster ovary cells) and a microtubule-disrupting agent monomethyl auristatin E (MMAE) connected by a protease cleavable valine-citrulline dipeptide linker.65 MMAE is a synthetic analog of dolastatin 10 (originally extracted from the Indian Ocean sea hare, Dolabela auricularia) with high antitumor potency via inhibition of tubulin polymerization. Brentuximab vedotin has a molecular weight of approximately 153 kDa, and is composed of approximately four molecules of MMAE, which are bound to an anti-human CD30 antibody (see Figure 1). After binding to CD30-positive cells, brentuximab vedotin is internalized by receptor-mediated endocytosis into a lysosomal vesicle containing proteolytic enzymes. While stable in the plasma, the dipeptide linker is rapidly hydrolyzed by lysosomal enzymes, and MMAE is subsequently released into the cytoplasm upon peptide cleavage.66,67 This results in MMAE activity, which induces growth arrest selectively in the G2/M phase, followed by apoptosis.65 In addition, MMAE accumulates in the extracellular space due to its inherent membrane permeability and has potential cytotoxic effects on surrounding CD30-negative bystander cells.68 Another potential mechanism of action is interruption of CD30/CD30 ligand interaction. It is speculated that the CD30 ligand (CD153) present on bystander cells recruited by HRS cells can regulate tumor cell proliferation.69,70 By interfering with CD30/CD30 L interaction, brentuximab vedotin may potentially inhibit tumor growth. Furthermore, treatment with brentuximab vedotin led to decreased levels of chemokines and cytokines, particularly TARC, which resolve the inflammatory infiltrate and disrupt the microenvironment, subsequently facilitating antitumor immune response.71
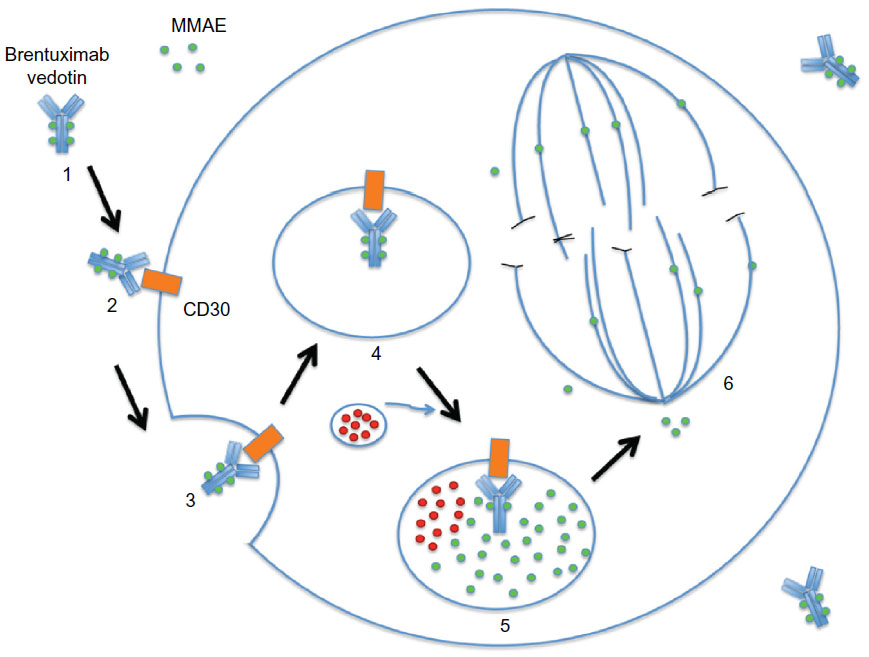 | Figure 1 Brentuximab vedotin mechanism of action. |
Preclinical studies
The preclinical studies convincingly proved that brentuximab vedotin was stable, with only a 2% release of MMAE following 10-day incubation in human plasma.65 In vitro studies using a variety of ALCL (Karpas 299 and SU-DHL-1) and HL (L540cy) cell lines demonstrated that brentuximab vedotin was highly effective and selective against CD30-positive tumor cells with a half maximal inhibitory concentration (IC50) value less than 10 ng/mL, while, in the CD30-negative cells (Raji, Ramos and Daudi; Burkitt lymphoma B-cell lineage), the drug was more than 300-fold less active. Released MMAE in the cytoplasm selectively induced growth arrest in the G2/M phase of the cell cycle, with subsequent apoptotic cell death. In subcutaneous xenograft models of HL and ALCL with Karpas 299 or L540cy cells implanted into flanks of severe combined immunodeficiency (SCID) mice, respectively, doses of brentuximab vedotin up to 30 mg/kg were well tolerated with no significant toxicity.65,72,73 In the ALCL model, 1 mg/kg brentuximab vedotin induced complete remission in all five mice, and in the HL model, partial tumor regression was observed in all mice receiving brentuximab vedotin at 1 mg/kg.65 In a disseminated model of ALCL, with Karpas 299 cells injected into the tail vein of SCID mice, four of five mice showed no apparent signs of disease for more than 130 days.65 In another preclinical study using a xenograft mouse model of HL (L540cy cells), administration of brentuximab vedotin induced a durable response in four of nine mice.74 In addition, the combination of brentuximab vedotin with conventional regimens for HL (ABVD) resulted in durable tumor regression in all nine mice and induced delay in tumor growth. Similarly, combination treatment of brentuximab vedotin with gemcitabine, a pyrimidine antimetabolite that inhibits DNA synthesis, induced durable responses in all five mice, as well as significant delays in tumor growth. The promising results of these preclinical studies indicated the clinical potential of brentuximab vedotin in treating HL and ALCL.
Phase I studies
In an initial phase I, open-label, multicenter, dose-escalation study, brentuximab vedotin was administered at a dose of 0.1–3.6 mg/kg once every 3 weeks to 45 patients with refractory or relapsed CD30-positive hematologic neoplasms including HL (n=42), systemic ALCL (n=2), and angioimmunoblastic T-cell lymphoma (n=1).75 Patients had received a median of three previous chemotherapy regimens (range 1–7), and 33 patients (73%) had undergone auto-SCT. A dose of 2.7 mg/kg had caused unacceptable toxic effects, and 1.8 mg/kg was considered the maximum tolerated dose (MTD) with acceptable toxic effects, which primarily included grade 1 or 2 (mild to moderate) toxic effects, such as fatigue (36%), pyrexia (33%), diarrhea (22%), nausea (22%), neutropenia (22%), and peripheral neuropathy (22%). Objective responses were observed in 17 patients (38%), including eleven (24%) with complete remission. The two patients with ALCL achieved complete remission. For patients who received the MTD (1.8 mg/kg), the objective response rate was 50% (six of 12 patients). Remissions were durable in these patients who had refractory or relapsed disease, with a median duration of at least 9.7 months. In addition, tumor regression was observed on computed tomography (CT) in 36 of 42 patients who could be evaluated.
In the second phase I dose-escalation study, brentuximab vedotin was administered at a dose of 0.4–1.4 mg/kg on a weekly schedule for 3 weeks of a 4-week cycle to 44 patients (38 with HL, five with systemic ALCL, and one with peripheral T-cell lymphoma).76 The hypothesis was that the weekly dosing could enhance antitumor activity without compromising its safety, with patients able to receive up to 12 cycles if they were obtaining clinical benefit with acceptable safety. Patient characteristics were similar to those enrolled in the prior phase I study; patients had received a median of three prior chemotherapy regimens, and 30 patients (68%) had undergone auto-SCT. During the treatment, the nature of the adverse events was similar to those seen in the prior study, but the incidence was higher. The most common adverse events were peripheral sensory neuropathy (66%), fatigue (52%), nausea (50%), diarrhea (32%), arthralgia (27%), and pyrexia (25%); the majority of events were grade I–II in severity. Overall, 32 (73%) patients experienced neuropathy, with grade III events in six (14%) patients; eight patients discontinued treatment because of peripheral neuropathy. Of the 41 evaluable patients, the overall response rate was 59% (n=24), with 34% (n=14) achieving complete remission. For patients treated at the MTD (1.2 mg/kg), the overall objective response rate was 58% (seven of 12 patients); three (25%) achieved complete remission. The objective response rate was 54% for HL patients and 80% for ALCL patients. The best clinical response in patients with peripheral T-cell lymphoma was partial remission. Taken together, the antitumor activity of brentuximab vedotin administered on a weekly schedule was similar to that observed with dosing once every 3 weeks utilized in the first phase I dose-escalation study;75 however, due to the significant peripheral neuropathy that occurred with weekly dosing, the alternative schedule of once every 3 weeks was adopted for phase II studies.
Phase II studies/pivotal clinical trials
Given the encouraging results from the phase I studies, two single-arm, single-agent, multicenter phase II studies were conducted. Brentuximab vedotin was administered at 1.8 mg/kg once every 3 weeks for up to 16 cycles in patients with refractory or relapsed HL and systemic ALCL.77,78 In both studies, the primary endpoint was overall objective response rate, defined as the sum of complete remission and partial remission and determined by the independent central review facility using the revised response criteria for malignant lymphomas.79
The phase II study in HL enrolled 102 patients who had relapsed disease after auto-SCT.78 The median number of prior chemotherapy regimens excluding auto-SCT was 3.5 (range 1–13). Patients continued treatment until either disease progression or unacceptable toxicity occurred, up to a maximum of 16 cycles. Tumor reductions were observed in 94% (n=96) of 102 patients. The overall objective response rate was 75% (n=76), with complete remission in 34% (n=35) and partial remission in 40% (n=41) of patients. The overall disease control rate (complete remission + partial remission + stable disease) was 96%. The median duration of objective response was 6.7 months, and of complete remission was 20.5 months. The most frequent (≥10%) treatment-related adverse events were peripheral sensory neuropathy (42%), nausea (35%), fatigue (34%), neutropenia (19%), diarrhea (18%), pyrexia (14%), vomiting (13%), arthralgia (12%), pruritus (12%), myalgia (11%), peripheral motor neuropathy (11%), and alopecia (10%). A significant number of patients (55%; n=56) experienced adverse events of grade 3 or higher, including non-febrile neutropenia (20%), peripheral sensory neuropathy (8%), thrombocytopenia (8%), and anemia (6%). A total of 20 patients discontinued treatment, and 47% of patients delayed dosing due to adverse events. There were no deaths within 30 days from the last brentuximab vedotin administration, and no drug-related deaths occurred. Overall, brentuximab vedotin showed remarkable therapeutic efficacy in heavily pretreated HL patients, with tolerable toxicities.
The phase II study in ALCL enrolled a total of 58 patients with relapsed or refractory disease, including 28% (n=16) ALK-positive and 72% (n=42) ALK-negative patients, who received 1.8 mg/kg of brentuximab vedotin intravenously once every 3 weeks.77 The median number of prior chemotherapy regimens excluding auto-SCT was two (range 1–6), and 26% (n=15) of patients had failed an auto-SCT before study enrollment. Tumor reductions were observed in 97% (n=57) of patients. The overall objective response rate was 86% (n=50), with 57% (n=33) of patients achieving complete remission and 29% (n=17) achieving partial remission. The median duration of objective responses was 12.6 months, and of complete remission was 13.2 months. Among patients with ALK-negative disease, the overall response rate was 88% (n=37), and complete remission rate was 52% (n=22). The response rate is similar to that among patients with ALK-positive disease, who had an overall objective response rate of 81% (n=13) and a complete remission rate of 69% (n=11). The duration of response did not differ between these two groups. The most frequent treatment-emergent adverse events were peripheral sensory neuropathy (41%), nausea (40%), fatigue (38%), pyrexia (34%), diarrhea (29%), rash (24%), constipation (22%), and neutropenia (21%). Adverse events of grade 3 or higher occurred in 60% (n=35) of the patients, including non-febrile neutropenia (21%), thrombocytopenia (14%), peripheral sensory neuropathy (12%), and anemia (7%). Adverse events led to treatment discontinuation in 24% (n=14) and delayed dosing in 40% (n=23) of the patients. Six deaths occurred within 30 days of the last brentuximab vedotin administration; none of these deaths were drug related. In summary, brentuximab vedotin demonstrated significant antitumor activity in both ALK-positive and ALK-negative ALCL, with a high overall response rate and manageable toxicities.
Therapeutic indications
Based on these two non-comparative, multicenter phase II trials, brentuximab vedotin received accelerated approval from the US FDA in August 2011 for the treatment of patients with HL for whom either auto-SCT or at least two prior multi-agent chemotherapy regimens have failed and who are not candidates for auto-SCT. Brentuximab vedotin is also approved for patients with systemic ALCL after failure of at least one prior multi-agent chemotherapy regimen.80 Given the rationale that patients with HL who are refractory to second-line chemotherapy should not proceed to auto-SCT, National Comprehensive Cancer Network (NCCN) guidelines (version 2.2013) included brentuximab vedotin as an option for HL patients who have failed auto-SCT or at least two prior chemotherapy regimens, regardless of their eligibility for auto-SCT.
Experience with brentuximab vedotin in different clinical settings
Use in HL and ALCL outside of studies
To investigate the clinical activity of brentuximab vedotin outside of studies, the German Hodgkin Study Group (GHSG) reported their experience with 45 patients with HL treated in Germany, Switzerland, and Austria.81 In these heavily pretreated patients, the overall objective response rate was 60%, with 22% achieving complete remission, supporting the therapeutic potency reported previously. The toxicity profile was similar to the previously published data, and no patients had to discontinue treatment because of adverse events. A single UK center evaluated 24 patients with HL (n=18), ALCL (n=5), or CD30-positive T-cell lymphoma (n=1) refractory to at least two lines of chemotherapy or auto-SCT (33%; n=8), who were treated with brentuximab vedotin.82 The overall objective response rate was 67% (72% in HL and 60% in ALCL), and complete remission rate was 25% (17% in HL and 60% in ALCL). The median progression-free survival was 5.1 months. In the majority of cases, the adverse events were mild to moderate. Following treatment, six (27%) patients proceeded to allo-SCT. In these non-trial settings, the overall results were similar to the published phase II studies.
Use as a bridge to allo-SCT
To study the impact of brentuximab vedotin on future reduced-intensity allo-SCT, a retrospective study examined 18 patients with refractory or relapsed HL who were treated with brentuximab vedotin.83 A total of 17 patients had previous auto-SCT; before the allo-SCT, six patients were in complete remission and eight were in partial remission. A total of eleven patients required unrelated or mismatched donors. All patients achieved engraftment, with 1-year overall survival of 100%, progression-free survival of 92.3%, and non-relapse mortality of 0%. The study demonstrated that brentuximab vedotin prior to allo-SCT successfully provided adequate disease control to improve post-transplantation disease-free survival.
Use in relapsed HL following allo-SCT
There is no effective therapy and outcomes are poor for patients with relapsed HL following allo-SCT. To explore the potential utility of brentuximab vedotin in this population, a cohort of 25 HL patients with recurrent disease after allo-SCT were treated with 1.2 or 1.8 mg/kg brentuximab vedotin once every 3 weeks (median 8 cycles, range 1–16 cycles).84 Among 24 evaluable patients, the overall objective response rate was 50%, with complete response rate of 38%. The median progression-free survival was 7.8 months. The adverse events were manageable, but more patients had to discontinue treatment (n=9; 36%) than reported from the Phase II study, which excluded patients with prior allo-SCT.78 This study suggested a potential benefit of brentuximab vedotin in selected patients with recurrent HL after allo-SCT.
Use as a salvage treatment prior to SCT
There are only limited data on the effect of brentuximab vedotin in transplant-naïve, refractory, or relapsed patients with HL. A retrospective study analyzed 14 patients with refractory or relapsed HL who did not receive high-dose chemotherapy and auto-SCT prior to brentuximab vedotin treatment as a single agent.85 The overall objective response rate was 71%, with a complete remission rate of 36%. After treatment, five patients proceeded to high-dose chemotherapy followed by auto-SCT or allo-SCT. The median progression-free survival for all patients was 9 months. Of the 14 patients, ten were alive at the time of analysis, and the 12-month estimate for overall survival was 69%. The toxicity was tolerable, and none of the patients had to discontinue therapy. This study demonstrated the therapeutic efficacy of brentuximab vedotin, which can be used as salvage therapy prior to transplantation in patients with refractory or relapsed HL who might otherwise not qualify for transplant intervention.
Use in CD30-positive solid tumors
In addition to HL and ALCL, CD30 positivity has been reported on other malignant tumors, such as germ cell tumors. A phase II, open-label, multicenter study to evaluate the efficacy of brentuximab vedotin in CD30-positive non-lymphomatous malignancies is ongoing.86 Three patients with refractory or relapsed CD30-positive testicular cancer who received brentuximab vedotin at 1.8 mg/kg once every 21 days as part of the phase II study were reported.87 The treatment was well tolerated, with no grade 3–4 toxicities. After two cycles, the first patient had serologic response with radiologic partial response that lasted 2 months, followed by serologic and radiographic progression in the liver while on therapy. The second patient was serologic marker negative and had a radiographic partial response in the lung and mediastinum after cycle two and continued on therapy. The third patient had serologic response after cycle two and continued to have serologic response. The initial experience suggested the potential benefit of brentuximab vedotin in other CD30-positive tumors.
Ongoing studies of brentuximab vedotin
Several randomized phase III trials are ongoing to evaluate the therapeutic efficacy of brentuximab vedotin in various clinical settings. A randomized, double-blind, placebo-controlled, multicenter phase III study in patients at high risk of residual HL after auto-SCT is currently ongoing, and is expected to be completed in 2016.88 To evaluate the clinical benefit of combination of brentuximab and multi-agent chemotherapy regimens in patients with newly diagnosed CD30-positive lymphomas, randomized phase III trials are also being performed with inclusion of brentuximab vedotin in one of the first-line therapy arms. In advanced stage HL,89 comparison of ABVD alone versus brentuximab vedotin in combination with doxorubicin, vinblastine, and dacarbazine (AVD) as first-line therapy is currently underway. In newly diagnosed CD30-positive mature T- and NK-cell lymphomas,90 comparison of CHOP alone versus brentuximab vedotin in combination with cyclophosphamide, doxorubicin, and prednisone (CHP) as first-line therapy is also being evaluated. The data from these studies will provide the basis for full US FDA approval.
Clinical trials in pediatric and younger adult patients with relapsed or refractory HL and ALCL are underway. A phase I/II trial is currently evaluating the combination of brentuximab vedotin and gemcitabine hydrochloride in younger patients (13 months to 30 years) with relapsed or refractory HL.91 A pilot phase II trial was just opened to study the efficacy of brentuximab vedotin, combination chemotherapy, and radiation therapy in treating younger patients (up to 18 years) with stage IIB, IIIB, and IV HL.92 A phase I/II, open-label, single-agent, multicenter, dose-escalation study is ongoing to investigate brentuximab vedotin in treating pediatric patients (2–18 years) with refractory or relapsed systemic ALCL.93 Given the relative high frequency of HL and systemic ALCL in the pediatric population, data from these trials may provide additional evidence for developing treatment strategies to improve the long-term outcome.
As other malignancies besides HL and ALCL may express CD30, phase II studies are currently evaluating the potential of brentuximab vedotin in other CD30-positive neoplasms. Brentuximab vedotin is being investigated in phase II studies for patients with CD30-positive lymphomas other than HL and ALCL,94 with cutaneous T-cell lymphomas, including cutaneous ALCL, MF, and extensive lymphomatoid papulosis,95 with CD30-positive non-lymphomatous malignancies (including acute lymphoid leukemia, acute myeloid leukemia, chronic lymphocytic leukemia, multiple myeloma, and solid tumors),86 with diffuse large B-cell lymphoma in combination with rituximab-CHOP (R-CHOP),96 and with CD30-positive germ cell tumors.97 Pilot studies evaluating brentuximab vedotin are underway in patients with MF with variable CD30 expression level,98 with advanced systemic mastocytosis or mast cell leukemia,99 and with mismatched unrelated allo-SCT to prevent graft-versus-host disease.100 Finally, a multicenter, open-label, Phase I study is also ongoing to evaluate the safety profile (including MTD, immunogenicity, and antitumor activity) of brentuximab vedotin given sequentially and combined with a multi-agent chemotherapy regimen as front-line therapy for newly diagnosed CD30-positive mature T- and NK-cell lymphomas, including systemic ALCL.101
Conclusion and future perspectives
Brentuximab vedotin combines several key mechanisms, including interference with the NF-κB signaling pathway,44 cytotoxic effect of MMAE in tumor cells and bystander cells, disruption of microenvironment through chemokine modulation, and sensitizing tumor cells to other chemotherapy agents, which contribute to its potent antitumor activity.65,68 Despite limited data, the observed antitumor activity in phase II studies, as demonstrated by high overall response rate and duration of responses in refractory or relapsed HL and systemic ALCL, is considered clinically relevant, prompting the accelerated approval by the US FDA (Table 1). However, given the single-arm design of the two phase II trials and the limited number of patients, reliable interpretation of the treatment effect on time-to-event endpoints (such as progression-free survival and overall survival) cannot be rendered and the safety profile cannot be fully characterized.80 Conversion to regular approval will require completion of clinical trials to verify the safety and efficacy of brentuximab vedotin in comparison with standard therapy, including randomized controlled trials to compare ABVD alone versus brentuximab vedotin in combination with AVD as first-line therapy for advanced HL, and CHOP alone versus brentuximab vedotin in combination with CHP as first-line therapy for newly diagnosed CD30-positive mature T- and NK-cell lymphomas.
 | Table 1 Clinical studies with anti-CD30 monoclonal antibodies |
Given the relatively manageable toxicity profile and substantial treatment effects in refractory or relapsed HL and systemic ALCL demonstrated by both phase II studies, clinical trials to explore the therapeutic potential of brentuximab vedotin in other CD30-positive neoplasms are currently underway. The success of brentuximab vedotin is a milestone for innovative ADCs and encourages the application of ADC technology in other antigen targets and cancer treatments. With the emerging clinical data from ongoing clinical trials using ADC strategy, it continues to enhance the field of immune-based cancer treatment.
Disclosure
Dr Jonathan Fromm received research funding from Seattle Genetics. The authors report no other conflicts of interest in this work.
References
Senter PD. Potent antibody drug conjugates for cancer therapy. Curr Opin Chem Biol. 2009;13(3):235–244. | |
Evens AM, Hutchings M, Diehl V. Treatment of Hodgkin lymphoma: the past, present, and future. Nat Clin Pract Oncol. 2008;5(9):543–556. | |
Vose J, Armitage J, Weisenburger D; International T-Cell Lymphoma Project. International peripheral T-cell and natural killer/T-cell lymphoma study: pathology findings and clinical outcomes. J Clin Oncol. 2008;26(25):4124–4130. | |
Ansell SM, Horwitz SM, Engert A, et al. Phase I/II study of an anti-CD30 monoclonal antibody (MDX-060) in Hodgkin’s lymphoma and anaplastic large-cell lymphoma. J Clin Oncol. 2007;25(19):2764–2769. | |
Forero-Torres A, Leonard JP, Younes A, et al. A phase II study of SGN-30 (anti-CD30 mAb) in Hodgkin lymphoma or systemic anaplastic large cell lymphoma. Br J Haematol. 2009;146(2):171–179. | |
Siegel R, DeSantis C, Virgo K, et al. Cancer treatment and survivorship statistics, 2012. CA Cancer J Clin. 2012;62(4):220–241. | |
Kelly KM, Hodgson D, Appel B, et al. Children’s Oncology Group’s 2013 blueprint for research: Hodgkin lymphoma. Pediatr Blood Cancer. 2013;60(6):972–978. | |
Kanzler H, Küppers R, Hansmann ML, Rajewsky K. Hodgkin and Reed-Sternberg cells in Hodgkin’s disease represent the outgrowth of a dominant tumor clone derived from (crippled) germinal center B cells. J Exp Med. 1996;184(4):1495–1505. | |
Küppers R. The biology of Hodgkin’s lymphoma. Nat Rev Cancer. 2009;9(1):15–27. | |
Eichenauer DA, Engert A, Dreyling M; ESMO Guidelines Working Group. Hodgkin’s lymphoma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2011;22 Suppl 6:vi55–vi58. | |
Linch DC, Winfield D, Goldstone AH, et al. Dose intensification with autologous bone-marrow transplantation in relapsed and resistant Hodgkin’s disease: results of a BNLI randomised trial. Lancet. 1993;341(8852):1051–1054. | |
Majhail NS, Weisdorf DJ, Defor TE, et al. Long-term results of autologous stem cell transplantation for primary refractory or relapsed Hodgkin’s lymphoma. Biol Blood Marrow Transplant. 2006;12(10):1065–1072. | |
Schmitz N, Pfistner B, Sextro M, et al. Aggressive conventional chemotherapy compared with high-dose chemotherapy with autologous haemopoietic stem-cell transplantation for relapsed chemosensitive Hodgkin’s disease: a randomised trial. Lancet. 2002;359(9323):2065–2071. | |
Burroughs LM, O’Donnell PV, Sandmaier BM, et al. Comparison of outcomes of HLA-matched related, unrelated, or HLA-haploidentical related hematopoietic cell transplantation following nonmyeloablative conditioning for relapsed or refractory Hodgkin lymphoma. Biol Blood Marrow Transplant. 2008;14(11):1279–1287. | |
Corradini P, Dodero A, Farina L, et al. Allogeneic stem cell transplantation following reduced-intensity conditioning can induce durable clinical and molecular remissions in relapsed lymphomas: pre-transplant disease status and histotype heavily influence outcome. Leukemia. 2007;21(11):2316–2323. | |
Ram R, Gooley TA, Maloney DG, et al. Histology and time to progression predict survival for lymphoma recurring after reduced-intensity conditioning and allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2011;17(10):1537–1545. | |
Sarina B, Castagna L, Farina L, et al. Allogeneic transplantation improves the overall and progression-free survival of Hodgkin lymphoma patients relapsing after autologous transplantation: a retrospective study based on the time of HLA typing and donor availability. Blood. 2010;115(18):3671–3677. | |
Daw S, Wynn R, Wallace H. Management of relapsed and refractory classical Hodgkin lymphoma in children and adolescents. Br J Haematol. 2011;152(3):249–260. | |
Friedman DI, Wolden S, Constine DL, et al. AHOD0031: a phase III study of dose-intensive therapy for intermediate risk Hodgkin lymphoma: a report from the children’s oncology group. Blood (ASH Annu Meeting Abstr). 2010;116:766. | |
Keller FG, Nachman J, Constine LS, et al. A phase III study for the treatment of children and adolescents with newly diagnosed low risk Hodgkin lymphoma (HL). Blood (ASH Annu Meeting Abstr). 2010;116:767. | |
Kelly KM, Sposto R, Hutchinson R, et al. BEACOPP chemotherapy is a highly effective regimen in children and adolescents with high-risk Hodgkin lymphoma: a report from the Children’s Oncology Group. Blood. 2011;117(9):2596–2603. | |
Schwartz CL, Constine LS, Villaluna D, et al. A risk-adapted, response-based approach using ABVE-PC for children and adolescents with intermediate- and high-risk Hodgkin lymphoma: the results of P9425. Blood. 2009;114(10):2051–2059. | |
Wolden SL, Chen L, Kelly KM, et al. Long-term results of CCG 5942: a randomized comparison of chemotherapy with and without radiotherapy for children with Hodgkin’s lymphoma – a report from the Children’s Oncology Group. J Clin Oncol. 2012;30(26):3174–3180. | |
Merkel O, Hamacher F, Sifft E, Kenner L, Greil R; European Research Initiative on Anaplastic Large Cell Lymphoma. Novel therapeutic options in anaplastic large cell lymphoma: molecular targets and immunological tools. Mol Cancer Ther. 2011;10(7):1127–1136. | |
Webb TR, Slavish J, George RE, et al. Anaplastic lymphoma kinase: role in cancer pathogenesis and small-molecule inhibitor development for therapy. Expert Rev Anticancer Ther. 2009;9(3):331–356. | |
Ferreri AJ, Govi S, Pileri SA, Savage KJ. Anaplastic large cell lymphoma, ALK-negative. Crit Rev Oncol Hematol. 2013;85(2):206–215. | |
Lowe EJ, Gross TG. Anaplastic large cell lymphoma in children and adolescents. Pediatr Hematol Oncol. 2013;30(6):509–519. | |
Lamant L, de Reyniès A, Duplantier MM, et al. Gene-expression profiling of systemic anaplastic large-cell lymphoma reveals differences based on ALK status and two distinct morphologic ALK+ subtypes. Blood. 2007;109(5):2156–2164. | |
Savage KJ, Harris NL, Vose JM, et al; International Peripheral T-Cell Lymphoma Project. ALK-anaplastic large-cell lymphoma is clinically and immunophenotypically different from both ALK+ ALCL and peripheral T-cell lymphoma, not otherwise specified: report from the International Peripheral T-Cell Lymphoma Project. Blood. 2008;111(12):5496–5504. | |
Swerdlow SH, Campo E, Harris NL, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, Fourth Edition. Lyon: IARC Press; 2008. | |
Foss FM, Zinzani PL, Vose JM, Gascoyne RD, Rosen ST, Tobinai K. Peripheral T-cell lymphoma. Blood. 2011;117(25):6756–6767. | |
Sibon D, Fournier M, Brière J, et al. Long-term outcome of adults with systemic anaplastic large-cell lymphoma treated within the Groupe d’Etude des Lymphomes de l’Adulte trials. J Clin Oncol. 2012;30(32):3939–3946. | |
Schmitz N, Trümper L, Ziepert M, et al. Treatment and prognosis of mature T-cell and NK-cell lymphoma: an analysis of patients with T-cell lymphoma treated in studies of the German High-Grade Non-Hodgkin Lymphoma Study Group. Blood. 2010;116(18):3418–3425. | |
Suzuki R, Kagami Y, Takeuchi K, et al. Prognostic significance of CD56 expression for ALK-positive and ALK-negative anaplastic large-cell lymphoma of T/null cell phenotype. Blood. 2000;96(9):2993–3000. | |
O’Connor OA, Pro B, Pinter-Brown L, et al. Pralatrexate in patients with relapsed or refractory peripheral T-cell lymphoma: results from the pivotal PROPEL study. J Clin Oncol. 2011;29(9):1182–1189. | |
Mosse YP, Balis FM, Lim MS, et al. Efficacy of crizotinib in children with relapsed/refractory ALK-driven tumors including anaplastic large cell lymphoma and neuroblastoma: a Children’s Oncology Group phase I consortium study. J Clin Oncol (ASCO Annu Meeting Abstr). 2012;30:9500. | |
Gambacorti-Passerini C, Messa C, Pogliani EM. Crizotinib in anaplastic large-cell lymphoma. N Engl J Med. 2011;364(8):775–776. | |
Pogliani EM, Dilda I, Villa F, et al. High response rate to crizotinib in advanced, chemoresistant ALK+ lymphoma patients. J Clin Oncol (ASCO Annu Meeting Abstr). 2011;29:e18507. | |
Brugières L, Le Deley MC, Rosolen A, et al. Impact of the methotrexate administration dose on the need for intrathecal treatment in children and adolescents with anaplastic large-cell lymphoma: results of a randomized trial of the EICNHL Group. J Clin Oncol. 2009;27(6):897–903. | |
Lamant L, McCarthy K, d’Amore E, et al. Prognostic impact of morphologic and phenotypic features of childhood ALK-positive anaplastic large-cell lymphoma: results of the ALCL99 study. J Clin Oncol. 2011;29(35):4669–4676. | |
Le Deley MC, Reiter A, Williams D, et al; European Intergroup for Childhood Non-Hodgkin Lymphoma. Prognostic factors in childhood anaplastic large cell lymphoma: results of a large European intergroup study. Blood. 2008;111(3):1560–1566. | |
Duckett CS, Gedrich RW, Gilfillan MC, Thompson CB. Induction of nuclear factor kappaB by the CD30 receptor is mediated by TRAF1 and TRAF2. Mol Cell Biol. 1997;17(3):1535–1542. | |
Lee SY, Park CG, Choi Y. T cell receptor-dependent cell death of T cell hybridomas mediated by the CD30 cytoplasmic domain in association with tumor necrosis factor receptor-associated factors. J Exp Med. 1996;183(2):669–674. | |
Mir SS, Richter BW, Duckett CS. Differential effects of CD30 activation in anaplastic large cell lymphoma and Hodgkin disease cells. Blood. 2000;96(13):4307–4312. | |
Younes A, Aggarwall BB. Clinical implications of the tumor necrosis factor family in benign and malignant hematologic disorders. Cancer. 2003;98(3):458–467. | |
Nadali G, Tavecchia L, Zanolin E, et al. Serum level of the soluble form of the CD30 molecule identifies patients with Hodgkin’s disease at high risk of unfavorable outcome. Blood. 1998;91(8):3011–3016. | |
Visco C, Nadali G, Vassilakopoulos TP, et al. Very high levels of soluble CD30 recognize the patients with classical Hodgkin’s lymphoma retaining a very poor prognosis. Eur J Haematol. 2006;77(5):387–394. | |
Zanotti R, Trolese A, Ambrosetti A, et al. Serum levels of soluble CD30 improve International Prognostic Score in predicting the outcome of advanced Hodgkin’s lymphoma. Ann Oncol. 2002;13(12):1908–1914. | |
Zinzani PL, Pileri S, Bendandi M, et al. Clinical implications of serum levels of soluble CD30 in 70 adult anaplastic large-cell lymphoma patients. J Clin Oncol. 1998;16(4):1532–1537. | |
Horie R, Watanabe T. CD30: expression and function in health and disease. Semin Immunol. 1998;10(6):457–470. | |
Tarkowski M. Expression and a role of CD30 in regulation of T-cell activity. Curr Opin Hematol. 2003;10(4):267–271. | |
Kempf W, Pfaltz K, Vermeer MH, et al. EORTC, ISCL, and USCLC consensus recommendations for the treatment of primary cutaneous CD30-positive lymphoproliferative disorders: lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma. Blood. 2011;118(15):4024–4035. | |
Younes A, Carbone A. CD30/CD30 ligand and CD40/CD40 ligand in malignant lymphoid disorders. Int J Biol Markers. 1999;14(3):135–143. | |
Edinger JT, Clark BZ, Pucevich BE, Geskin LJ, Swerdlow SH. CD30 expression and proliferative fraction in nontransformed mycosis fungoides. Am J Surg Pathol. 2009;33(12):1860–1868. | |
Gerber HP. Emerging immunotherapies targeting CD30 in Hodgkin’s lymphoma. Biochem Pharmacol. 2010;79(11):1544–1552. | |
Borchmann P, Treml JF, Hansen H, et al. The human anti-CD30 antibody 5F11 shows in vitro and in vivo activity against malignant lymphoma. Blood. 2003;102(10):3737–3742. | |
Heuck F, Ellermann J, Borchmann P, et al. Combination of the human anti-CD30 antibody 5F11 with cytostatic drugs enhances its antitumor activity against Hodgkin and anaplastic large cell lymphoma cell lines. J Immunother. 2004;27(5):347–353. | |
Wahl AF, Klussman K, Thompson JD, et al. The anti-CD30 monoclonal antibody SGN-30 promotes growth arrest and DNA fragmentation in vitro and affects antitumor activity in models of Hodgkin’s disease. Cancer Res. 2002;62(13):3736–3742. | |
Cardarelli PM, Moldovan-Loomis MC, Preston B, et al. In vitro and in vivo characterization of MDX-1401 for therapy of malignant lymphoma. Clin Cancer Res. 2009;15(10):3376–3383. | |
Hammond PW, Vafa O, Jacinto J, et al. A humanized anti-CD30 monoclonal antibody, XmAbTM2513, with enhanced in vitro potency against CD30-positive lymphomas mediated by high affinity Fc-receptor binding. Blood (ASH Annu Meeting Abstr). 2005;106:1470. | |
Lawrence CE, Hammond PW, Zalevsky J, et al. XMAbTM2513, an Fc engineering humanized anti-CD30 monoclonal antibody, has potent in vitro and in vivo activities, and has the potential for treating hematologic malignancies. Blood (ASH Annu Meeting Abstr). 2007;110:2340. | |
Thertulien R, Frankel A, Evens AM, et al. A phase I, open-label, dose-escalation, multidose study of MDX-1401 (defucosylated human anti-CD30 monoclonal antibody) in patients with CD30-positive refractory/relapsed Hodgkin’s lymphoma. AACR Meeting Abstr. 2009;1227. | |
Blum KA, Smith M, Fung H, et al. Phase I study of an anti-CD30 Fc engineered humanized monoclonal antibody in Hodgkin lymphoma (HL) or anaplastic large cell lymphoma (ALCL) patients: safety, pharmacokinetics (PK), immunogeninicty, and efficacy. J Clin Oncol (ASCO Annu Meeting Abstr). 2009;27(15S):8531. | |
Younes A, Zalevsky J, Blum KA, et al. Evaluation of the pharmacokinetics, immunogenicity, and safety of XmAb(R)2513 in the ongoing study XmAb2513-2501: a phase 1 study of every other week XmAb2513 to evaluate the safety, tolerability, and pharmacokinetics in patients with Hodgkin lymphoma or anaplastic large cell lymphoma. Blood (ASH Annu Meeting Abstr). 2008;112: 5012. | |
Francisco JA, Cerveny CG, Meyer DL, et al. cAC10-vcMMAE, an anti-CD30-monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood. 2003;102(4):1458–1465. | |
Dubowchik GM, Mosure K, Knipe JO, Firestone RA. Cathepsin B-sensitive dipeptide prodrugs. 2. Models of anticancer drugs paclitaxel (Taxol), mitomycin C and doxorubicin. Bioorg Med Chem Lett. 1998;8(23):3347–3352. | |
Sanderson RJ, Hering MA, James SF, et al. In vivo drug-linker stability of an anti-CD30 dipeptide-linked auristatin immunoconjugate. Clin Cancer Res. 2005;11(2 Pt 1):843–852. | |
Okeley NM, Miyamoto JB, Zhang X, et al. Intracellular activation of SGN-35, a potent anti-CD30 antibody-drug conjugate. Clin Cancer Res. 2010;16(3):888–897. | |
Gruss HJ, Pinto A, Gloghini A, et al. CD30 ligand expression in nonmalignant and Hodgkin’s disease-involved lymphoid tissues. Am J Pathol. 1996;149(2):469–481. | |
Pinto A, Aldinucci D, Gloghini A, et al. Human eosinophils express functional CD30 ligand and stimulate proliferation of a Hodgkin’s disease cell line. Blood. 1996;88(9):3299–3305. | |
Theurich S, Malcher J, Wennhold K, et al. Brentuximab vedotin combined with donor lymphocyte infusions for early relapse of Hodgkin lymphoma after allogeneic stem-cell transplantation induces tumor-specific immunity and sustained clinical remission. J Clin Oncol. 2013;31(5):e59–e63. | |
Hamblett KJ, Senter PD, Chace DF, et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin Cancer Res. 2004;10(20):7063–7070. | |
McDonagh CF, Turcott E, Westendorf L, et al. Engineered antibody-drug conjugates with defined sites and stoichiometries of drug attachment. Protein Eng Des Sel. 2006;19(7):299–307. | |
Oflazoglu E, Kissler KM, Sievers EL, Grewal IS, Gerber HP. Combination of the anti-CD30-auristatin-E antibody-drug conjugate (SGN-35) with chemotherapy improves antitumour activity in Hodgkin lymphoma. Br J Haematol. 2008;142(1):69–73. | |
Younes A, Bartlett NL, Leonard JP, et al. Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N Engl J Med. 2010;363(19):1812–1821. | |
Fanale MA, Forero-Torres A, Rosenblatt JD, et al. A phase I weekly dosing study of brentuximab vedotin in patients with relapsed/refractory CD30-positive hematologic malignancies. Clin Cancer Res. 2012;18(1):248–255. | |
Pro B, Advani R, Brice P, et al. Brentuximab vedotin (SGN-35) in patients with relapsed or refractory systemic anaplastic large-cell lymphoma: results of a phase II study. J Clin Oncol. 2012;30(18):2190–2196. | |
Younes A, Gopal AK, Smith SE, et al. Results of a pivotal phase II study of brentuximab vedotin for patients with relapsed or refractory Hodgkin’s lymphoma. J Clin Oncol. 2012;30(18):2183–2189. | |
Cheson BD, Pfistner B, Juweid ME, et al; International Harmonization Project on Lymphoma. Revised response criteria for malignant lymphoma. J Clin Oncol. 2007;25(5):579–586. | |
de Claro RA, McGinn K, Kwitkowski V, et al. U.S. Food and Drug Administration approval summary: brentuximab vedotin for the treatment of relapsed Hodgkin lymphoma or relapsed systemic anaplastic large-cell lymphoma. Clin Cancer Res. 2012;18(21):5845–5849. | |
Rothe A, Sasse S, Goergen H, et al. Brentuximab vedotin for relapsed or refractory CD30+ hematologic malignancies: the German Hodgkin Study Group experience. Blood. 2012;120(7):1470–1472. | |
Gibb A, Jones C, Bloor A, et al. Brentuximab vedotin in refractory CD30+ lymphomas: a bridge to allogeneic transplantation in approximately one quarter of patients treated on a Named Patient Programme at a single UK center. Haematologica. 2013;98(4):611–614. | |
Chen R, Palmer JM, Thomas SH, et al. Brentuximab vedotin enables successful reduced-intensity allogeneic hematopoietic cell transplantation in patients with relapsed or refractory Hodgkin lymphoma. Blood. 2012;119(26):6379–6381. | |
Gopal AK, Ramchandren R, O’Connor OA, et al. Safety and efficacy of brentuximab vedotin for Hodgkin lymphoma recurring after allogeneic stem cell transplantation. Blood. 2012;120(3):560–568. | |
Sasse S, Rothe A, Goergen H, et al. Brentuximab vedotin (SGN-35) in patients with transplant-naive relapsed/refractory Hodgkin lymphoma. Leuk Lymphoma. 2013;54(10):2144–2148. | |
Seattle Genetics, Inc. Brentuximab Vedotin in Patients With CD30-positive Nonlymphomatous Malignancies. Available from: http://clinicaltrials.gov/show/NCT01461538. NLM identifier: NCT01461538. Accessed November 20, 2013. | |
Albany C, Feldman DR, Garbo LE, Einhorn LH. Antitumor activity of brentuximab vedotin in CD30 positive refractory germ cell tumors. J Clin Oncol. 2013;31 Suppl 6:abstr 327. | |
Seattle Genetics, Inc. A Phase 3 Study of Brentuximab Vedotin (SGN-35) in Patients at High Risk of Residual Hodgkin Lymphoma Following Stem Cell Transplant (The AETHERA Trial). Available from: http://clinicaltrials.gov/show/NCT01100502. NLM identifier: NCT01100502. Accessed November 20, 2013. | |
Millennium Pharmaceuticals, Inc. Phase 3 Frontline Therapy Trial in Patients With Advanced Classical Hodgkin Lymphoma. Available from: http://clinicaltrials.gov/show/NCT01712490. NLM identifier: NCT01712490. Accessed November 20, 2013. | |
Seattle Genetics, Inc. ECHELON-2: A Comparison of Brentuximab Vedotin and CHP With Standard-of-care CHOP in the Treatment of Patients With CD30-positive Mature T-cell Lymphomas. Available from: http://clinicaltrials.gov/show/NCT01777152. NLM identifier: NCT01777152. Accessed November 20, 2013. | |
National Cancer Institute (NCI). Brentuximab Vedotin and Gemcitabine Hydrochloride in Treating Younger Patients With Relapsed or Refractory Hodgkin Lymphoma. Available from: http://clinicaltrials.gov/show/NCT01780662. NLM identifier: NCT01780662. Accessed November 20, 2013. | |
St. Jude Children’s Research Hospital. Adcetris (Brentuximab Vedotin), Combination Chemotherapy, and Radiation Therapy in Treating Younger Patients With Stage IIB, IIIB and IV Hodgkin Lymphoma. Available from: http://clinicaltrials.gov/show/NCT01920932. NLM identifier: NCT01920932. Accessed November 20, 2013. | |
Millennium Pharmaceuticals, Inc. Study of Brentuximab Vedotin (SGN-35) in Pediatric Patients With Relapsed or Refractory Systemic Anaplastic Large-Cell Lymphoma or Hodgkin Lymphoma. Available from: http://clinicaltrials.gov/show/NCT01492088. NLM identifier: NCT01492088. Accessed November 20, 2013. | |
Seattle Genetics, Inc. A Study of Brentuximab Vedotin in Relapsed or Refractory Non-Hodgkin Lymphoma. Available from: http://clinicaltrials.gov/show/NCT01421667. NLM identifier: NCT01421667. Accessed November 20, 2013. | |
M.D. Anderson Cancer Center. SGN-35 in CD30-positive Lymphoproliferative Disorders (ALCL), Mycosis Fungoides (MF), and Extensive Lymphomatoid Papulosis (LyP). Available from: http://clinicaltrials.gov/show/NCT01352520. NLM identifier: NCT01352520. Accessed November 20, 2013. | |
Seattle Genetics, Inc. Study of Brentuximab Vedotin Combined With RCHOP in Front-line Treatment of Patients With Diffuse Large B-cell Lymphoma (DLBCL). Available from: http://clinicaltrials.gov/show/NCT01925612. NLM identifier: NCT01925612. Accessed November 20, 2013. | |
Fondazione Michelangelo. Brentuximab Vedotin (SGN-35) as Salvage Treatment for CD30-positive Germ Cell Tumors. Available from: http://clinicaltrials.gov/show/NCT01851200. NLM identifier: NCT01851200. Accessed November 20, 2013. | |
Youn Kim. Pilot Study of Brentuximab Vedotin (SGN-35) in Patients With MF With Variable CD30 Expression Level. Available from: http://clinicaltrials.gov/show/NCT01396070. NLM identifier: NCT01396070. Accessed November 20, 2013. | |
Stanford University. Brentuximab Vedotin in Treating Patients With Advanced Systemic Mastocytosis or Mast Cell Leukemia. Available from: http://clinicaltrials.gov/show/NCT01807598. NLM identifier: NCT01807598. Accessed November 20, 2013. | |
Washington University School of Medicine. Brentuximab Vedotin Prevention of (GVHD) After Mismatched Unrelated Allogeneic Stem Cell Transplantation. Available from: http://clinicaltrials.gov/show/NCT01700751. NLM identifier: NCT01700751. Accessed November 20, 2013. | |
Seattle Genetics, Inc. A Phase 1 Study of Brentuximab Vedotin Given Sequentially and Combined With Multi-Agent Chemotherapy for CD30-Positive Mature T-Cell and NK-Cell Neoplasms. Available from: http://clinicaltrials.gov/show/NCT01309789. NLM identifier: NCT01309789. Accessed November 20, 2013. |
 © 2013 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2013 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.