")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Simvastatin prevents isoproterenol-induced cardiac hypertrophy through modulation of the JAK/STAT pathway
Authors Al-Rasheed N, Al-Oteibi M, Al-Manee RZ, Al-Shareef SA, Al-Rasheed N, Hasan I, Mohamad R, Mahmoud A , Al-Amin M, Al-Ajmi H
Received 10 April 2015
Accepted for publication 2 May 2015
Published 23 June 2015 Volume 2015:9 Pages 3217—3229
DOI https://doi.org/10.2147/DDDT.S86431
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Shu-Feng Zhou
Nouf M Al-Rasheed,1 Maha M Al-Oteibi,1 Reem Z Al-Manee,1 Sarah A Al-Shareef,1 Nawal M Al-Rasheed,1 Iman H Hasan,1 Raeesa A Mohamad,2 Ayman M Mahmoud3
1Department of Pharmacology, Faculty of Pharmacy, 2Department of Anatomy, College of Medicine, King Saud University, Riyadh, Saudi Arabia; 3Physiology Division, Department of Zoology, Faculty of Science, Beni-Suef University, Egypt
Abstract: Simvastatin (SIM) is a lipid-soluble inhibitor of hydroxy-3-methylglutaryl coenzyme A reductase with multiple reported therapeutic benefits. The present study was designed to investigate the effect of pretreatment with SIM on isoproterenol (ISO)-induced cardiac hypertrophy in rats. Twenty-four male albino Wistar rats weighing 180–200 g were divided into four groups. Groups I and III received normal saline while groups II and IV received SIM (10 mg/kg body weight) for 30 days per gavage. In the last 7 days, rats of groups III and IV were administered ISO (5 mg/kg) intraperitoneally to induce cardiac hypertrophy. Administration of ISO induced an increase in heart-to-body weight (HW/BW) ratio, an increase in serum interleukin-6, and elevated systolic and diastolic blood pressure. Serum levels of lipids, cardiovascular risk indices, and cardiac troponin I and creatine phosphokinase-MB showed significant increase in ISO-induced hypertrophic rats. Histopathological examination of heart tissue revealed focal areas of subendocardium degeneration, mononuclear cellular infiltrations, fibrous tissue deposition, and increased thickness of the myocardium of left ventricle. In addition, ISO-administered rats exhibited significant upregulation of cardiac Janus kinase, phosphorylated signal transducer and activator of transcription, and nuclear factor-kappa B. Pretreatment with SIM significantly prevented ISO-induced cardiac hypertrophy, alleviated the altered biochemical parameters, and improved the heart architecture. In conclusion, our study provides evidence that SIM prevented the development of cardiac hypertrophy via modulation of the Janus kinase/signal transducer and activator of transcription-signaling pathway in the heart of ISO-administered animals.
Keywords: simvastatin, cardiac hypertrophy, JAK/STAT pathway, IL-6, isoproterenol
Introduction
Cardiac hypertrophy is one of the major causes of morbidity and mortality in the world.1 It is initially compensatory for an increased workload in response to sustained exercise and during development and pregnancy. However, prolongation and prevalence of this process leads to congestive heart failure (HF) and sudden death.2 HF is a major health burden accounting for approximately 25% of all deaths in developing countries and HF patients have a 50% mortality rate within 4 years.3,4 Also, cardiomyocyte hypertrophy has been reported to often occur after myocardial infarction (MI) as an adaptive response.5,6 MI is a complication of cardiovascular disease and its prevalence is growing rapidly in developing countries probably due to the acquisition of a western lifestyle.7,8
Numerous scientific reviews and studies demonstrate that various signal transduction pathways are implicated in the development of cardiomyocyte hypertrophy.9–12 The Janus kinase (JAK)/signal transducer and activator of transcription (STAT) pathway mediates the transduction of stress signals from the plasma membrane to the nucleus through the STAT proteins.13 STAT proteins are translocated into the nucleus and bind to the promoter region of target genes, thereby regulating their transcription.13 In the heart, STAT proteins regulate the expression of genes encoding proteins involved in inflammation, angiogenesis, extracellular matrix composition, apoptosis, and cellular signaling.14–16 Proteins of the interleukin (IL)-6 family transduce their signals via glycoprotein 130 (gp130) predominantly to STAT3.17 The IL-6-gp130-JAK/STAT-signaling pathway is a key player in the development of cardiac hypertrophy and HF.17–19 Thus, modulation of this signaling pathway is an important strategy for the treatment of cardiovascular diseases.
Statins are known to prevent hypercholesterolemia, which is a major risk factor in the development of coronary heart disease and stroke.20 Recent data have indicated that statins are able to protect the myocardium against ischemic injury.21 In addition, multiple studies have demonstrated that early and chronic pretreatment with statins can improve myocardial perfusion and decrease the sizes of no-reflow and infarction areas after ischemic reperfusion. These effects were mainly through the inhibition of myocardial inflammation and apoptosis and the improvement of endothelial function.22–25 Simvastatin (SIM), a lipid-soluble inhibitor of 3-hydroxy-3-methylglutaryl coenzyme A reductase, has been reported to inhibit noradrenaline-induced hypertrophy of cultured neonatal rat cardiomyocytes.26 Wu et al27 have suggested that SIM attenuates hypertrophic responses induced by cardiotrophin-1 via JAK/STAT pathway in cultured cardiomyocytes. More recently, Liu et al28 reported that SIM prevents cardiac hypertrophy in vitro and in rat with pressure overload due to an abdominal aortic constriction. Therefore, the current study was designed to investigate the protective role of SIM against isoproterenol (ISO)-induced cardiac hypertrophy in rats focusing on its modulatory effect on the JAK/STAT pathway.
Materials and methods
Chemicals
SIM and ISO were purchased from Sigma-Aldrich (St Louis, MO, USA). Rabbit polyclonal anti-JAK, mouse antinuclear factor-kappa B (NF-κB) p65, and goat antiphospho-STAT3 were obtained from Santa Cruz Biotechnology (Inc., Dallas, TX, USA). All other chemicals were of analytical grade and obtained from standard commercial supplies.
Experimental animals
Adult male albino Wistar rats weighing 180–200 g were supplied by the Experimental Animal Center at the College of Pharmacy at King Saud University (Riyadh, Saudi Arabia). The rats were maintained in controlled environment (25°C±1°C) on a 12-hour light/dark cycle and were provided access to standard laboratory chow and tap water ad libitum. Rats were kept under observation for 1 week before the onset of the experiment for acclimatization and to exclude any intercurrent infection. All animal procedures were undertaken with the approval of Institutional Research Ethics Committee of the College of Pharmacy at King Saud University (Riyadh, Saudi Arabia).
Experimental design
Twenty four rats were randomly allocated into four groups having six in each as follows:
Group I (Control): Rats received normal saline orally for 30 days and intraperitoneally (ip) for the last 7 days.
Group II (SIM): Rats received SIM (10 mg/kg body weight [BW]) dissolved in saline by oral gavage for 30 days and ip saline for the last 7 days.
Group III (ISO): Rats received normal saline orally for 30 days and daily ISO (5 mg/kg, ip) for the last 7 days.
Group IV (ISO + SIM): Rats received SIM (10 mg/kg BW) dissolved in saline by oral gavage for 30 days and ISO (5 mg/kg BW, ip) for the last 7 days.
The doses were adjusted consistently as indicated by any change in BW to maintain comparable dosage over the study period. At the end of the experiment, animals were sacrificed and blood samples were collected and centrifuged to separate serum. Sera were then kept at −20°C for subsequent biochemical assays. Hearts were immediately excised, perfused with ice-cold phosphate-buffered saline, and fixed in 10% formalin or homogenized in phosphate-buffered saline.
Assessment of general parameters and blood pressure
The final BW along with the weight of heart was recorded, and the heart-to-body weight (HW/BW) ratio was calculated and used to estimate the degree of cardiac hypertrophy. The arterial blood pressure (BP) was measured using the tail cuff method (UGO BASILE, Italy) before and after the administration of ISO with minimal stress and restraint. Systolic and diastolic BP were measured before and at the end of treatment period.
Assay of cardiac troponin I and creatine kinase MB
Heart left ventricle samples (10% w/v) were homogenized in chilled phosphate-buffered saline and the homogenates were centrifuged for 5 minutes at 5,000× g. The clear homogenates were collected and used for the assays. Troponin I and creatine kinase MB (CK-MB) were measured in the cardiac homogenates using specific rat enzyme-linked immunosorbent assay kits purchased from EIAab (Wuhan, People’s Republic of China) following the manufacturer instructions. In brief, standards or samples were added to the appropriate microtiter plate wells with a biotin-conjugated polyclonal antibody preparation specific for troponin I or CK-MB. After incubation and washing, avidin conjugated to horseradish peroxidase was added to each well and incubated. Thereafter, a 3,3′,5,5′-tetramethylbenzidine (TMB) substrate solution was added to each well. The enzyme–substrate reaction was terminated by the addition of a sulfuric acid solution and the color change was measured spectrophotometrically at a wavelength of 450 nm. Troponin I and CK-MB in the samples were determined from the prepared standard curves.
Determination of lipid profile and cardiovascular risk indices
Serum total cholesterol, triglycerides, and high-density lipoprotein (HDL)-cholesterol were assayed using commercial diagnostic kits (United Diagnostics Industry, Riyadh, KSA), according to the methods of Allain et al,29 Fossati and Prencipe,30 and Burnstein et al31 respectively. Serum very low-density lipoprotein (vLDL)-cholesterol concentration was calculated according to the following formula:32 vLDL-cholesterol = triglycerides/5. Serum low-density lipoprotein (LDL)-cholesterol level was calculated from the formula:33 LDL-cholesterol = total cholesterol – {(triglycerides/5) + HDL-cholesterol}. Cardiovascular risk indices were calculated according to Ross34 as follows: cardiovascular risk index 1= total cholesterol/HDL-cholesterol and cardiovascular risk index 2= LDL-cholesterol/HDL-cholesterol.
Assay of serum IL-6
Serum levels of IL-6 were determined using specific enzyme-linked immunosorbent assay kits (R&D Systems, Inc., Minneapolis, MN, USA) following the manufacturer’s instructions. The concentration of assayed cytokine was measured specrophotometrically at 450 nm. Standard curves were constructed by using standard cytokine and concentrations of the unknown samples were determined from the standard plots.
Histopathological study
The excised hearts were fixed in 10% buffered formalin at 4°C for 24 hours and processed to prepare transverse, mid-ventricular, 5-μm-thick paraffin sections. These sections were stained with hematoxylin and eosin (H&E) and Masson’s Trichrome stains. The thickness of the wall of the left ventricle was measured using image analysis by scanning H&E-stained slides. High-resolution, whole-slide digital scans of all stained slides were created with a ScanScope scanner (Aperio Technologies, Vista, CA, USA). The digital slide images were viewed and analyzed using Aperio’s viewing and image analysis tools. Left ventricular thickness was measured at ×20 magnification using the linear measurement tool of Aperio’s Image Scope software (Aperio Technologies). To minimize error, thickness was measured at five randomly chosen points per heart section and averages were calculated.
Immunohistochemical examination
Immunostaining of the heart sections for detection of pSTAT3, JAK, and NF-κB was performed using streptavidin-biotinylated horseradish peroxidase method (Novalink Max Polymer detection system; Novocastra Laboratories, Newcastle, UK). The procedure involved the following steps: endogenous peroxidase activity was inhibited by 3% H2O2 in distilled water for 5 minutes, and then the sections were washed in Tris-buffered saline (pH 7.6) (Sigma-Aldrich) for 10 minutes. Nonspecific binding of antibodies was blocked by incubation with protein block for 5 minutes (Novocastra). Sections were incubated with pSTAT3 goat polyclonal antibody (Santa Cruz Biotechnology), rabbit polyclonal JAK primary antibody (Santa Cruz Biotechnology), and mouse monoclonal NF-κB p65 (Santa Cruz Biotechnology), diluted 1:100 for 1 hour at room temperature. Sections were washed three times in Tris-buffered saline and then incubated with biotinylated IgG (Novocastra) for 30 minutes, followed by washing in Tris-buffered saline and incubation with Novolink polymer (Novocastra) for 30 minutes. Peroxidase was detected with working solution of diaminobenzedine substrate (Novocastra) for 10 minutes. Sections were then washed in distilled water for 10 minutes, nuclei were stained with Mayer’s hematoxylin, and sections were mounted in DPX. For negative control sections, the same procedure was followed with omission of incubation in primary antibodies. Image analysis of the immunostaining was performed using the freeware ImageJ (NIH, USA), and values were presented as % relative to control.
Statistical analysis
Statistical analysis was performed using GraphPad Prism (GraphPad Software, San Diego, CA, USA), and all statistical comparisons were made by means of the one-way analysis of variance test followed by Tukey’s test post hoc analysis. Results were expressed as mean ± standard error of the mean and a P-value <0.05 was considered significant.
Results
Effect of SIM on BW, HW, and BP
BW of control and treated rats showed nonsignificant (P>0.05) changes throughout the experiment (Table 1). On the other hand, ISO-administered rats exhibited significant (P<0.01) increase in HW and HW/BW ratio when compared with the control rats. Treatment of the ISO-administered rats with SIM significantly (P<0.01) ameliorated both the HW and HW/BW ratio as compared to ISO-injected rats (Table 1). SIM produced a nonsignificant (P>0.05) effect on the mentioned parameters when compared with control rats.
 | Table 1 Effect of SIM on cardiac biomarkers in ISO-induced cardiac hypertrophy in rats |
Similarly, ISO administration produced a significant (P<0.001) elevation in systolic and diastolic BP (Table 1). Oral supplementation of SIM significantly (P<0.01) alleviated the altered BP levels when compared with the ISO-induced rats.
Effect of SIM on cardiac troponin I and CK-MB
ISO-administered rats exhibited significant (P<0.01) increase in cardiac troponin I levels when compared with the control rats, as represented in Figure 1. Treatment of ISO-induced rats with SIM markedly (P<0.05) decreased cardiac troponin I levels. More or less similar, ISO administration produced a significant (P<0.001) increase in cardiac CK-MB activity. Oral treatment of the ISO-induced rats with SIM potentially (P<0.05) ameliorated the altered CK-MB activity (Figure 2). SIM supplementation to the normal rats produced a nonsignificant (P>0.05) effect on either troponin I level or CK-MB activity when compared with the corresponding control rats.
 | Figure 1 Effect of SIM on cardiac troponin I levels. |
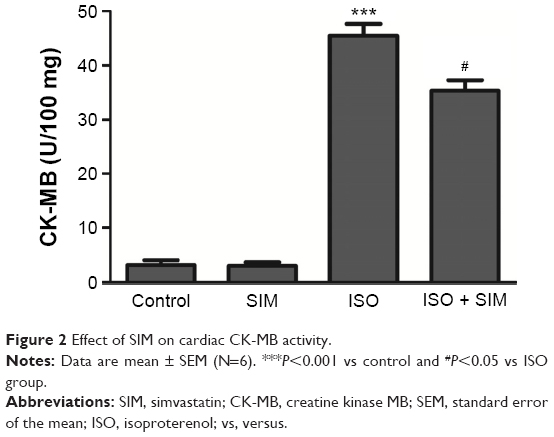 | Figure 2 Effect of SIM on cardiac CK-MB activity. |
Effect of SIM on serum IL-6 levels
Serum levels of the proinflammatory cytokine IL-6 were significantly (P<0.01) elevated in ISO-administered rats when compared with the control group (Figure 3). SIM significantly (P<0.01) decreased the elevated serum cytokine level when supplemented to ISO-administered rats. However nonsignificant, SIM supplementation to normal rats slightly decreased serum IL-6 levels when compared with the control rats.
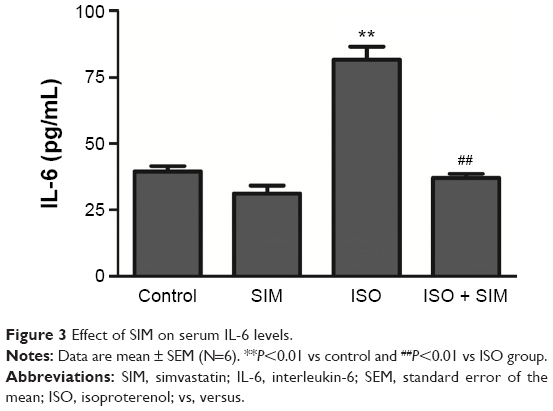 | Figure 3 Effect of SIM on serum IL-6 levels. |
Effect of SIM on serum lipid profile and cardiovascular risk indices
Data represented in Figure 4 show the effect of SIM on lipid profile and cardiovascular risk indices of normal and ISO-induced hypertrophic rats. Compared to the control group, rats supplemented with SIM exhibited significant (P<0.05) decrease in serum total and LDL-cholesterol, while the effect on serum triglycerides, HDL-, and LDL-cholesterol levels was nonsignificant (P>0.05). On the contrary, ISO-induced rats exhibited significant increase in serum total cholesterol (P<0.001), triglycerides (P<0.01), LDL-cholesterol (P<0.05), and vLDL-cholesterol (P<0.01) when compared with their corresponding control rats. HDL-cholesterol showed nonsignificant (P>0.05) variation between all studied groups. In addition, ISO-administered rats showed significant (P<0.01) increase in total cholesterol/HDL-cholesterol and LDL-cholesterol/HDL-cholesterol. Oral supplementation of SIM to the ISO-induced rats potentially ameliorated the altered serum lipid profile parameters as well as cardiovascular risk indices.
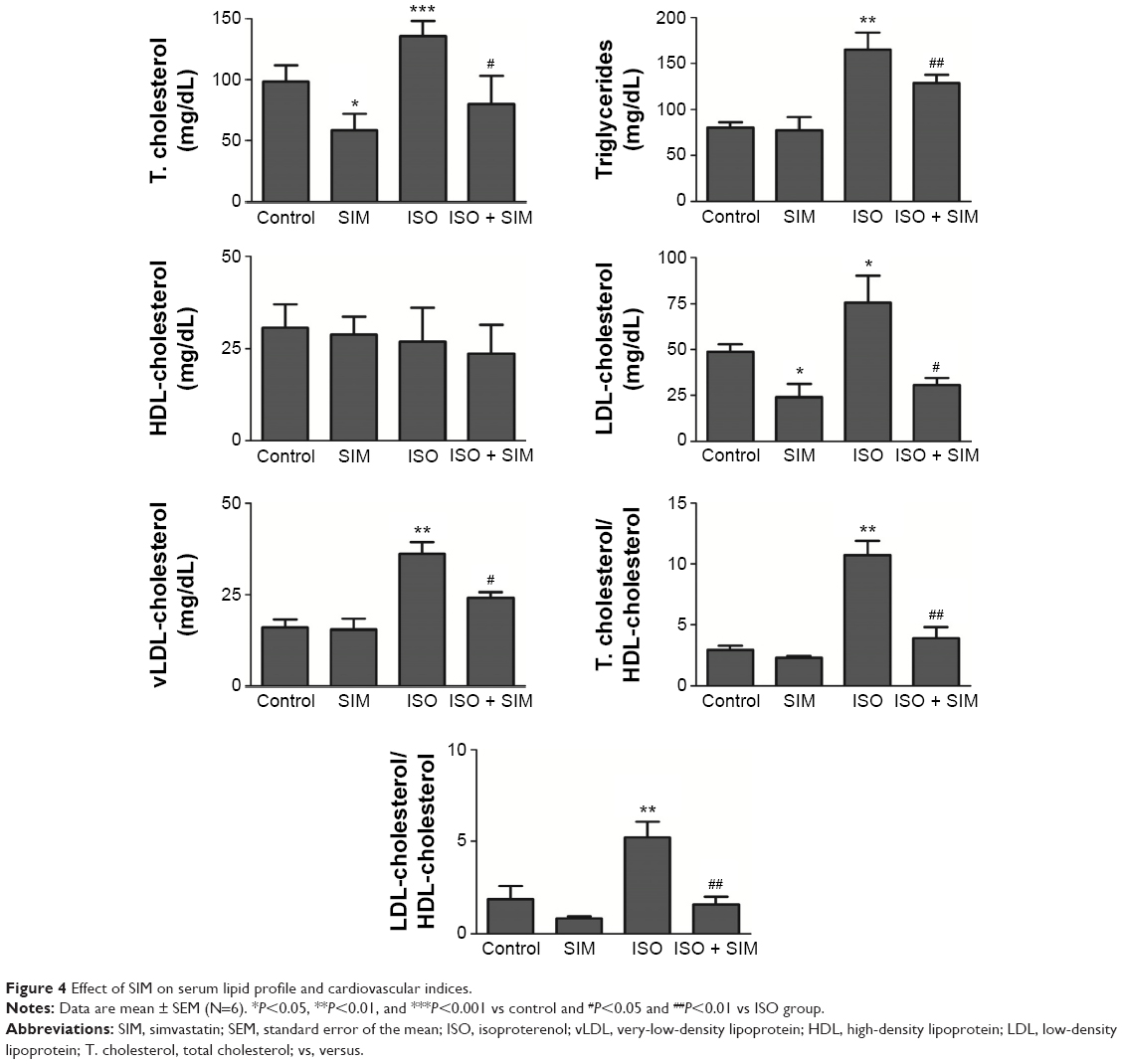 | Figure 4 Effect of SIM on serum lipid profile and cardiovascular indices. |
Histopatholigical changes
Microscopic investigation of H&E-stained heart tissue, from control and SIM received rats, demonstrated typical normal endocardium, myocardium, and normal amount and distribution of the vascular endomysium between cardiac cells (Figure 5A, B). Heart tissue from ISO-induced rats showed focal areas of subendocardium degenerations, mainly in the left ventricle, with many focal areas of mononuclear cellular infiltrations and there was an increase in fibrous tissue of the endomysium and thickness of the myocardium of left ventricle (Figure 5C). Myocardium of left ventricle from ISO-induced rats treated with SIM (Figure 5D) revealed marked decrease in the left ventricular myocardium thickness and inflammatory cellular infiltration when compared with the ISO control rats.
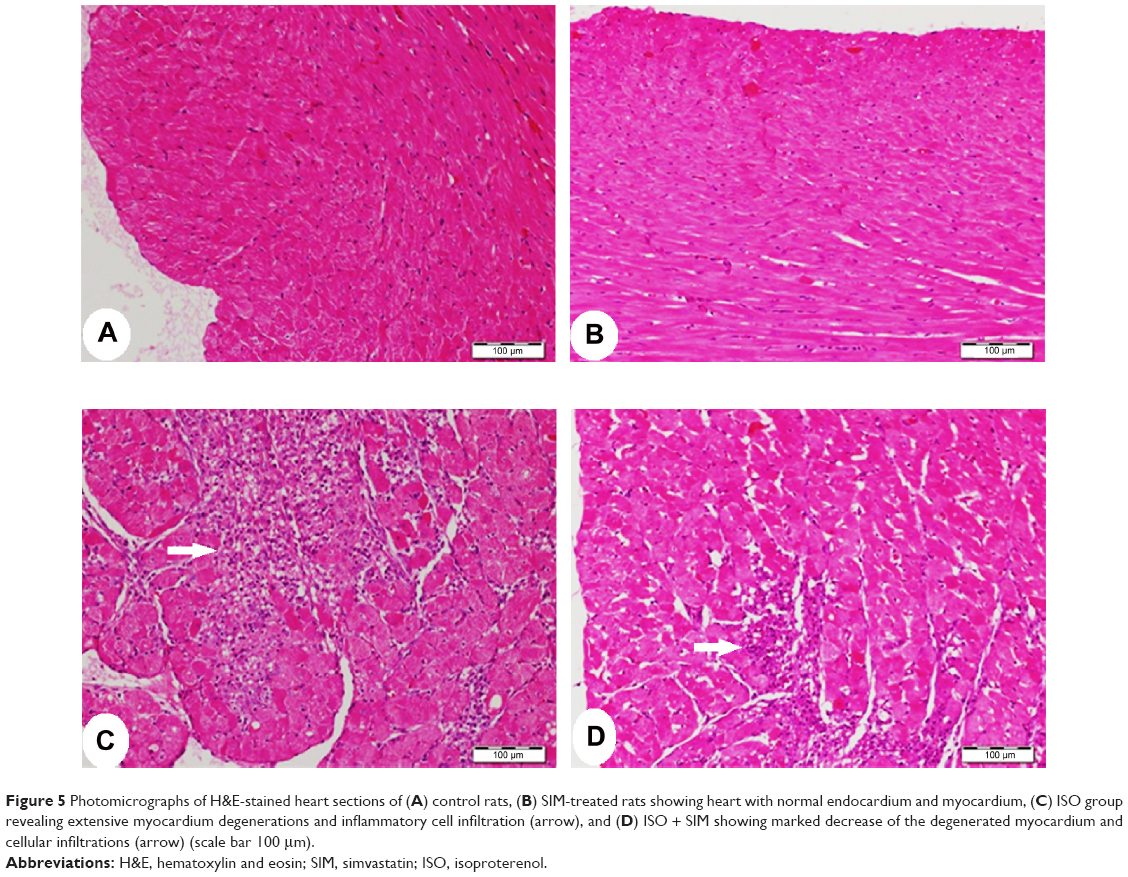 | Figure 5 Photomicrographs of H&E-stained heart sections of (A) control rats, (B) SIM-treated rats showing heart with normal endocardium and myocardium, (C) ISO group revealing extensive myocardium degenerations and inflammatory cell infiltration (arrow), and (D) ISO + SIM showing marked decrease of the degenerated myocardium and cellular infiltrations (arrow) (scale bar 100 μm). |
Heart sections stained with Masson’s Trichrome showed normal distribution and normal amount of cardiac fibrous tissue in both control and SIM-treated rats, as represented in Figure 6A, B. On the other hand, ISO-administered rats exhibited excessive fibrous tissue deposition in the endomysium between cardiomyocytes of the left ventricle, especially in the subendothelial areas (Figure 6C). Treatment of the ISO-administered rats with SIM produced a marked decrease in fibrosis, which is restricted to small patches under endothelium of the left ventricle (Figure 6D).
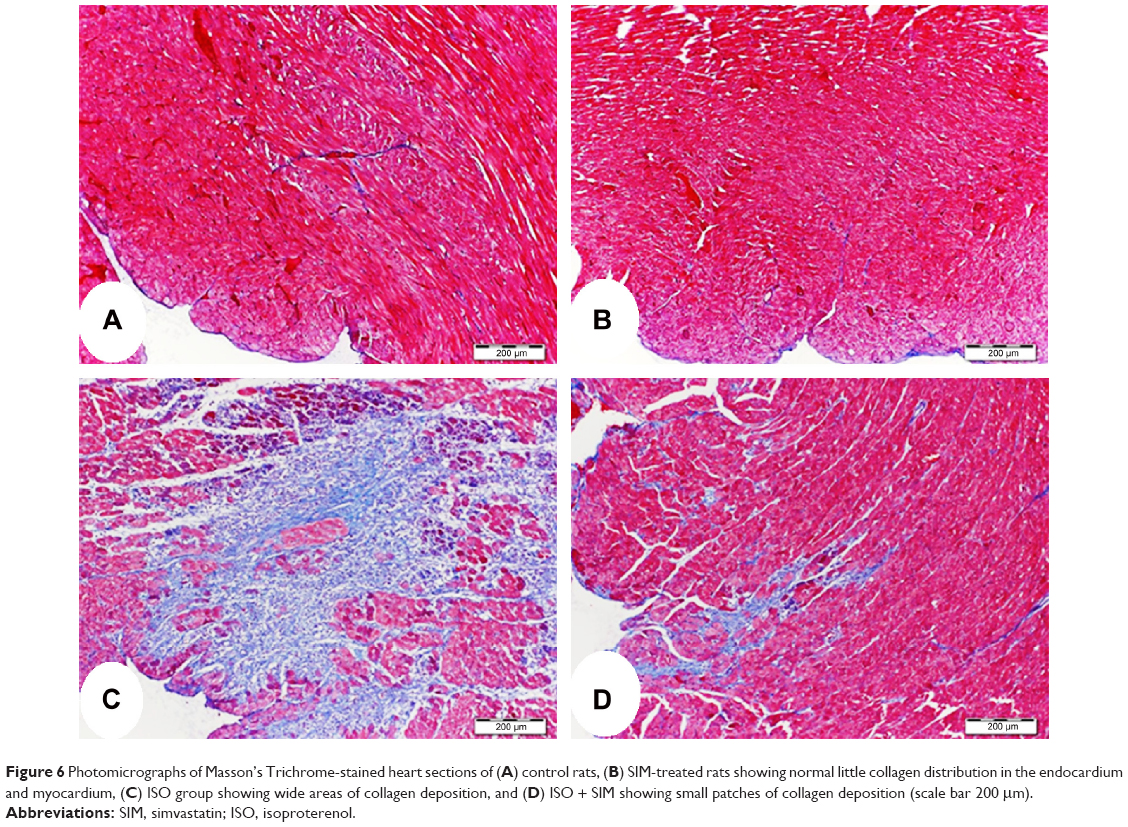 | Figure 6 Photomicrographs of Masson’s Trichrome-stained heart sections of (A) control rats, (B) SIM-treated rats showing normal little collagen distribution in the endocardium and myocardium, (C) ISO group showing wide areas of collagen deposition, and (D) ISO + SIM showing small patches of collagen deposition (scale bar 200 μm). |
Effect of SIM on cardiac pSTAT3, JAK, and NF-κB
The immunohistochemical staining of the heart with pSTAT3 primary antibody revealed normal weak immune staining of cytoplasm of few myocardium cells and cells in the endomysium of heart from normal control (Figure 7A) and SIM-administered rats (Figure 7B). ISO-administered rats showed diffuse strong positive immune reaction of the nuclei of the inflammatory cells in the endomysium and the cytoplasm of degenerated heart cells (Figure 7C). ISO-induced rats treated with SIM showed marked decrease of the immune reaction, especially the cardiac cells cytoplasm (Figure 7D).
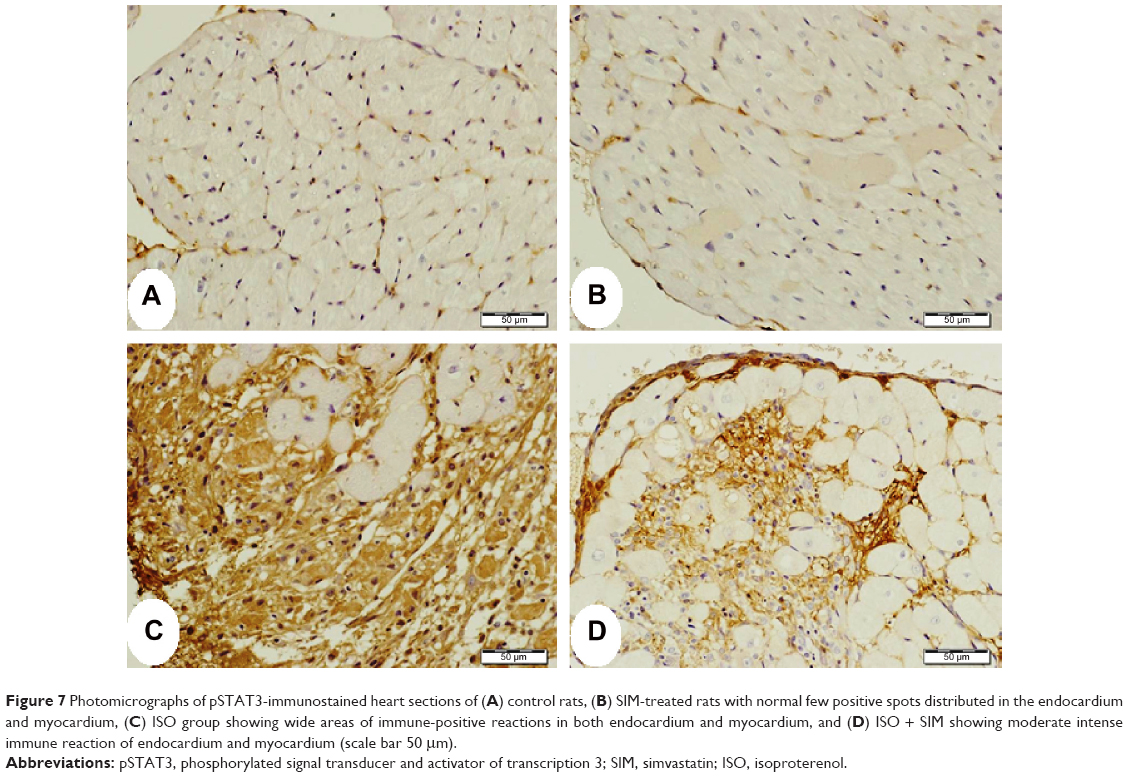 | Figure 7 Photomicrographs of pSTAT3-immunostained heart sections of (A) control rats, (B) SIM-treated rats with normal few positive spots distributed in the endocardium and myocardium, (C) ISO group showing wide areas of immune-positive reactions in both endocardium and myocardium, and (D) ISO + SIM showing moderate intense immune reaction of endocardium and myocardium (scale bar 50 μm). |
Heart sections from both control and SIM-administered rats, immunostained with JAK primary antibody, revealed few endocardium cells with weak positive reaction (Figure 8A, B). ISO-induced rats showed large strongly immunostained patches (Figure 8C), which exhibited marked decrease and restriction to the endocardium and few myocardium in the areas of degeneration in SIM-treated rats (Figure 8D).
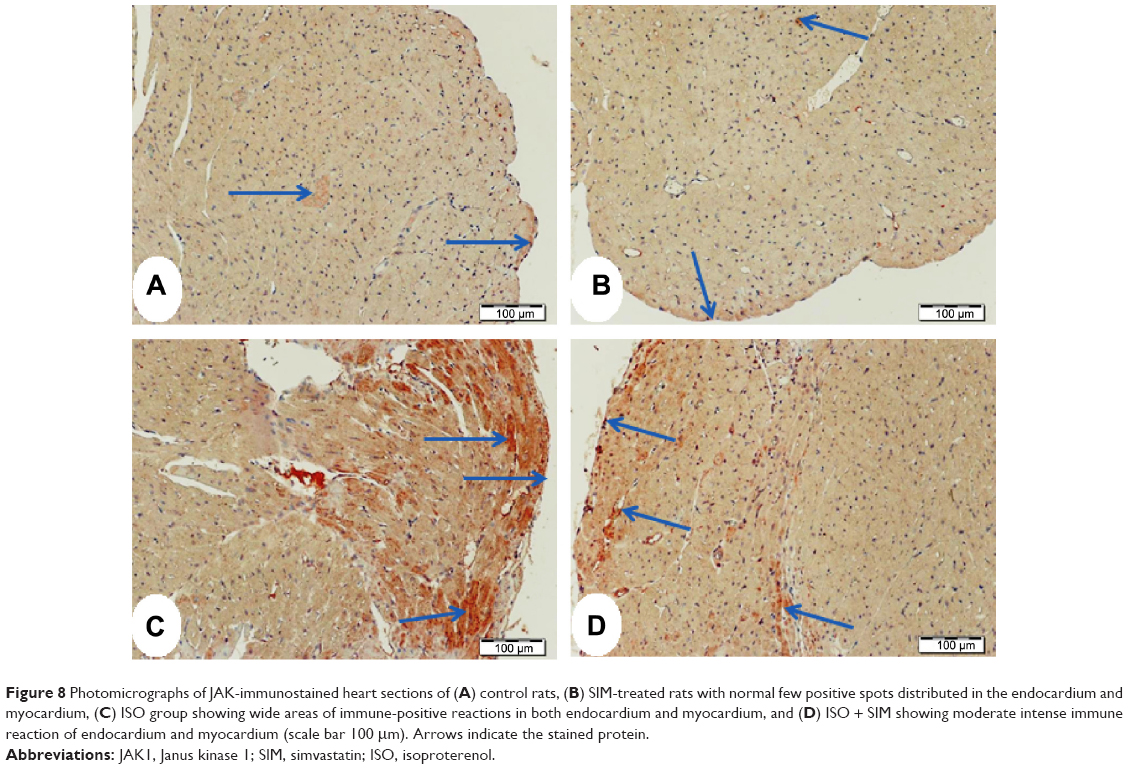 | Figure 8 Photomicrographs of JAK-immunostained heart sections of (A) control rats, (B) SIM-treated rats with normal few positive spots distributed in the endocardium and myocardium, (C) ISO group showing wide areas of immune-positive reactions in both endocardium and myocardium, and (D) ISO + SIM showing moderate intense immune reaction of endocardium and myocardium (scale bar 100 μm). Arrows indicate the stained protein. |
Similarly, NF-κB showed massive strong immune reactivity especially the inflammatory cells and the endothelium of the endocardium in the heart sections of ISO-administered rats (Figure 9) when compared with either control (Figure 9A) or SIM-treated group (Figure 9B). Treatment of the ISO-induced rats with SIM produced a marked decrease in immunostained cells but still the endothelial cells of the endocardium had strong immune reaction (Figure 9D).
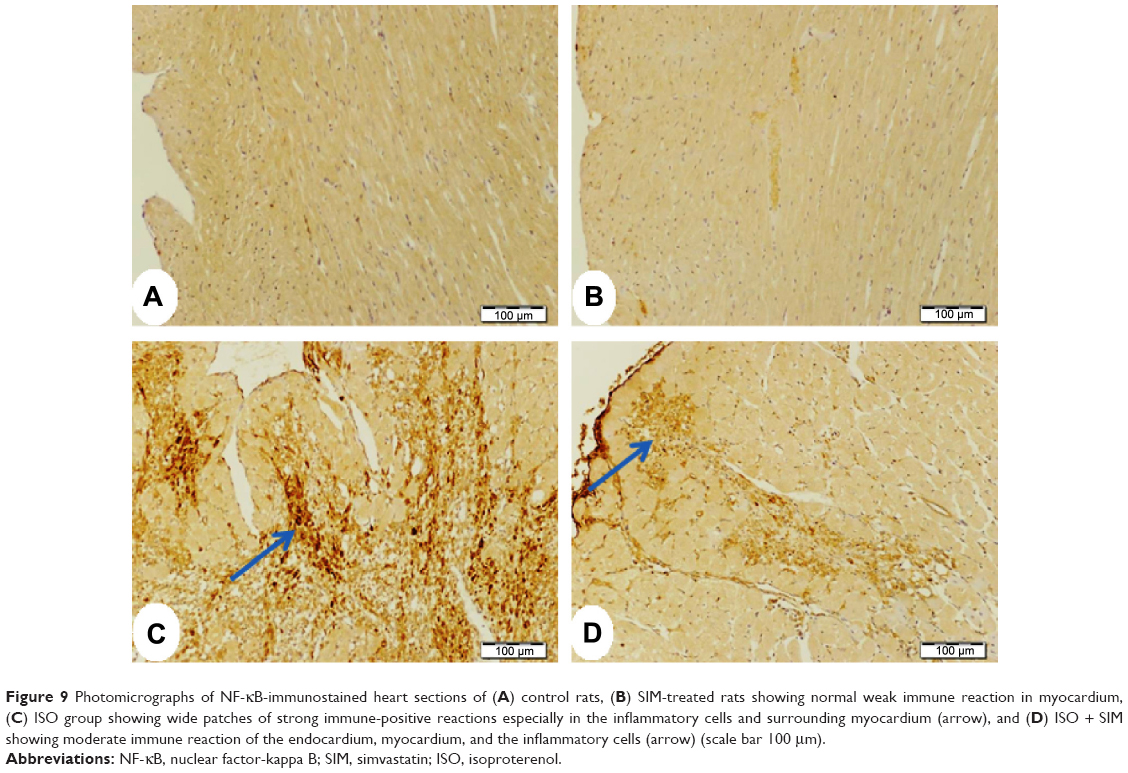 | Figure 9 Photomicrographs of NF-κB-immunostained heart sections of (A) control rats, (B) SIM-treated rats showing normal weak immune reaction in myocardium, (C) ISO group showing wide patches of strong immune-positive reactions especially in the inflammatory cells and surrounding myocardium (arrow), and (D) ISO + SIM showing moderate immune reaction of the endocardium, myocardium, and the inflammatory cells (arrow) (scale bar 100 μm). |
Image analysis demonstrated significant increase in pSTAT3 (P<0.01), JAK (P<0.05), and NF-κB (P<0.001) expression in ISO-administered rats when compared with the control rats (Figure 10). Treatment with SIM induced significant decrease in immunostaining of pSTAT3 (P<0.05), JAK (P<0.05), and NF-κB (P<0.001) compared to the ISO-administered rats. SIM supplementation to normal rats produced nonsignificant effect on the immunostained proteins (Figure 10).
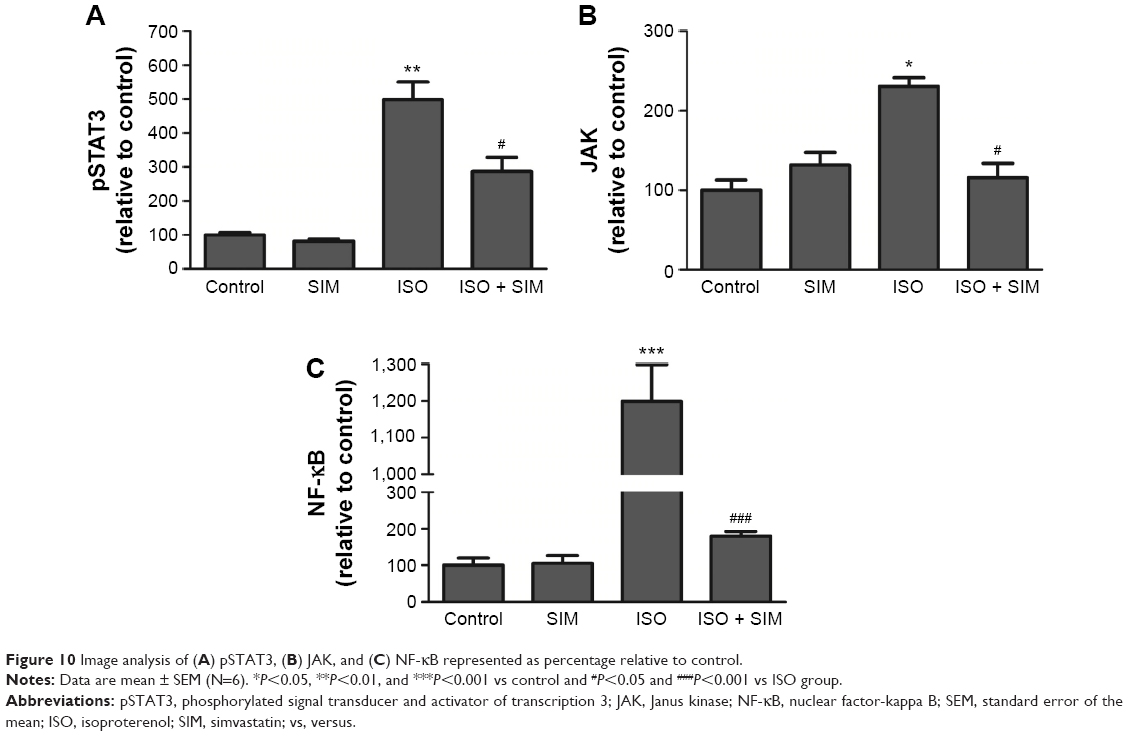 | Figure 10 Image analysis of (A) pSTAT3, (B) JAK, and (C) NF-κB represented as percentage relative to control. |
Discussion
Cardiac hypertrophy is a common compensatory response of the heart to acute myocardial injury, infection, or hemodynamic stress.35 Sustained adrenergic stimulation is an important hallmark of the maladaptive cardiac hypertrophy. The β1-adrenergic receptor agonist, ISO, induced cardiac hypertrophy mimics this sustained adrenergic stimulation and it represents the widely used model.36 ISO has been reported to induce infarct-like necrosis of the heart muscle.37 It is also known to generate free radicals and to provoke lipid peroxidation leading to irreversible damage to the myocardial membrane.38 In addition, the activation of β-adrenergic signaling induces different mechanisms including enhanced protein synthesis and stimulation of mitogen-activated protein kinases and phosphotidyl inositol-3 kinases, which contribute to the hypertrophic phenotype.39 Therefore, the present study aimed at evaluating potential protective effects of SIM against ISO-induced cardiac hypertrophy in rats.
Our results show that on the eighth day of ISO administration, rats exhibited myocardial hypertrophy as evidenced by the significant increase in HW and HW/BW ratio. In this context, Taylor and Tang40 reported that maximum HW can be reached on the eighth day of subcutaneous ISO administration. These findings are in agreement with multiple studies demonstrating increased HW in ISO-administered rats.41,42 The observed increased HW in ISO-administered rats might be attributed to the increase in water content, edematous intramuscular space, and infiltrations by inflammatory cells following extensive necrosis of cardiac muscle.43 The ISO-induced cardiac hypertrophy is further confirmed by the noticed mononuclear cellular infiltrations, increase in fibrous tissue of the endomysium and thickness of the myocardium of left ventricle, and the excessive fibrous tissue deposition in the endomysium between cardiomyocytes of the left ventricle. Pretreatment with SIM significantly decreased the HW in ISO-induced rats, which is consistent with the observations of Liu et al28 who reported that SIM prevents cardiac hypertrophy in vitro and in rat with pressure overload due to an abdominal aortic constriction.
There is considerable controversy about the BP changes in ISO-induced rats. The study of Upaganlawar and Balaraman41 reported decreased systolic and diastolic BP on the second day of ISO-induced MI in rats. In addition, multiple studies have demonstrated declined pumping ability44–46 and depressed contractile ability of the heart in ISO-induced hypertrophy,47–49 while others reported contractile function enhancement.50–52 In the current study, hypertrophic rats exhibited significant elevation in both systolic and diastolic BP, which is in agreement with Papparella et al53 who reported increased BP in angiotensin II–treated hypertrophic rats. On the other hand, treatment of the induced rats with SIM potentially decreased systolic and diastolic BP. This alleviation might be directly connected to its protective effect against ISO-induced cardiac perturbations. In addition, ISO has been reported to be associated with increases in serum and myocardial lipids, which in turn lead to coronary heart disease.54 Therefore, the ameliorative effect of SIM against ISO-induced cardiac hypertrophy might be partially connected to its hypolipidemic and myocardial-enhancing effects. On the contrary, Adameová et al55 revealed that SIM alleviates myocardial contractile dysfunction in rat heart independent of its cholesterol-lowering effects. However, the study of Morikawa-Futamatsu et al56 demonstrated that fluvastatin protects against angiotensin II-induced hypertrophy via cyclin D1 inhibition and Rho kinase in vivo and in vitro. Moreover, cerivastatin treatment has been reported to improve the hypertrophic and cardiomyopathic phenotype of transgenic rats.57 Thus, the positive therapeutic effects of statins are not limited to their cholesterol-lowering effects. Furthermore, the protective effects of SIM against ISO-induced cardiac hypertrophy in the present study were also confirmed by the declined levels of cardiac troponin I and CK-MB. These cardiac markers have been reported to increase in heart tissue in response to myofibrillar disintegration and increased permeability caused by inflammation.58
Serum IL-6 levels showed significant increase in ISO-administered rats. In post-MI and HF patients, IL-6 has been reported to be a strong prognostic marker for the morbidity and mortality.59–61 Multiple studies have demonstrated that IL-6 levels are elevated in patients with unstable angina, after MI, and in patients with HF.62,63 In addition, elevated levels of IL-6 were detected in isolated cardiomyocytes subjected to hypoxic stress.64 Also, Hirota et al65 investigated the involvement of IL-6 in the progression of cardiac hypertrophy in mice overexpressing both IL-6 and IL-6 receptors. At the age of 5 months, the reported mice developed ventricular hypertrophy and ventricular wall thickening. Moreover, upregulation of IL-6 was noticed in the viable border zone adjacent to infarcted tissue66 and in left ventricle of ISO-induced cardiac hypertrophy in mice.67 Consistent with the elevated levels of serum IL-6, NF-κB was upregulated in the heart of ISO-induced rats, confirming the close link between inflammation and cardiac hypertrophy. NF-κB induces the transcription of IL-6, inducible nitric oxide synthase, and cyclooxygenase-2.68–70 Therefore, upregulation of NF-κB in the current study may reflect the degree of inflammation in the ISO-induced rats. On the other hand, SIM-pretreated rats exhibited significant decrease in serum IL-6 and downregulation of cardiac tissue NF-κB. These results suggest that SIM inhibits cardiac hypertrophy through attenuating ISO-induced inflammation. In a recent study, inhibition of NF-κB was found to protect against ISO-induced cardiac hypertrophy in cardiomyocytes as well as in rats.71
IL-6 transduces a hypertrophic signal via gp130 predominantly to STAT317 in cardiac myocytes and in satellite cells,72 demonstrating the key role of IL-6-gp130-JAK/STAT-signaling pathway in cardiac hypertrophy. In conjunction with increased IL-6, the current study shows upregulation of JAK and STAT in the heart of ISO-induced hypertrophic rats. Also, cardiac hypertrophy was associated with increased levels of IL-6 and STAT3 in a model of long-term intermittent hypoxia in rat hearts.73 Recent studies have shown that the JAK/STAT pathway plays a crucial role in pressure overload-induced cardiac hypertrophy and remodeling and ischemia/reperfusion-induced cardiac dysfunction.27,74,75 The current results show upregulation of the IL-6-JAK/STAT-signaling pathway in ISO-induced cardiac hypertrophy. SIM treatment significantly inhibited cardiac hypertrophy via downregulation of the JAK/STAT pathway. Accordingly, the studies of Wu et al27 and Liu et al28 suggested that SIM attenuates hypertrophic responses induced by cardiotrophin-1 in cultured cardiomyocytes and in rat with pressure overload, respectively, via JAK/STAT pathway. Downregulation of the JAK/STAT pathway and decreased IL-6 levels prove the role of this pathway in the mechanisms underlying the inhibitory effects of SIM on ISO-induced cardiac hypertrophy.
In conclusion, our findings demonstrate that SIM prevents ISO-induced cardiac hypertrophy in rats through its lipid-lowering effect and attenuating inflammation. The study provides further evidence that the ameliorative effect of SIM on ISO-induced cardiac hypertrophy is related to downregulation of the JAK/STAT-signaling pathway. Thus, SIM might be considered as a potential candidate for the prevention and treatment of cardiac hypertrophy.
Acknowledgment
This research project was supported by a grant from the Research Center of the Female Scientific and Medical Colleges, Deanship of Scientific Research, King Saud University, Saudi Arabia.
Disclosure
The authors report no conflicts of interest in this work.
References
Lorell BH, Carabello BA. Left ventricular hypertrophy: pathogenesis, detection, and prognosis. Circulation. 2000;102:470–479. | ||
Yung CK, Halperin VL, Tomaselli GF, Winslow RL. Gene expression profiles in end-stage human idiopathic dilated cardiomyopathy: altered expression of apoptotic and cytoskeletal genes. Genomics. 2004;83:281–297. | ||
Lopez AD. Assessing the burden of mortality from cardiovascular diseases. World Health Stat Q. 1993;46(2):91–96. | ||
Dickstein K, Cohen-Solal A, Filippatos G, et al. ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2008: the Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2008 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association of the ESC (HFA) and endorsed by the European Society of Intensive Care Medicine (ESICM). Eur Heart J. 2008;29(19):2388–2442. | ||
Pfeffer MA, Braunwald E. Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications. Circulation. 1990;81(4):1161–1172. | ||
Richey PA, Brown SP. Pathological versus physiological left ventricular hypertrophy: a review. J Sports Sci. 2001;16(2):129–141. | ||
Steyn K, Sliwa K, Hawken S, et al; INTERHEART Investigators in Africa. Risk factors associated with myocardial infarction in Africa: the INTERHEART Africa study. Circulation. 2005;112(23):3554–3561. | ||
Longo DL, Kasper DL, Jameson JL, Fauci AS, Hauser SL, Loscalzo J. Mortality and the Global Burden of Disease. Harrison’s Principles of Internal Medicine. 18th ed. New York: McGraw Hill; 2012. | ||
Usui S, Yeh I, Tian B, Sadoshima J. Global changes in gene expression during cardiac hypertrophy: a new direction of cardiac signaling research. J Mol Cell Cardiol. 2006;41(2):219–222. | ||
Guinamard R, Bois P. Involvement of transient receptor potential proteins in cardiac hypertrophy. Biochim Biophys Acta. 2007;1172(8):885–894. | ||
Padmasekar M, Nandiqama R, Wartenberq M, Schluter KD, Sauer H. The acute phase protein alpha2-macroglobulin induces rat ventricular cardiomyocyte hypertrophy via ERK1,2 and PI3-kinase/Akt pathways. Cardiovasc Res. 2007;75(1):118–128. | ||
Takeishi Y, Goto K, Kubota I. Role of diacylglycerol kinase in cellular regulatory processes: a new regulator for cardiomyocyte hypertrophy. Pharmacol Ther. 2007;115(3):352–359. | ||
Boengler K, Buechert A, Heinen Y, et al. Cardioprotection by ischemic postconditioning is lost in aged and STAT3-deficient mice. Circ Res. 2008;102:131–135. | ||
Hilfiker-Kleiner D, Hilfiker A, Fuchs M, et al. Signal transducer and activator of transcription 3 is required for myocardial capillary growth, control of interstitial matrix deposition, and heart protection from ischemic injury. Circ Res. 2004;95:187–195. | ||
Hilfiker-Kleiner D, Hilfiker A, Drexler H. Many good reasons to have STAT3 in the heart. Pharmacol Ther. 2005;107:131–137. | ||
Snyder M, Huang XY, Zhang JJL. Identification of novel direct Stat3 target genes for control of growth and differentiation. J Biol Chem. 2008;283:3791–3798. | ||
Fischer P, Hilfiker-Kleiner D. Survival pathways in hypertrophy and heart failure: the gp130-STAT3 axis. Basic Res Cardiol. 2007;102:279–297. | ||
Booz GW, Day JNE, Baker KM. Interplay between the cardiac renin angiotensin system and JAK-STAT signaling: role in cardiac hypertrophy, ischemia/reperfusion dysfunction, and heart failure. J Mol Cell Cardiol. 2002;34:1443–1453. | ||
Terrell AM, Crisostomo PR, Wairiuko GM, Wang M, Morrell ED, Meldrum DR. JAK/STAT/SOCS signaling circuits and associated cytokine-mediated inflammation and hypertrophy in the heart. Shock. 2006;26:226–234. | ||
Kapur NK, Musunuru K. Clinical efficacy and safety of statins in managing cardiovascular risk. Vasc Health Risk Manag. 2008;4:341–453. | ||
Balakumar P, Mahadevan N. Interplay between statins and PPARs in improving cardiovascular outcomes: a double-edged sword? Br J Pharmacol. 2012;165:373–379. | ||
Iwakura K, Ito H, Kawano S, et al. Chronic pre-treatment of statins is associated with the reduction of the no-reflow phenomenon in the patients with reperfused acute myocardial infarction. Eur Heart J. 2006;27(5):534–539. | ||
Zhao JL, Yang YJ, Cui CJ, You SJ, Gao RL. Pretreatment with simvastatin reduces myocardial no-reflow by opening mitochondrial K(ATP) channel. Br J Pharmacol. 2006;149(3):243–249. | ||
Manickavasagam S, Ye Y, Lin Y, et al. The cardioprotective effect of a statin and cilostazol combination: relationship to Akt and endothelial nitric oxide synthase activation. Cardiovasc Drugs Ther. 2007;21(5):321–330. | ||
Merla R, Ye Y, Lin Y, et al. The central role of adenosine in statin-induced ERK1/2, Akt, and eNOS phosphorylation. Am J Physiol Heart Circ Physiol. 2007;293(3):H1918–H1928. | ||
Luo JD, Xie F, Zhang WW, Ma XD, Guan JX, Chen X. Simvastatin inhibits noradrenaline-induced hypertrophy of cultured neonatal rat cardiomyocytes. Br J Pharmacol. 2001;132(1):159–164. | ||
Wu L, Zhao L, Zheng Q, et al. Simvastatin attenuates hypertrophic responses induced by cardiotrophin-1 via JAK-STAT pathway in cultured cardiomyocytes. Mol Cell Biochem. 2006;284(1–2):65–71. | ||
Liu J, Shen Q, Wu Y. Simvastatin prevents cardiac hypertrophy in vitro and in vivo via JAK/STAT pathway. Life Sci. 2008;82:991–996. | ||
Allain CC, Poon LS, Chan CS, Richmond W, Fu PC. Enzymatic determination of total serum cholesterol. Clin Chem. 1974;20(4):470–475. | ||
Fossati P, Prencipe L. Serum triglycerides determined colorimetrically with an enzyme that produces hydrogen peroxide. Clin Chem. 1982;28(1):2077–2080. | ||
Burnstein M, Selvenick HR, Morfin R. Rapid method for isolation of lipoprotein from human serum with polyanions. J Lipid Res. 1970;11:583–595. | ||
Nobert WT. Clinical Guide to Laboratory Tests. 3rd ed. Philadelphia: W.B. Saunders Company; 1995. | ||
Friedewald WT. Estimation of concentration of low-density lipoprotein cholesterol in plasma without use of the preparative ultracentrifuge. Clin Chem. 1972;18(7):499–502. | ||
Ross R. The pathogenesis of atherosclerosis. In: Braunwald E, editor. Heart Disease. 4th ed. Philadelphia, PA: W. B. Saunders Company; 1992;1106–1124. | ||
Rosamond W, Flegal K, Friday G, et al. Heart disease and stroke statistics – 2007 update. A report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2007;115:e69–e171. | ||
Molojavyi A, Lindecke A, Raupach A, Moellendorf S, Köhrer K, Gödecke A. Myoglobin-deficient mice activate a distinct cardiac gene expression program in response to isoproterenol-induced hypertrophy. Physiol Genomics. 2010;41:137–145. | ||
Ennis IL, Escudero EM, Console GM, et al. Regression of isoproterenol-induced cardiac hypertrophy by Na+/H+ exchanger inhibition. Hypertension. 2003;41:1324–1329. | ||
Sathish V, Ebenezar KK, Devaki T. Synergistic effect of nicorandil and amlodipine on tissue defense system during experimental myocardial infarction in rats. Mol Cell Biochem. 2003;243:133–138. | ||
Weihua Z, Vijayan E, Mohinder SN, Suresh KG, Naranjan SD. Role of mitogen-activated protein kinase in cardiac hypertrophy and heart failure. Exp Clin Cardiol. 2003;8:173–183. | ||
Taylor PB, Tang Q. Development of isoproterenol-induced cardiac hypertrophy. Can J Physiol Pharmacol. 1984;62:384–389. | ||
Upaganlawar A, Balaraman R. Protective effects of Lagenaria siceraria (Molina) fruit juice in isoproterenol induced myocardial infarction. Int J Pharmacol. 2010;6(5):645–651. | ||
Li H, Xie YH, Yang Q, et al. Cardioprotective effect of paeonol and danshensu combination on isoproterenol-induced myocardial injury in rats. PLoS One. 2012;7(11):e48872. | ||
Patel V, Upaganlawar A, Zalawadia R, Balaraman R. Cardioprotective effect of melatonin against isoproterenol induced myocardial infarction in rats: a biochemical, electrocardiographic and histoarchitectural evaluation. Eur J Pharmacol. 2010;644:160–168. | ||
Beznak M, Hacker P. Hemodynamics during the chronic stage of myocardial damage caused by isoproterenol. Can J Cardiol. 1964;42:269–274. | ||
Lin YC. Hemodynamics in the rat with isoproterenol induced cardiac hypertrophy. Res Commun Chem Pathol Pharmacol. 1973;6:213–220. | ||
Baldwin KM, Ernst SB, Mullin WJ, Schrader LF, Herrick RE. Exercise capacity and cardiac function of rats with drug-induced cardiac enlargement. J Appl Physiol. 1982;52:591–595. | ||
Vassalo DV, Vasquez EC, Cabral AM. Contractile performance of papillary muscles of renovascular hypertensive and isoproterenol pretreated rats. Pharmacol Res Commun. 1988;20:61–72. | ||
Bowling N, Wyss VL, Gengo PJ, Utterback B, Kauffman RF, Hayes IN. Cardiac inotropic responses to calcium and foskolin are not altered by prolonged isoproterenol infusion. Eur J Pharmacol. 1990;187:155–164. | ||
Stein B, Bartel S, Kirchhefer U, et al. Relation between contractile function and regulatory cardiac proteins in hypertrophied hearts. Am J Physiol. 1996;270:H2021–H2028. | ||
Cihák R, Kolar F, Pelouch V, Prochazka J, Ostadal B, Widimsky J. Functional changes in the right and left ventricle during development of cardiac hypertrophy and after its regression. Cardiovasc Res. 1992;26:845–850. | ||
Arthur GD, Belcastro AN. A calcium stimulated cysteine protease involved in isoproterenol induced cardiac hypertrophy. Mol Cell Biochem. 1997;176:241–248. | ||
Tang Q, Taylor PB. Altered contractile function in isoproterenol-induced hypertrophied rat heart. J Hypertens. 1996;14:751–757. | ||
Papparella I, Ceolotto G, Montemurro D, et al. Green tea attenuates angiotensin II induced cardiac hypertrophy in rats by modulating reactive oxygen species production and the Src/epidermal growth factor receptor/Akt signaling pathway. J Nutr. 2008;138(9):1596–1601. | ||
Prince PS, Rajadurai M. Preventive effect of Aegle marmelos leaf extract on isoprenaline induced myocardial infarction in rats: biochemical evidence. J Pharm Pharmacol. 2005;57:1353–1357. | ||
Adameova A, Harcarova A, Matejikova J, et al. Simvastatin alleviates myocardial contractile dysfunction and lethal ischemic injury in rat heart independent of cholesterol-lowering effects. Physiol Res. 2009;58:449–454. | ||
Morikawa-Futamatsu K, Adachi S, Maejima Y, et al. HMG-CoA reductase inhibitor fluvastatin prevents angiotensin II-induced cardiac hypertrophy via Rho kinase and inhibition of cyclin D1. Life Sci. 2006;79(14):1380–1390. | ||
Dechend R, Fiebeler A, Park JK, et al. Amelioration of angiotensin II-induced cardiac injury by a 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitor. Circulation. 2001;104(5):576–581. | ||
Apple FS. Tissue specificity of cardiac troponin I, cardiac troponin T and creatine kinase-MB. Clin Chim Acta. 1999;284:151–159. | ||
Tsutamoto T, Hisanaga T, Wada A, et al. Interleukin-6 spillover in the peripheral circulation increases with the severity of heart failure, and the high plasma level of interleukin-6 is an important prognostic predictor in patients with congestive heart failure. J Am Coll Cardiol. 1998;31:391–398. | ||
Orús J, Roig E, Perez-Villa F, et al. Prognostic value of serum cytokines in patients with congestive heart failure. J Heart Lung Transplant. 2000;19:419–425. | ||
Rattazzi M, Puato M, Faggin E, Bertipaglia B, Zambon A, Pauletto P. C reactive protein and interleukin-6 in vascular disease: culprits or passive bystanders? J Hypertens. 2003;21:1787–1803. | ||
Manginas A, Bei E, Chaidaroglou A, et al. Peripheral levels of matrix metalloproteinase-9, interleukin-6, and C-reactive protein are elevated in patients with acute coronary syndromes: correlations with serum troponin I. Clin Cardiol. 2005;28:182–186. | ||
Shu J, Ren N, Du JB, Zhang M, Cong HL, Huang TG. Increased levels of interleukin-6 and matrix metalloproteinase-9 are of cardiac origin in acute coronary syndrome. Scand Cardiovasc J. 2007;41:149–154. | ||
Yamauchi-Takihara K, Ihara Y, Ogata A, Yoshizaki K, Azuma J, Kishimoto T. Hypoxic stress induces cardiac myocyte-derived interleukin-6. Circulation. 1995;91:1520–1524. | ||
Hirota H, Yoshida K, Kishimoto T, Taga T. Continuous activation of Gp130, a signal-transducing receptor component for interleukin 6-related cytokines, causes myocardial hypertrophy in mice. Proc Natl Acad Sci U S A. 1995;92:4862–4866. | ||
Gwechenberger M, Mendoza LH, Youker KA, et al. Cardiac myocytes produce interleukin-6 in culture and in viable border zone of reperfused infarctions. Circulation. 1999;99:546–551. | ||
Cha HN, Choi JH, Kim YW, Kim JY, Ahn MW, Park SY. Metformin inhibits isoproterenol-induced cardiac hypertrophy in mice. Korean J Physiol Pharmacol. 2010;14:377–384. | ||
Maihçfner C, Charalambous MP, Bhambra U, Lightfoot T, Geisslinger G, Gooderham NJ. Expression of cyclooxygenase-2 parallels expression of interleukin-1beta, interleukin-6 and NF kappa B in human colorectal cancer. Carcinogenesis. 2003;24:665–671. | ||
Pikarsky E, Porat RM, Stein I, et al. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431:461–466. | ||
Lee SJ, Lim KT. Chemopreventive effect of plant originated glycoprotein on colitis-mediated colorectal cancer in A/J mice. J Biomed Sci. 2008;15:111–121. | ||
Chen X, Zeng S, Zou J, et al. Rapamycin attenuated cardiac hypertrophy induced by isoproterenol and maintained energy homeostasis via inhibiting NF-κB activation. Mediators Inflamm. 2014;2014:868753. | ||
Serrano AL, Baeza-Raja B, Perdiguero E, Jardi M, Munoz-Cinoves P. Interleukin-6 is an essential regulator of satellite cell-mediated skeletal muscle hypertrophy. Cell Metab. 2008;7:33–44. | ||
Chen LM, Kuo WW, Yang JJ, et al. Eccentric cardiac hypertrophy was induced by long term intermittent hypoxia in rats. Exp Physiol. 2007;92:409–416. | ||
Oshima Y, Fujio Y, Nakanishi T, et al. STAT3 mediates cardioprotection against ischemia/reperfusion injury through metallothionein induction in the heart. Cardiovasc Res. 2005;65(2):428–435. | ||
Barry SP, Townsend PA, Latchman DS, Stephanou A. Role of the JAK-STAT pathway in myocardial injury. Trends Mol Med. 2007;13(2):82–89. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.