")
Back to Journals » Clinical Pharmacology: Advances and Applications » Volume 7
Severe iatrogenic bradycardia related to the combined use of beta-blocking agents and sodium channel blockers
Authors Kawabata M, Yokoyama Y, Sasaki T, Tao S, Ihara K, Shirai Y, Sasano T, Goya M, Furukawa T, Isobe M, Hirao K
Received 5 November 2014
Accepted for publication 9 December 2014
Published 16 February 2015 Volume 2015:7 Pages 29—36
DOI https://doi.org/10.2147/CPAA.S77021
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Arthur E. Frankel
Mihoko Kawabata,1 Yasuhiro Yokoyama,1 Takeshi Sasaki,1 Susumu Tao,1 Kensuke Ihara,1 Yasuhiro Shirai,1 Tetsuo Sasano,2 Masahiko Goya,1 Tetsushi Furukawa,3 Mitsuaki Isobe,4 Kenzo Hirao1
1Heart Rhythm Center, Tokyo Medical and Dental University, Tokyo, Japan; 2Department of Biofunctional Informatics, Graduate School of Health Care Sciences, Tokyo Medical and Dental University, Tokyo, Japan; 3Department of Bio-informational Pharmacology, Medical Research Institute, Tokyo Medical and Dental University, Tokyo, Japan; 4Department of Cardiovascular Medicine, Tokyo Medical and Dental University, Tokyo, Japan
Purpose: Drug-induced bradycardia is common during antiarrhythmic therapy; the major culprits are beta-blockers. However, whether other antiarrhythmic drugs are also a significant cause of this, alone or in combination with beta-blockers, is not well known.
Methods: We retrospectively investigated the records of all patients hospitalized at our institution for drug-related bradycardia from the years 2004 to 2012. Patients with cardiac disease and electrolytic or hormonal abnormalities that could cause bradyarrhythmias were excluded.
Results: Eight patients were identified (mean age, 79±5 years; range, 71–85 years; 6 women). Three patients were taking only beta-blockers (hereafter referred to as the BB group), while five patients were on both beta-blockers and Na channel blockers (hereafter referred to as the BB + Na group). Heart rates ranged from 20~49 beats/minute on arrival. The initial electrocardiogram showed sinus bradycardia (n=6) or sinus arrest with escape beats (n=2). QRS duration was 80–100 ms. The clinical presentation of the BB + Na group was considerably worse than that of the BB group, and included cardiogenic shock and heart failure. Four of the BB + Na patients had been on their medications for over 300 days. The BB group recovered solely with drug discontinuation, while 4 of the 5 patients in the BB + Na group needed additional treatments, such as intravenous administration of atropine or adrenergic agonist and temporary pacing. Bradycardia did not recur during follow-up (median, 687 days).
Conclusion: Although wide QRS ventricular tachyarrhythmia is a better known proarrhythmic effect of Na channel blockers, life-threatening bradycardia may also occur in combination with beta-blockers in the elderly, even months after the start of medication, and at plasma concentrations that do not prolong QRS width.
Keywords: proarrhythmia, elderly, QRS duration
Introduction
Drug-induced bradycardia is an important and common but poorly characterized clinical problem. Clinically, severe bradycardia can be induced during therapy with beta-adrenergic-blockers (beta-blockers), non-dihydropyridine calcium channel antagonists, and though rare, with any of the other classes of antiarrhythmic drugs.1–8
Beta-blockers are the most common culprits in causing bradycardia, interfering with slow action potential generation and atrioventricular conduction. They target the sympathetic nervous system and have negative chronotropic and inotropic effects. In chronic heart failure, beta-blocker therapy protects the heart against cardiotoxic overstimulation by the catecholamines, improving left ventricular function and performance. Such therapy also improves survival and reduces risk of arrhythmias (sudden cardiac death). On the other hand, beta-blockers are contraindicated in acute heart failure because they decrease cardiac output acutely. Their negative chronotropic effects could slow down the heart rhythm to an unexpected degree.9 The purpose of this study was to examine drugs that cause marked bradycardia.
Methods
Study subjects
In this retrospective study, we reviewed all patients that visited the emergency department of our hospital, the Tokyo Medical and Dental University, or were hospitalized between January 2004 and August 2012 with a diagnosis of emergent bradycardia associated with drug side effects. Patients were excluded if their bradycardia was attributable to cardiac disease, eg, acute myocardial infarction, vasovagal syncope, cardiac myopathy, myocarditis, or failure of a previously implanted pacemaker device. We also excluded patients who had electrolyte imbalances or hormonal abnormalities. The cause-and-effect relationship between medication use and bradycardia was determined from the response to drug withdrawal. This study was approved by our institutional ethics committee.
Follow-up
Medications suspected of being the cause of bradycardia were discontinued in all subjects soon after admission. Whether or not clinically significant and symptomatic bradycardia recurred after discharge was assessed during the follow-up period. Because of the small number of patients, we did not conduct statistical analyses. All values are expressed as mean ± standard deviation.
Results
Eight patients were found who fulfilled our criteria for inclusion. Baseline characteristics of the study population are shown in Table 1. The mean age was 79±5 years (range, 71–85), and six patients were women. With regard to antiarrhythmic medications, three patients were taking beta-blockers only (in the BB group), while five patients were on both beta-blockers and Na channel blockers (in the BB + Na group). The beta-blockers were carvedilol (n=3), metoprolol (n=2), betaxolol, bisoprolol, and carteolol (n=1 each). The Na channel blockers were pilsicainide, which is a pure Na channel blocker (n=2), and disopyramide, flecainide, mexiletine, and cibenzoline (n=1 each). One patient was also on a calcium channel antagonist, but it was a dihydropyridine that generally had no chronotropic effects.10 The duration that the patients had been on these medications before their bradycardic event ranged from 6 hours to 6 years (median, 332.5 days). The indications for treatment with these drugs were tachyarrhythmia (n=7), hypertension (n=5), and stable angina pectoris (n=1).
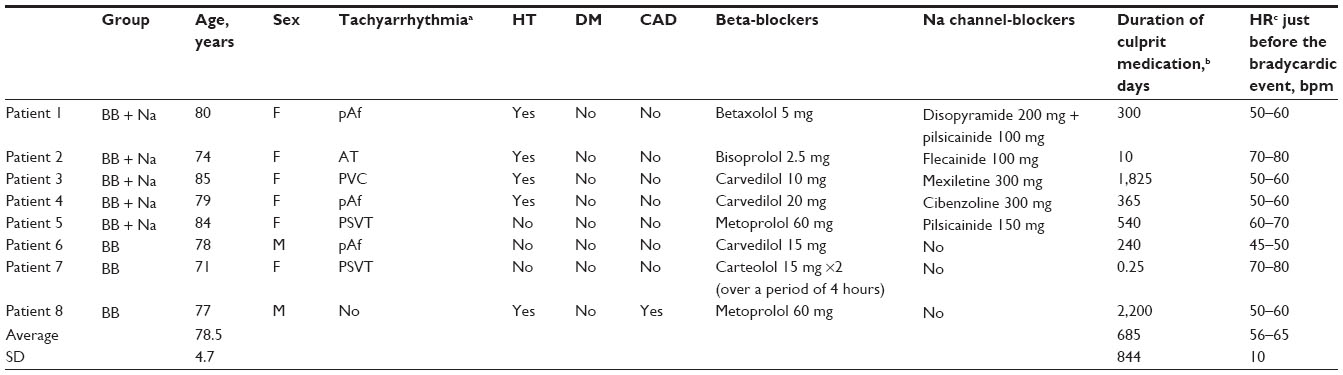 | Table 1 Baseline characteristics of the study population |
The most common presenting symptom was syncope (Table 2). The patients’ heart rates ranged from 20–49 beats/minute on arrival. The initial electrocardiogram (ECG) showed sinus bradycardia in six patients and sinus arrest with escape beats in two patients (Figures 1–4). The mean QRS duration was 91.5±8.2 ms (range, 80–100 ms). Two-dimensional echocardiography was performed in all patients and showed a mean left ventricular ejection fraction of 72±9% (range, 60–84%). The clinical presentation of the BB + Na group was considerably worse than that of the BB group, and included three patients with cardiogenic shock and one patient with heart failure. All patients in the BB group presented with syncope and recovered solely with drug discontinuation. In contrast, in the BB + Na group, only one patient recovered solely with drug discontinuation, and the remaining four patients needed supportive medical treatment that included intravenous administration of atropine or adrenergic agonist; two of these patients required temporary pacing as well. Of these two patients, one also required mechanical ventilation and needed transfer to the intensive care unit (Patient 1). The duration for full recovery after medication withdrawal was longer in the BB + Na group (11.3±8.6 hours in the BB + Na group vs 5.2±2.5 hours in the BB group).
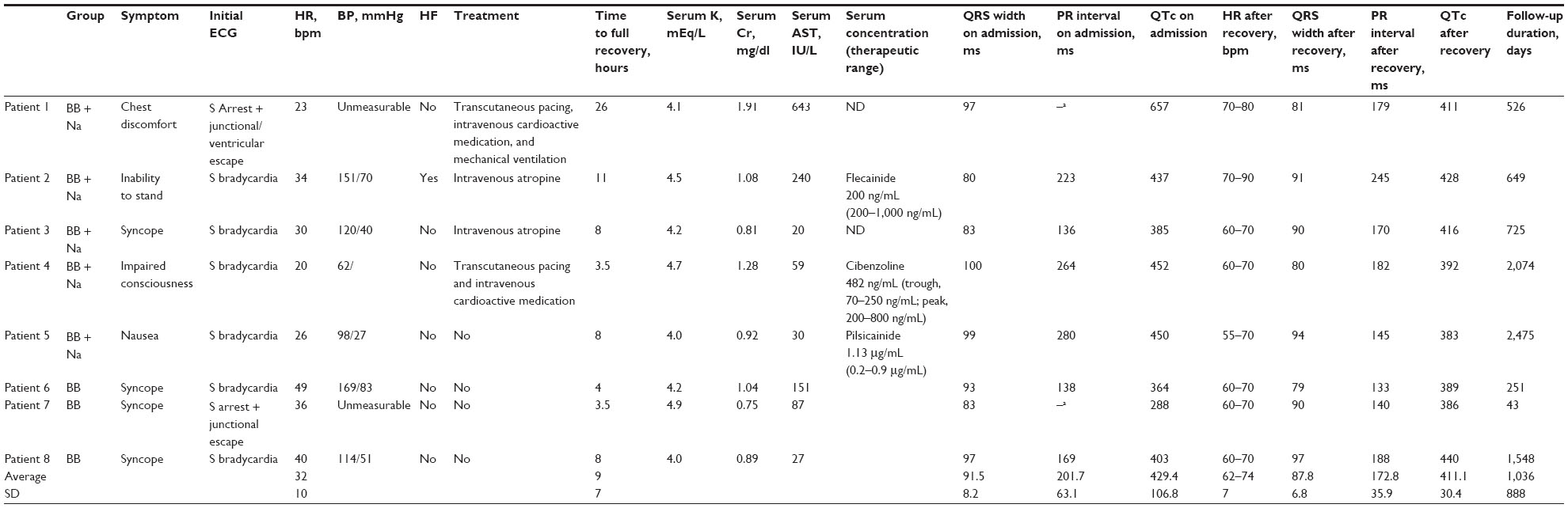 | Table 2 Clinical presentation on arrival and after recovery |
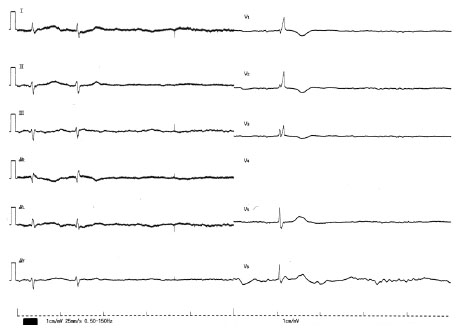 | Figure 1 The 12-lead ECG on admission in patient 1. The 12-lead ECG shows sinus arrest and ventricular and junctional escape beats with a pause of 4.7 seconds. The QRS width of the junctional escape beat was 97 ms. The V4 lead was disconnected. |
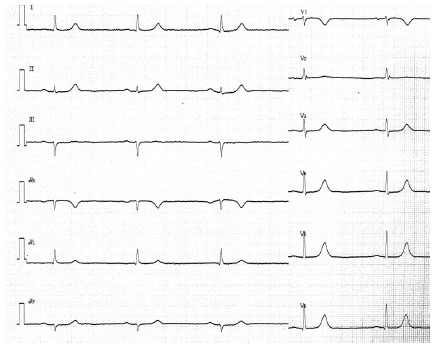 | Figure 2 The 12-lead ECG on admission in patient 2. The 12-lead ECG shows sinus bradycardia of 34 beats/minute with QRS width of 80 ms. |
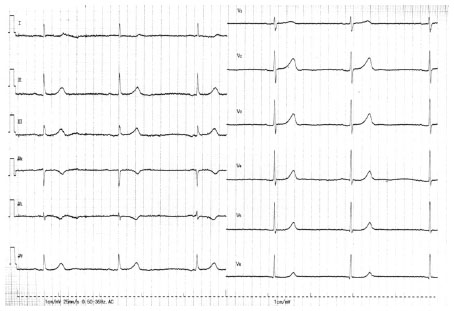 | Figure 3 The 12-lead ECG on admission in patient 4. The 12-lead ECG shows sinus bradycardia of 20 beats/minute with QRS width of 100 ms. |
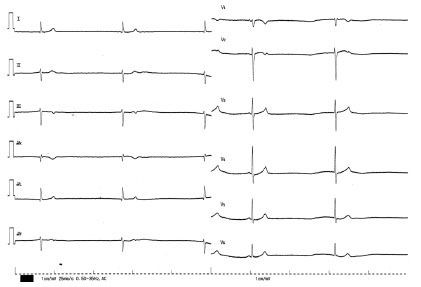 | Figure 4 The 12-lead ECG on admission in patient 7. The 12-lead ECG shows sinus arrest and junctional escape beats of 36 beats/minute with QRS width of 93 ms. The 4th QRS complex is thought of as a sinus beat with the preceding P wave. |
Serum concentration of the Na channel blockers was measured in three patients. The serum concentration exceeded the therapeutic range in one of those three patients. The median follow-up duration was 687 days (range, 43–2,475 days). Holter monitoring was performed in two patients during the follow-up period, resulting in no remarkable findings, including bradycardia. None of the patients developed bradycardia again during the follow-up period. Therefore, no permanent pacemaker implantation was performed.
At our hospital, beta-blockers were prescribed to 2,964 patients and Na channel blockers to 946 patients during the past year. Of these, 415 patients took both. In this study, we found eight patients demonstrating life-threatening drug-induced bradycardia over 8 years. According to a rough estimate, the annual incidence of severe drug-induced bradycardia would be 0.0015% in patients taking beta-blockers alone and 0.15% in those taking the combination of Na channel- and beta-blockers.
We describe in more detail patient 1, who had the worst clinical presentations of the patients in our study. Patient 1 was an 80-year-old woman, who was referred to our hospital for chest discomfort lasting 5 hours. She had been diagnosed with paroxysmal atrial fibrillation (AF) and hypertension. As it had been difficult to control her paroxysms of AF, various drugs had been added incrementally. When she was admitted, she had been taking betaxolol 5 mg, disopyramide 200 mg, pilsicainide 100 mg, and azelnidipine 8 mg for 10 months.
At the time of admission, the patient was drowsy and her blood pressure was unmeasurable. Her last ECG recorded at a prior routine hospital visit had shown sinus rhythm of 50–60 beats/minute, while ECG on admission demonstrated sinus arrest and ventricular/junctional escape beats with pauses of 4.7 seconds (Figure 1). Mechanical ventilation and transcutaneous pacing were performed immediately and dopamine, noradrenaline, adrenaline, and atropine sulfate were administered intravenously. Laboratory examination revealed creatinine levels at 1.91 mg/dL, aspartate aminotransferase at 643 IU/L, alanine transaminase at 396 IU/L, and K (potassium) at 4.1 mEq/L. Echocardiogram showed no specific findings with normal left ventricular contraction. Emergent coronary angiogram disclosed no stenosis. All drugs were discontinued. Her ECG returned to sinus rhythm with a heart rate of 60 beats/minute 2 hours later. Her blood pressure also improved to 114/62 mmHg. All therapies were terminated 26 hours later. Her renal and liver function indices gradually returned to normal range. Her heart rate returned to approximately 80 beats/minute (Figure 5), at which point paroxysmal AF with rapid ventricular response recurred. Aprindine 20 mg and verapamil 80 mg were therefore started with caution. No bradycardia recurred during follow-up.
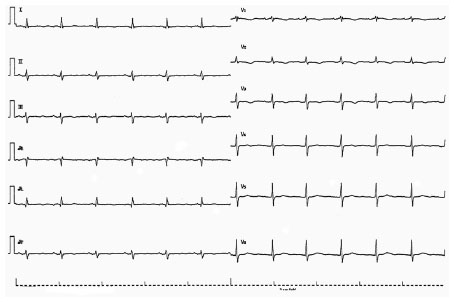 | Figure 5 The 12-lead ECG recorded 2 days after initial recording in patient 1. The 12-lead ECG shows sinus rhythm with a heart rate of 73 beats/minute. The QRS width was 81 ms. |
Discussion
Major findings
We present data on eight patients who had suffered clinically significant and symptomatic drug-induced bradycardia, responsible for the development of syncope, cardiogenic shock, and heart failure. All of our patients were over 70 years old and taking beta-blockers at the time of presentation. Their ECGs exhibited heart rates of 20–49 beats/minute, sinus bradycardia or sinus arrest with escape rhythms, and normal QRS duration. Four of the five patients who were also on Na channel-blockers required more than drug discontinuation; they required cardioactive medication, temporary pacing, and in one patient, mechanical ventilation. None of the patients had recurrence of bradycardia during the follow-up period.
Bradycardia due to proarrhythmic effects
Proarrhythmia is a potential problem that may contribute to increased mortality during antiarrhythmic therapy. Bradycardia is a possible side effect of all classes of antiarrhythmic drugs.1,2 However, it is known that drug-induced bradycardia usually does not appear in patients with normal sinus node function and normal atrioventricular conduction.11,12 Previous reports found that advanced age, combined drug therapy, the period during the initial 24 hours after initiation of drug therapy, decreased systolic performance, and female were predictors of proarrhythmia,1,2 while preexisting conduction disturbance,11 ventricular arrhythmia,11 and combined drug therapy5 were associated with drug-induced bradycardia in particular.
Beta-blockers cause bradycardia by antagonizing the actions of catecholamines produced by the sympathetic nerves at the cell receptor. Five of our patients were on the beta-blockers metoprolol or carvedilol. Emergent bradycardia associated with the use of metoprolol and carvedilol in clinical practice was 18 per 1000 person-years in one study.13 In patients with heart failure, incidence of life-threatening bradycardia induced by carvedilol was 0.9% (Carvedilol Prospective Randomized Cumulative Survival [COPERNICUS] study),14 while incidence of bradycardia necessitating withdrawal of metoprolol was very similar at 0.8% in the Metoprolol controlled release/extended release Randomized Intervention Trial (MERIT-HF).15 None of our patients had heart failure or any other structural heart disease.
The Na channel-blockers our patients were taking included disopyramide, pilsicainide, mexiletine, flecainide, and cibenzoline. During therapy with antiarrhythmic drugs, the incidence of significant drug-induced bradycardia has been reported as 0%–32%,1–6,16,17 and it is more common with amiodarone (1%–17%),3,5 sotalol (1%–16.7%),3,6 propafenone (10.9%),3 or flecainide (14%)3 than with procainamide (6.3%),3 quinidine (7.3%),3 or disopyramide (5%).3 In a series of patients undergoing drug trials for AF and atrial flutter, the risk of a bradycardic adverse event was 3.5% during the 10-day monitoring period.5 It was 6.8% among those who received amiodarone. The antiarrhythmic drugs used in that study were procainamide, quinidine, disopyramide, propafenone, flecainide, sotalol, and amiodarone. Amiodarone use and the use of any rate-controlling medication of beta-blocker, calcium channel-blocker, or digoxin were associated with bradycardia.5 In another report of patients undergoing drug trials for AF, bradyarrhythmia was the most common manifestation of proarrhythmia, occurring in 7.9% of the drug trials.3 Among the bradyarrhythmias, 53% were classified as clinically severe. Interestingly, no differences in the use of beta-blockers were found between trials that did and did not have severe bradyarrhythmias.3
To our knowledge, ours is the first report that focuses on bradycardia due to the combined use of Na channel- and beta-blockers. There were no significant bradycardic events caused by Na channel-blockers alone in our report. As they have potential for directly suppressing sinus nodes, as described in the “Mechanism of sinus node dysfunction caused by sodium channel blockers” section, the combined use of Na channel- and beta-blockers would enhance the effects of both medications.
Mechanism of sinus node dysfunction caused by sodium channel blockers
The pacemaker activity of the sinus node cells arises from spontaneous diastolic depolarization (the pacemaker potential) that relies on the concerted action of many cellular elements and processes, including ion channels, ion pumps and exchangers, cyclic adenosine monophosphate and Ca2 homeostasis, and a variety of modulatory factors. It is known that Na channel blockers may suppress sinus node function.7,18 However, the ionic mechanisms responsible for this suppression have not been conclusively defined yet. We discuss several possible mechanisms.
Ca channel blockade by Na channel-blockers
In dogs, cibenzoline had no effect, but produced sinus bradycardia when combined with beta-blockers, suggesting calcium channel antagonism.19 One other study suggested that mexiletine had considerable Ca channel blocking action at therapeutic concentration,20 while pilsicainide, despite being a pure Na channel blocker, inhibited Ca current at a plasma concentration higher than is therapeutic in vitro.21
Na currents may be necessary for conduction of sinus node impulses
Studies show that the sinus node action potential is initiated in the center of the node (Ca channel-dependent) and conducts to the surrounding atrial muscle via the peripheral regions (Na channel-dependent).22,23 Na channel block with tetrodotoxin has been shown to cause sinus node dysfunction, ie, a slowing of sinus node pacemaking, a slowing of sinus node conduction, and sinus node exit block.24,25
Inhibition of ryanodine receptor-mediated Ca2+ release by Na channel-blockers
Spontaneous local submembrane Ca2+ release through the ryanodine receptor (RyRs) in the sinus node cells activates an inward Na–Ca2+ exchange current, which contributes to accelerating diastolic depolarization rate.26 Flecainide and propafenone have been reported to suppress such Ca2+ release by inhibiting the ryanodine receptor, which may contribute to a decelerated pacemaking rate.27,28 Aprindine and cibenzoline have been reported to inhibit Na–Ca2+ exchange current in a concentration-dependent manner in vitro, which would likewise result in suppression of pacemaking rate.29,30
QRS duration, sex, age, and drug duration
It is well-known that Na channel-blockers can cause ventricular arrhythmias, namely, sine-wave-shaped ventricular tachycardia with wide QRS duration. QRS width is commonly used as a marker of magnitude of Na channel-blocking effects. Therefore, we measured QRS duration in our patients’ admission ECGs and after antiarrhythmic drug discontinuation. Although we found narrowing of QRS in some patients following drug discontinuation, the absolute value of QRS duration at admission was normal and not suggestive of Na channel-blocker overdose. We are aware of only one study reporting QRS width lengthening during drug-induced bradycardia, for the drug pilsicainide.7
Six of our eight patients were female. The number of patients was too small to say whether females were more likely to suffer from drug-induced bradycardia, but other known sex differences do exist in proarrhythmia, eg, for torsade de pointes.31 In torsade de pointes, the sex difference arises from differences in potassium current density. A significant association between amiodarone-associated bradycardia and female sex has been reported,9 but we know of no sex difference studies for Na current density.
It is well-known that older age is a risk factor for proarrhythmia.1,2 In our series, all patients were over 70 years old. This is likely due to several factors: increased fibrosis and a decrease of pacemaker cells in the nodal area, likelihood of being on multiple drugs, and diminished mental capacity leading to poor adherence. Many of our patients were taking antiarrhythmic medications for tachyarrhythmia that needed to be controlled. It may be important to consider non-pharmacological treatment for tachycardia, even in the elderly.
Proarrhythmia is said to occur usually within several days of beginning a drug or increasing the dose.32 However, most of our patients exhibited bradycardia only after long-term drug treatment. Of the five BB+Na group patients, there was one patient who had been taking their medication for 10 days, but the other four patients had been taking their medication for 300 days or more. The occurrence of proarrhythmia later in drug therapy might suggest progression of disease or may result from other precipitating factors. However, none of our patients had any identifiable precipitating causes of their bradycardia. Furthermore, their symptoms were of rapid onset. Therefore, it is not clear what signs the physician could have been watching for in predicting and preventing bradyarrhythmia.
Limitations
The limitations of this study were several. First, this is a retrospective study done at a single site with small sample size. Second, not all patients underwent evaluation of sinus node function by EPS or Holter monitoring. Patients might have had asymptomatic bradycardia that would have been identified during 24-hour ECG monitoring. Although they might have been predisposed to latent sinus node dysfunction, their sinus rates were within normal limits both before start and after discontinuation of their medications. Therefore, it is thought that their severe bradycardia was newly caused by antiarrhythmic drugs. In any case, they need careful and continuous follow-up for sinus node function. Third, the plasma concentration of Na channel-blockers was not measured in all patients. The exact plasma concentrations of drugs that might cause significant bradycardia are unidentified. It is also unknown whether serum concentrations necessarily correlate with considerable bradycardia.
Conclusion
Although severe bradycardia due to proarrhythmic effects of Na channel-blockers is infrequent, it may occur in combination with beta-blockers, causing a possibly fatal outcome. We found this to occur with Na channel-blockers at a plasma concentration that did not prolong the QRS width. Caution should be exercised in prescribing, especially in elderly patients.
Disclosure
The authors report no conflicts of interest in this work.
References
Friedman PL, Stevenson WG. Proarrhythmia. Am J Cardiol. 1998; 82(8A):50N–58N. | |
Zipes DP. Proarrhythmic effect of antiarrhythmic drugs. Am J Cardiol. 1987;59(11):E26–E31. | |
Maisel WH, Kuntz KM, Reimond SC, et al. Risk of initiating antiarrhythmic drug therapy for atrial fibrillation in patients admitted to a university hospital. Ann Intern Med. 1997;127(4):281–284. | |
Stanton MS, Prystowsky EN, Fineberg NS, Miles WM, Zipes DP, Heger JJ. Arrhythmogenic effects of antiarrhythmic drugs: A study of 506 patients treated for VT or fibrillation. J Am Coll Cardiol. 1989;14(1):209–215. | |
Hauser TH, Pinto DS, Josephson ME, Zimetbaum P. Safety and feasibility of a clinical pathway for the outpatient initiation of antiarrhythmic medications in patients with atrial fibrillation or atrial flutter. Am J Cardiol. 2003;91(12):1437–1441. | |
Chung MK, Schweikert RA, Wilkoff BL, et al. Is hospital admission for initiation of antiarrhythmic therapy with sotalol for atrial fibrillation required? J Am Coll Cardiol. 1998;32(1):169–176. | |
Toeda T, Susa R, Saigawa T, et al. A case of sinus pause due to the proarrhythmia of pilsicainide. Jpn Heart J. 2000;41(3):405–410. | |
Lee JH, Ryu HM, Bae MH, et al. Prognosis and natural history of drug-related bradycardia. Korean Circ J. 2009;39(9):367–371. | |
Lymperopoulos A, Rengo G, Koch WJ. Adrenergic nervous system in heart failure: pathophysiology and therapy. Circ Res. 2013;113(6):739–753. | |
Taira N. Differences in cardiovascular profile among calcium antagonists. Am J Cardiol. 1987;59(3):24B–29B. | |
Essebag V, Hadjis T, Platt RW, Pilote L. Amiodarone and the risk of bradyarrhythmia requiring permanent pacemaker in elderly patients with atrial fibrillation and prior myocardial infarction. J Am Coll Cardiol. 2003;41(2):249–254. | |
Bigger JT Jr, Reiffel JA. Sick sinus syndrome. Annu Rev Med. 1979; 30(1):91–118. | |
Shin J, Pharm D, Pletcher M. Risk of emergent bradycardia associated with the use of carvedilol and metoprolol in routine clinical practice. Am J Med. 2013;126(9):805–810. | |
Krum H1, Roecker EB, Mohacsi P, et al; Carvedilol Prospective Randomized Cumulative Survival (COPERNICUS) Study Group. Effects of initiating carvedilol in patients with severe chronic heart failure: results from the COPERNICUS Study. JAMA. 2003;289(6):712–718. | |
Hjalmarson A, Goldstein S, Fagerberg B, et al. Effects of controlled- release metoprolol on total mortality, hospitalizations, and well-being in patients with heart failure. JAMA. 2000;283(10):1295–1302. | |
Israel CW, Ehrlich JR, Barold SS, Hohnloser SH. Treatment of tachyarrhythmias with pacing and antiarrhythmic drugs. In: Israel CW, Barold SS, editors. Advances in the Treatment of Atrial Tachyarrhythmias: Pacing, Cardioversion, and Defibrillation. Armonk: Futura; 2002:305–323. | |
Ovsyshcher IE, Barold SS. Drug induced bradycardia: to pace or not to pace? Pacing Clin Electrophysiol. 2004;27(8):1144–1147. | |
Ishikawa T, Sumita S, Kimura K, et al. Sinus node recovery time assessment by the overdrive suppression test employing an intravenous injection of disopyramide phosphate. Europace. 2000;2(1):54–59. | |
Sassine A, Massé C, Brugada J, et al. Evidence for a class 4 effect of cibenzoline “in vivo”. Arch Int Pharmacodyn Ther. 1987;287(1):78–88. | |
Ono K, Kiyosue T, Arita M. Comparison of the inhibitory effects of mexiletine and lidocaine on the calcium current of single ventricular cells. Life Sci. 1986;39(16):1465–1470. | |
Inomata N, Ishihara T, Akaike N. SUN 1165: a new antiarrhythmic Na current blocker in ventricular myocytes of guinea-pig. Comp Biochem Physiol C. 1987;87(2):237–243. | |
Bleeker WK, Mackaay AJ, Masson-Pévet M, Bouman LN, Becker AE. Functional and morphological organization of the rabbit sinus node. Circ Res. 1980;46(1):11–22. | |
Kodama I, Nikmaram MR, Boyett MR, Suzuki R, Honjo H, Owen JM. Regional differences in the role of the Ca2+ and Na+ currents in pacemaker activity in the sinoatrial node. Am J Physiol. 1997;272(6 Pt 2):H2793–H2806. | |
Maier SK, Westenbroek RE, Yamanushi TT, et al. An unexpected requirement for brain-type sodium channels for control of heart rate in the mouse sinoatrial node. Proc Natl Acad Sci U S A. 2003;100(6):3507–3512. | |
Lei M, Jones SA, Liu J, et al. Requirement of neuronal- and cardiac-type sodium channels for murine sinoatrial node pacemaking. J Physiol. 2004;559(Pt 3):835–848. | |
Bogdanov KY, Vinogradova TM, Lakatta EG. Sinoatrial nodal cell ryanodine receptor and Na+-Ca2+ exchanger: molecular partners in pacemaker regulation. Circ Res. 2001;88(12):1254–1258. | |
Watanabe H, Chopra N, Laver D, et al. Flecainide prevents catecholaminergic polymorphic ventricular tachycardia in mice and humans. Nat Med. 2009;15(4):380–383. | |
Hwang HS, Hasdemir C, Laver D, et al. Inhibition of cardiac Ca2+ release channels (RyR2) determines efficacy of class I antiarrhythmic drugs in catecholaminergic polymorphic ventricular tachycardia. Circ Arrhythm Electrophysiol. 2011;4(2):128–135. | |
Kimura J, Watanabe Y, Li L, Watano T. Pharmacology of Na+/Ca2+ exchanger. Ann NY Acad Sci. 2002;976(1):513–519. | |
Watanabe Y, Koide Y, Kimura J. Topics on the Na+/Ca2+ exchanger: pharmacological characterization of Na+/Ca2+ exchanger inhibitors. J Pharmacol Sci. 2006;102(1):7–16. | |
Lehmann MH, Hardy S, Archibald D, Quart B, MacNeil DJ. Sex difference in risk of torsade de pointes with d,l-sotalol. Circulation. 1996;94(10):2534–2541. | |
Minardo JD, Heger JJ, Miles WM, Zipes DP, Prystowsky EN. Clinical characteristics of patients with ventricular fibrillation during antiarrhythmic drug therapy. N Engl J Med. 1988;319(5):257–262. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.