")
Back to Journals » OncoTargets and Therapy » Volume 7
Role of calcium-dependent protein kinases in chronic myeloid leukemia: combined effects of PKC and BCR-ABL signaling on cellular alterations during leukemia development
Authors Mencalha A, Corrêa S, Abdelhay E
Received 18 March 2014
Accepted for publication 29 April 2014
Published 8 July 2014 Volume 2014:7 Pages 1247—1254
DOI https://doi.org/10.2147/OTT.S64303
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
André L Mencalha,1 Stephany Corrêa,2 Eliana Abdelhay2
1Biophysics and Biometry Department, Roberto Alcântara Gomes Biology Institute, Rio de Janeiro's State University (UERJ), Rio de Janeiro, Brazil; 2Bone Marrow Transplantation Unit (CEMO), National Cancer Institute (INCA), Rio de Janeiro, Brazil
Abstract: Calcium-dependent protein kinases (PKCs) function in a myriad of cellular processes, including cell-cycle regulation, proliferation, hematopoietic stem cell differentiation, apoptosis, and malignant transformation. PKC inhibitors, when targeted to these pathways, have demonstrated efficacy against several types of solid tumors as well as leukemia. Chronic myeloid leukemia (CML) represents 20% of all adult leukemia. The aberrant Philadelphia chromosome has been reported as the main cause of CML development in hematopoietic stem cells, due to the formation of the BCR-ABL oncogene. PKCs and BCR-ABL coordinate several signaling pathways that are crucial to cellular malignant transformation. Experimental and clinical evidence suggests that pharmacological approaches using PKC inhibitors may be effective in the treatment of CML. This mini review summarizes articles from the National Center for Biotechnology Information website that have shown evidence of the involvement of PKC in CML.
Keywords: PKC signaling, chronic myeloid leukemia, pharmacological inhibitors, malignant transformation
Introduction
The calcium-dependent protein kinase (PKC) superfamily represents a group of eleven serine/threonine-kinase isoforms.1 Initially thought to be exclusively calcium (Ca2+)- and diacylglycerol (DAG)-dependent,2 PKCs can also be regulated by different lipid secondary messengers, including phosphatidylserine (PS) and sphingolipids. PKCs are subdivided into three classes that comprise different isoforms with particular features: 1) the classic PKCs (PKCα, PKCβI, PKCβII, PKCγ), which are Ca2+-dependent and activated by both PS and DAG; 2) the novel PKCs (PKCδ, PKCε, PKCη, PKCμ, PKCθ), which are only regulated by PS and DAG; and 3) atypical PKCs (PKCζ, PKCί/λ), which are Ca2+- and DAG-independent. These different classes are grouped according to protein-domain similarities and/or different activation signals (Figure 1).3
 | Figure 1 Scheme of protein kinase C (PKC) superfamily. This family comprises different isoforms grouped into three classes (classic, novel, and atypical) that share structural features. |
The different PKC isoforms are responsible for modifying the activities of a broad range of cellular proteins, including receptors, metabolic enzymes, cytoskeletal proteins, and transcription factors.4 PKC activation is mediated by phosphorylation, which plays a crucial role in the regulation of all PKCs and results in subcellular translocations to or from specific anchoring/scaffolding proteins.5 Once activated, PKC proteins remain active for a long time until the cytoplasmic Ca2+ signal is diminished.6 PKCs are involved in several cellular processes, such as cell proliferation, differentiation, polarity, and survival, and they are also important for the establishment and progression of malignant disorders, such as cancer.6 More importantly, they can present antagonistic functions, increasing their complexity.7 This review aims to summarize the existing data on the role of PKC in leukemia, specifically in CML. Through this review, we intend not only to show evidence for the involvement of PKC in CML development but also to raise new questions on whether PKC plays similar roles in other cancers.
Overall role of PKC in cancer
The aberrant regulation of different PKC isoforms has been associated with the development of many human diseases.8,9 Augmented levels and/or increased activation of PKCs have been linked not only to the malignant transformation of various cancer cell lines and tumors – including breast, ovarian, skin, lung, and gastric carcinomas10–14 – but also to aggressive and/or resistant subtypes. The classic PKCs, including PKCα, PKCβI, and PKCβII, have been shown to be overexpressed in early stage colon cancer, and have been associated with the proliferation of human breast cancer cell lines.15,16 PKCε and PKCί have been reported to be overexpressed in primary tumors and cell lines that were derived from human non-small-cell lung cancers, and they also contribute to cellular proliferation in squamous cell carcinoma.11,17–19 With regard to aggressiveness, PKC· has been linked to prostate cancer development and metastasis.20 PKCδ has been correlated to cell growth, invasion and metastasis in breast cancer.21 Also in breast cancer, PKCε expression was correlated with important factors, such as HER2 levels or low survival rates, demonstrating that this PKC isoform could also be used as a prognostic factor.22
PKCs have also been implicated in the alteration of hematopoietic stem cell (HSC) maturation.3,23 In addition, deregulation in PKC signaling represents an important feature during leukemia development.23 Survival of acute leukemic blasts was found to be mediated by PKC signaling through PDK1 activation.24 Moreover, the classic PKC isoforms are also associated with survival and poor prognosis in acute myeloid leukemia.25 A large number of studies indicate that PKC plays a central role in chronic lymphocytic leukemia development, making it a potential target for chronic lymphocytic leukemia therapy.26–30 PKCε was reported to play a crucial role in the pathogenesis of hairy cell leukemia.31 Moreover, PKCβ overexpression has been correlated with worsened prognosis in patients with lymphoid malignancies.32 In addition, the alpha and theta isoforms of PKC have been studied in the context of bone marrow transplantation.33 These examples indicate that alteration in PKC regulation may be important for the development of both solid tumors and hematological malignancies.
Chronic myeloid leukemia: a brief review
Chronic myeloid leukemia (CML) is characterized by the clonal hyperproliferation of immature blood cells. CML represents approximately 20% of all adult leukemia cases.34 Depending on the amount of immature cells in the peripheral blood, CML can be clinically divided into three phases: chronic, accelerated, and blastic crisis.35 The course of CML development has also been molecularly divided into chronic and advanced phases based on gene-expression changes.35 The rearrangement of the chromosomes 9 and 22 is a hallmark of CML.36 This abnormality, known as the Philadelphia (Ph) chromosome, is present in approximately 95% of all CML patients.37
The t(9;22)(q34.1;q11.21) translocation that produces the Ph chromosome results in the fusion of the BCR and ABL genes. The chimeric gene BCR-ABL encodes a protein with constitutive tyrosine-kinase (TK) activity.38 The BCR-ABL oncoprotein constitutively activates several downstream pathways, including the phosphatidylinositol-3 kinase (PI3K)/Akt, Ras/Raf/MAPK and JAK/STAT pathways.39,40 These pathways are responsible for the induction of cellular proliferation, loss of adhesion, blockage of cellular differentiation, and inhibition of programmed cell death. Through these cellular effects, the BCR-ABL oncoprotein has been described as the main effector of CML cellular transformation.41 CML is considered an excellent model for studying cancer development, because it harbors only one biomarker responsible for disease establishment and evolution. Therefore, deciphering the role of PKC in this cancer model could help answer several unresolved questions on CML development and provide possible insights into the role of PKC in other cancers.
Involvement of PKC in CML cellular alterations
BCR-ABL has been reported to block important signaling pathways crucial for blood stem cell maturation.42 Patients with CML exhibit accumulation of immature blood cells known as blasts in the peripheral blood;43 these cells do not present their normal functions, as they are not fully differentiated. As a consequence, CML patients may present immunodeficiency, which is associated with several defects presented by the immature blasts, such as internalization of ligands, degranulation, microbicidal killing, and alterations in chemotaxis and adhesion. PKC has been described as playing a role in HSC maturation, specifically during the formation of mature granulocytes and dendritic cells.42 Additionally, deficiency in granulocytes and dendritic cells has been associated with decreased immunity, as evidenced primarily by antigen cell defense in the case of granulocyte deficiency and decreased T-cell activation in the case of dendritic cell deficiency.44 Although there is no evidence that directly links PKC to CML cellular events, several studies have indicated that both PKC-mediated and BCR-ABL-mediated signaling pathways may produce similar changes in cellular phenotypes. Therefore, further investigation is warranted to understand the role of PKC- and BCR-ABL-mediated signaling in CML cellular transformation.
CML patients exhibit high levels of PKC in erythrocytes compared with normal individuals.45 Moreover, PKC activity is also increased in CML patients, as shown by increased PKC phosphorylation.45 This alteration in PKC activity may be one of the factors responsible for altered thermal sensitivity and mechanical stability of CML erythrocytes.45 The in vitro proliferation of T cells can be stimulated by the direct activation of PKC together with intracellular calcium signaling, suggesting that PKC may play an important role in immune defense mechanisms in CML patients.46
A significant decrease in the amount of PKCα was demonstrated in CML cells relative to normal neutrophils, whereas PKCδ was highly expressed in CML neutrophils compared with controls.47 Indeed, alterations in protein phosphorylation have been observed in CML neutrophils under stimulation with phorbol 12-myristate 13-acetate, a direct activator of PKC, further supporting a role for PKC signaling in CML cells.48 Furthermore, the alpha, beta, iota, theta, and mu isoforms of PKC are present at low levels in the cytosol of CML cells, and their respective kinase activities are also significantly decreased.47 Taken together, these data suggest that PKC activity may affect the phenotypes of CML blood cells, mainly in their maturation and immunological functions.
PKC signaling in CML cells
The oncoprotein BCR-ABL is a constitutively active TK that triggers several signaling pathways in CML cells through uncontrolled phosphorylation activity.39 Together, PKCs also play an important role in numerous cancer signaling pathways, including those downstream of the BCR-ABL oncoprotein (Figure 2).40 PKCί is one of the BCR-ABL downstream targets, and has been demonstrated to confer resistance to apoptosis in vitro after treatment with the anticancer drug Taxol® (Bristol-Myers Squibb, New York, NY, USA). In addition, active PKCί triggers the NF-κB pathway through RelA transactivation to maintain cell survival in the presence of cytotoxic compounds.49
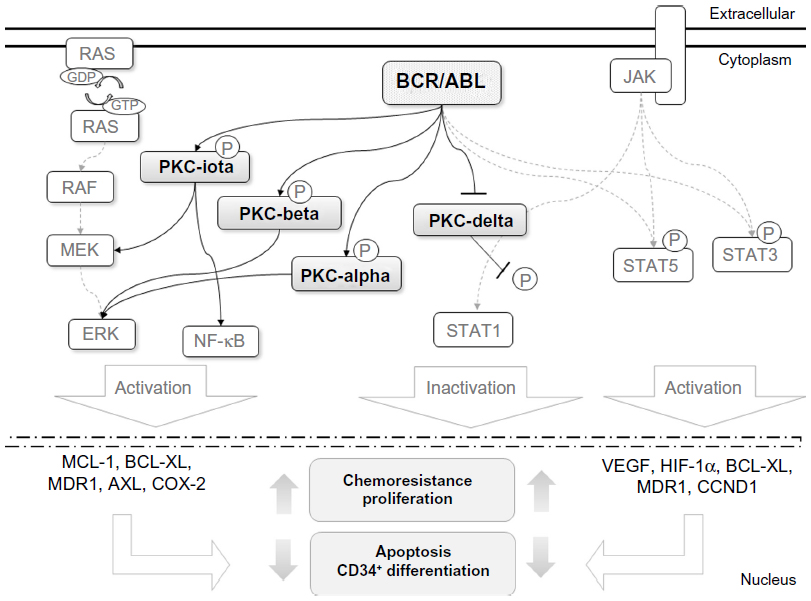 | Figure 2 Cross talk between BCR-ABL and protein kinase C (PKC) signaling. |
Ectopic expression of BCR-ABL can modulate the expression of PKC isoforms. These alterations have also been similarly found in the original CML cells.45,47 The induction of PKCί has been shown to be dependent on MEK1/2 activity, but not on PI3K or p38 MAPK activity.50 PKCί also participates in cellular differentiation by promoting the downregulation of BCR-ABL messenger ribonucleic acid transcription, which leads to decreased BCR-ABL protein expression. This downregulation of BCR-ABL appears to be driven by the MAPK pathway.44 IFNα administration was one of the earliest CML therapies that developed.51 IFNα is known to be an activator of distinct signaling pathways, such as JAK/STAT.52 IFNα treatment has been shown to induce the phosphorylation of PKCδ and the activation of its kinase domain in the CML-derived KT-1 cell line. Activated PKCδ was found to be crucial for the phosphorylation of STAT1, a member of the JAK/STAT pathway.53 Activation of STAT1 has been reported as a negative regulator of cell proliferation, in contrast to STAT3 and STAT5, which are constitutively activated in both CML patients and BCR-ABL-positive cell lines.54 Therefore, IFNα treatment induces an antileukemic process through PKCδ-mediated STAT1 activation.53
Alterations in gene-expression profiles by BCR-ABL TK activity have also been extensively studied to identify potential biomarker(s) and improve our knowledge of CML development.55,56 Several genes have already been described as BCR-ABL targets that are essential for the phenotypic changes of CML cells, such as VEGF, HIF1, MCL1, BCL2L1, CCND1, and COX2.57,58
PKC inhibition as a CML-treatment modality
Direct or indirect therapeutic inhibition of PKC isoforms alone or in conjunction with other therapeutic approaches has been the focus in many types of cancer. This is not surprising, since PKC isoforms are closely implicated in tumorigenesis. In vitro study of non-small-cell lung cancer showed that the indirect inhibition of PKCα and PKCβII promoted by glycyrrhetinic acid induced apoptosis in the NCI-H460 cell line.59 PKC inhibition together with Twist1 signaling contributes to castration and enzalutamide resistance in prostate cancer.60 In G-protein-mutant uveal melanoma, the use of specific targeting of PKC and PI3K/AKT pathways may be a future treatment.61
Regarding CML, recently the use of imatinib mesylate (IM; Gleevec®; Novartis, Basel, Switzerland), dasatinib, and nilotinib has been approved for this disease therapy.62 These drugs are TK inhibitors that target the kinase domain of the ABL protein. Clinically, their applications have led to disease remission, as evidenced by molecular, cytogenetic, and hematological data.63 During the early days of CML therapy, bryostatin was tested as a chemotherapeutic drug; it belongs to the family of macrocyclic lactones, and it is known as a potent activator of PKC.64 Experiments with bryostatin in CD34+ CML cells have demonstrated selective reduction of cell growth compared with control cells.65 Trichosanthin, a phytotherapeutic compound derived from the root of Radix trichosanthis, has been shown to synergistically promote IM-induced growth arrest and BCR-ABL downregulation in K562 cells.66 In addition, trichosanthin inhibits PKC activities as well as the PTK, procaspase-3, Hsp90 and NF-κB signaling pathways.67 The few reported studies67,68 evidence the existing gap in this field and the need for more studies focusing on PKCs’ role in CML.
Role of PKC in CML therapeutic resistance
Currently, a major focus of cancer research is to understand the cellular and molecular mechanisms underlying tumor-cell resistance to targeted molecular therapies. Despite advances in CML therapy, disease relapse still occurs in a subset of patients after IM treatment.68 Therapeutic resistance in CML has been divided into BCR-ABL-dependent and BCR-ABL-independent mechanisms. The first type is linked to mutations in the TK inhibitor-binding domains on BCR-ABL, whereas the second type is associated with parallel or constitutive signaling pathway activation that can be triggered by BCR-ABL activity as well as other mechanisms.68 The BCR-ABL-independent resistance mechanisms allow cancer cells to survive even under BCR-ABL inhibition.
Although the role of PKC in CML has not been extensively studied, recent evidence has shown a possible correlation between altered expression of PKC signaling proteins and therapeutic resistance in CML cells. Dufies et al observed the overexpression of AXL, a TK receptor and member of the TAM family, in IM-resistant CML cells, and showed that its overexpression is dependent on the alpha and beta isoforms of PKC, along with the constitutive activation of ERK1/2.69 Another study showed that the expression of ABCB1/MDR1, which is a biomarker of multidrug resistance, is dependent on PKC signaling and cyclooxygenase-2 induction.70
To minimize the development of IM-resistant CML, the efficacy of combination treatments with two or more drugs has been tested in vitro. A recent study reported that bryostatin treatment prior to IM had a more potent antiproliferative effect on a BCR-ABL-positive cell line and primary CML CD34+ cells compared with IM treatment alone.71 Furthermore, N-benzyladriamycin-14-valerate (AD198), a novel PKCδ activator, has been shown to induce apoptosis in IM-resistant BCR-ABL-positive cells. AD198 has shown effectiveness against IM-resistant cells both alone and in combination with IM.72
The occurrence of resistance in CML therapy is not only due to IM administration.62 It is known that cancer stem cells (CSCs) are more resistant to therapy. As expected, BCR-ABL-positive HSCs (CML HSCs) also have been described to be more resistant to IM therapy than mature cells, eg, due to enhanced self-renewal pathways.67 The compound TDZD-8, which inhibits both the PKC and FLT3 signaling pathways, promotes rapid induction of cell death in BCR-ABL-positive HSCs with low toxicity for normal HSCs and progenitor cells.73 This result could show great promise in overcoming IM resistance. Nevertheless, activation of several PKC isoforms in combination with antileukemic treatment has been shown to improve the desired antileukemic effect in CML cells. Robert et al observed an increase in autophagic cell death that was promoted by acadesine, which is an adenosine monophosphate-activated protein-kinase activator, when any one of the alpha, beta, or gamma isoforms of PKC was activated in CML cell lines.74 Pellicano et al also demonstrated that BMS-214662, a farnesyl-transferase inhibitor, induced apoptosis in primitive CML cells (CD34+, CD34+CD38−), and that this event was dependent on PKCβ upregulation.75 Therefore, these findings indicate the involvement of different PKC isoforms in CML cell drug resistance, thus highlighting the need for further studies to elucidate the complex role of PKC signaling in CML.
Present and future perspectives: the stem cell problem
Both the aforementioned contradictory studies72,73 bring attention to several relevant questions in cancer study today and for coming years. What are the differences and similarities that CSCs share with normal stem cells? How do they support tumors? How can we interfere to achieve the best cost–benefit result for sensibility and specificity? Since we do not have full comprehension regarding the normal stem cell, it is difficult to apply the knowledge acquired so far to complete this puzzle. We only have some pieces that still do not make total sense, but hopefully they will. With this in mind, it is clear that CML HSCs could share more features with normal HSCs or other stem cells (embryonic or pluripotent) than just self-renewal pathways. The answer to this question relies on future knowledge regarding normal stem cells and CSCs, and more importantly signaling from the microenvironment. Stem cells depend deeply on the microenvironment in order to present their normal function. Altered signaling from CML stroma has been proven to have a highly relevant role in disease development and maintenance,76 specially through mesenchymal stem cells (MSCs). Microenvironmental study has gained more importance over the years, but despite this, PKCs’ role in MSCs has only been studied in an MSC protective therapy context.77–79 It is certainly imperative that this field receive more attention in future.
Despite microenvironmental understanding, recently studies on stem cells have been trying to uncover more information in order to complete the puzzle. It has been described that some PKC isoforms are related to stem cell features, such as self-renewal and differentiation.80 In the normal context of HSCs, Sengupta et al showed that atypical PKCs are dispensable for the polarization and activity of mammalian HSCs in vivo, suggesting that an alternative tissue-specific and context-dependent signaling complex may be involved in controlling stem cell fate.81 This should be taken seriously, as the search for these other mechanisms could bring novel insights concerning normal stem cells.
In cancer, Chen et al demonstrated that CSC-like populations derived from multiple types of human primary tumors from human cancer cell lines and from transformed human cells required PKCδ activity, and PKC inhibition efficiently prevented tumor-sphere outgrowth from tumor-cell cultures, and also inhibited human CSC growth in vivo in a mouse xenograft model.82 Unfortunately, CML was not included in these models. Moreover, the authors highlighted that PKCδ is not required for the proliferation or survival of normal cells, suggesting the potential tumor-specificity of a PKCδ-targeted approach.82 This study was driven in CSC-like models; insofar as it was not performed in CSCs freshly isolated from tumors, the question about PKC involvement in this context still remains.
As presented, there are few studies focusing on PKCs’ role in CML HSCs, and no studies so far investigating PKCs in the CML microenvironment. However, taking other cancers as models, it is easy to identify the major challenge that is ahead of us.
Conclusion
Deregulation of PKC signaling has been linked to several types of cancer, including chronic myeloid leukemia. In vitro and in vivo data have shown that BCR-ABL and PKC function together to drive important molecular and cellular changes in CML cells. Initial studies revealed that administration of PKC inhibitors in conjunction with conventional CML therapy improved disease remission, as shown by molecular, cytogenetic, and hematological evidence. Nevertheless, recent reports have shown controversial findings, specially regarding CML resistance in more primitive cells. Taken together, the current findings demonstrate that further studies are necessary in order to achieve a better understanding of PKCs’ role in CML.
Author contributions
All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Nishizuka Y. Discovery and prospect of protein kinase C research: epilogue. J Biochem. 2003;133:155–158. | |
Nishizuka Y. The role of protein kinase C in cell surface signal transduction and tumour promotion. Nature. 1984;308:693–698. | |
Redig AJ, Platanias LC. The protein kinase C (PKC) family of proteins in cytokine signaling in hematopoiesis. J Interferon Cytokine Res. 2007;27:623–636. | |
Breitkreutz D, Braiman-Wiksman L, Daum N, Denning MF, Tennenbaum T. Protein kinase C family: on the crossroads of cell signaling in skin and tumor epithelium. J Cancer Res Clin Oncol. 2007;133:793–808. | |
Newton AC. Protein kinase C: ports of anchor in the cell. Curr Biol. 1996;6:806–809. | |
Mackay HJ, Twelves CJ. Targeting the protein kinase C family: are we there yet? Nature. 2007;7:554–562. | |
Halder K, Banerjee S, Bose A, Majumder S, Majumdar S. Overexpressed PKCδ downregulates the expression of PKCα in B16F10 melanoma: induction of apoptosis by PKCδ via ceramide generation. PLoS One. 2014;9:e91656. | |
Toker A. Signaling through protein kinase C. Front Biosci. 1998;3:1134–1147. | |
Kawakami T, Kawakami Y, Kitaura J. Protein kinase Cβ (PKCβ): normal functions and diseases. J Biochem. 2002;132:677–682. | |
O’Brian C, Vogel VG, Singletary SE, Ward NE. Elevated protein kinase C expression in human breast tumor biopsies relative to normal breast tissue. Cancer Res. 1989;49:3215–3217. | |
Uchida N, Okamura S, Kuwano H. Protein kinase C activity in human gastric carcinoma. Oncol Rep. 2000;7:793–796. | |
Bae KM, Wang H, Jiang G, Chen MG, Lu L, Xiao L. Protein kinase Cε is overexpressed in primary human non-small cell lung cancers and functionally required for proliferation of non-small cell lung cancer cells in a p21/Cip1-dependent manner. Cancer Res. 2007;67:6053–6063. | |
Carduner L, Picot CR, Leroy-Dudal J, Blay L, Kellouche S, Carreiras F. Cell cycle arrest or survival signaling through αv integrins, activation of PKC and ERK1/2 lead to anoikis resistance of ovarian cancer spheroids. Exp Cell Res. 2014;320:329–342. | |
Parker PJ, Justilien V, Riou P, Linch M, Fields AP. Atypical protein kinase Cί as a human oncogene and therapeutic target. Biochem Pharmacol. 2014;88:1–11. | |
Gökmen-Polar Y, Murray NR, Velasco MA, Gatalica Z, Fields AP. Elevated protein kinase C βII is an early promotive event in colon carcinogenesis. Cancer Res. 2001;61:1375–1381. | |
Li H, Weinstein IB. Protein kinase Cβ enhances growth and expression of cyclin D1 in human breast cancer cells. Cancer Res. 2006,66:11399–11408. | |
Regala RP, Weems C, Jamieson L, Copland JA, Thompson EA, Fields AP. Atypical protein kinase Cί plays a critical role in human lung cancer cell growth and tumorigenicity. J Biol Chem. 2005;280:31109–31115. | |
Aziz MH, Manoharan HT, Sand JM, Verma AK. Protein kinase Cε interacts with Stat3 and regulates its activation that is essential for the development of skin cancer. Mol Carcinog. 2007;46:646–653. | |
Felber M, Sonnemann J, Beck JF. Inhibition of novel protein kinase Cε augments TRAIL-induced cell death in A549 lung cancer cells. Pathol Oncol Res. 2007;13:295–301. | |
Benavides F, Blando J, Perez CJ, et al. Transgenic overexpression of PKCε in the mouse prostate induces preneoplastic lesions. Cell Cycle. 2011;10:268–277. | |
Hafeez BB, Zhong W, Weichert J, Dreckschmidt NE, Jamal MS, Verma AK. Genetic ablation of PKCε inhibits prostate cancer development and metastasis in transgenic mouse model of prostate adenocarcinoma. Cancer Res. 2011;71:2318–2327. | |
Zhu S, Yao F, Li WH, et al. KCδ-dependent activation of the ubiquitin proteasome system is responsible for high glucose-induced human breast cancer MCF-7 cell proliferation, migration and invasion. Asian Pac J Cancer Prev. 2013;14:5687–5692. | |
Rossi F, McNagny M, Smith G, Frampton J, Graf T. Lineage commitment of transformed haematopoietic progenitors is determined by the level of PKC activity. EMBO J. 1996;15:1894–1901. | |
Zabkiewicz J, Pearn L, Hills RK, et al. The PDK1 master kinase is overexpressed in acute myeloid leukemia and promotes PKC-mediated survival of leukemic blasts. Haematologica. 2014;99:858–864. | |
Ruvolo PP, Zhou L, Watt JC, et al. Targeting PKC-mediated signal transduction pathways using enzastaurin to promote apoptosis in acute myeloid leukemia-derived cell lines and blast cells. J Cell Biochem. 2011;112:1696–1707. | |
Kazi JU, Kabir NN, Rönnstrand L. Protein kinase C (PKC) as a drug target in chronic lymphocytic leukemia. Med Oncol. 2013;30:757. | |
Lutzny G, Kocher T, Schmidt-Supprian M, et al. Protein kinase Cβ-dependent activation of NF-κB in stromal cells is indispensable for the survival of chronic lymphocytic leukemia B cells in vivo. Cancer Cell. 2013;23:77–92. | |
Woloschak GE. Is PKC activation required for leukemia cell differentiation? Leuk Res. 1997;21:411–414. | |
Michie AM, Nakagawa R. Elucidating the role of protein kinase C in chronic lymphocytic leukaemia. Hematol Oncol. 2006;24:134–138. | |
Abrams ST, Lakum T, Lin K, et al. B-cell receptor signaling in chronic lymphocytic leukemia cells is regulated by overexpressed active protein kinase CβII. Blood. 2007;109:1193–1201. | |
Slupsky JR, Kamiguti AS, Harris RJ, Cawley JC, Zuzel M. Central role of protein kinase Cε in constitutive activation of ERK1/2 and Rac1 in the malignant cells of hairy cell leukemia. Am J Pathol. 2007;170:745–754. | |
Li S, Phong M, Lahn M, et al. Retrospective analysis of protein kinase Cβ (PKCβ) expression in lymphoid malignancies and its association with survival in diffuse large B-cell lymphomas. Biol Direct. 2007;21:8–16. | |
Fowler DH. Doubling down on PKC benefits allogeneic BMT. Blood. 2013;122:2298–2299. | |
Goldman JM, Melo JV. Chronic myeloid leukemia-advances in biology and new approaches to treatment. N Engl J Med. 2003;349:1451–1464. | |
Michor F. Chronic myeloid leukemia blast crisis arises from progenitors. Stem Cells. 2007;25:1114–1118. | |
Rowley JD. A new consistent chromosomal abnormality in chronic myelogenous leukemia identified by quinacrine fluorescence and Giemsa staining. Nature. 1973;243:290–293. | |
Medina J, Kantarjian H, Talpaz M, Cortes J, et al. Chromosomal abnormalities in Philadelphia chromosome-negative metaphases appearing during imatinib mesylate therapy in patients with Philadelphia chromosome-positive chronic myelogenous leukemia in chronic phase. Cancer. 2003;98:1905–1911. | |
Stam K, Heisterkamp N, Reynolds FH Jr, Groffen J. Evidence that the phl gene encodes a 160,000-dalton phosphoprotein with associated kinase activity. Mol Cell Biol. 1987;7:1955–1960. | |
Steelman LS, Pohnert SC, Shelton JG, Franklin RA, Bertrand FE, McCubrey JA. JAK/STAT, Raf/MEK/ERK, PI3K/Akt and BCR-ABL in cell cycle progression and leukemogenesis. Leukemia. 2004;18:189–218. | |
Hickey FB, England K, Cotter TG. Bcr-Abl regulates osteopontin transcription via Ras, PI-3K, aPKC, Raf-1, and MEK. J Leukoc Biol. 2005;78:289–300. | |
Lim YM, Wong S, Lau G, Witte ON, Colicelli J. BCR/ABL inhibition by an escort/phosphatase fusion protein. Proc Natl Acad Sci U S A. 2000;97:12233–12238. | |
Buckle AM, Mottram R, Pierce A, et al. The effect of Bcr-Abl protein tyrosine kinase on maturation and proliferation of primitive haematopoietic cells. Mol Med. 2000;6:892–902. | |
Melo JV. The diversity of BCR-ABL fusion proteins and their relationship to leukemia phenotype. Blood. 1996;88:2375–2384. | |
Lindner I, Kharfan-Dabaja MA, Ayala E, et al. Induced dendritic cell differentiation of chronic myeloid leukemia blasts is associated with down-regulation of BCR-ABL. J Immunol. 2003;171:1780–1791. | |
Kundu M, Basu J, Fujimagari M, Williamson P, Schlegel RA, Chakrabarti P. Altered erythrocyte protein kinase C activity and membrane protein phosphorylation in chronic myelogenous leukemia. Biochim Biophys Acta. 1991;1096:205–208. | |
Kharfan-Dabaja M, Ayala E, Lindner I, et al. Differentiation of acute and chronic myeloid leukemic blasts into the dendritic cell lineage: analysis of various differentiation-inducing signals. Cancer Immunol Immunother. 2005;54:25–36. | |
Balasubramanian N, Advani SH, Zingde SM. Protein kinase C isoforms in normal and leukemic neutrophils: altered levels in leukemic neutrophils and changes during myeloid maturation in chronic myeloid leukemia. Leuk Res. 2002;26:67–81. | |
Batliwalla F, Advani S, Gothoskar B, Zingde S. Differential phosphorylation in normal and leukemic granulocytes in response to phorbol 12-myristate 13-acetate. Leuk Res. 1994;18:327–336. | |
Lu Y, Jamieson L, Brasier AR, Fields AP. NF-κB/RelA transactivation is required for atypical protein kinase Cί-mediated cell survival. Oncogene. 2001;20:4777–4792. | |
Gustafson WC, Ray S, Jamieson L, Thompson EA, Brasier AR, Fields AP. Bcr-Abl regulates protein kinase Cί (PKCί) transcription via an Elk1 site in the PKCί promoter. J Biol Chem. 2004;279:9400–9408. | |
Homewood J, Watson M, Richards SM, Halsey J, Shepherd PC. Treatment of CML using IFN-alpha: impact on quality of life. Hematol J. 2003;4:253–262. | |
Darnell JE Jr, Kerr IM, Stark GR. JAK-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264:1415–1421. | |
Kaur S, Parmar S, Smith J, et al. Role of protein kinase C-delta (PKC-delta) in the generation of the effects of IFN-alpha in chronic myelogenous leukemia cells. Exp Hematol. 2005;33:550–557. | |
Benekli M, Baer MR, Baumann H, Wetzler M. Signal transducer and activator of transcription proteins in leukemias. Blood. 2003;101:2940–2954. | |
Hofmann W, de Vos S, Elashoff D, et al. Relation between resistance of Philadelphia-chromosome-positive acute lymphoblastic leukaemia to the tyrosine kinase inhibitor STI571 and gene-expression profiles: a gene-expression study. Lancet. 2002;359:481–486. | |
Salomon AR, Ficarro SB, Brill LM, et al. Profiling of tyrosine phosphorylation pathways in human cells using mass spectrometry. Proc Natl Acad Sci U S A. 2003;100:443–448. | |
Hickey FB, Cotter TG. BCR-ABL regulates phosphatidylinositol 3-kinase-p110γ transcription and activation and is required for proliferation and drug resistance. J Biol Chem. 2006;28:2441–2450. | |
Zheng C, Li L, Haak M, et al. Gene expression profiling of CD34+ cells identifies a molecular signature of chronic myeloid leukemia blast crisis. Leukemia. 2006;20:1028–1034. | |
Song J, Ko HS, Sohn EJ, et al. Inhibition of protein kinase C α/βII and activation of c-Jun NH2-terminal kinase mediate glycyrrhetinic acid induced apoptosis in non-small cell lung cancer NCI-H460 cells. Bioorg Med Chem Lett. 2014;15;24:1188–1191. | |
Shiota M, Yokomizo A, Takeuchi A, et al. Inhibition of protein kinase C/Twist1 signaling augments anticancer effects of androgen deprivation and enzalutamide in prostate cancer. Clin Cancer Res. 2014;15;20:951–961. | |
Musi E, Ambrosini G, De Stanchina E, Schwartz GK. The phosphoinositide 3-kinasea selective inhibitor, BYL719, enhances the effect of the protein kinase C inhibitor, AEB071, in GNAQ/GNA11 mutant uveal melanoma cells. Mol Cancer Ther. 2014;13:1044–1053. | |
Swords R, Alvarado Y, Giles F. Novel Abl kinase inhibitors in chronic myeloid leukemia in blastic phase and Philadelphia chromosome-positive acute lymphoblastic leukemia. Clin Lymphoma Myeloma. 2007;7 Suppl 3:S113–S119. | |
Hochhaus A, Kreil S, Corbin AS, et al. Molecular and chromosomal mechanisms of resistance to imatinib (STI571) therapy. Leukemia. 2002;16:2190–2196. | |
Szallasi Z, Denning MF, Smith CB, et al. Bryostatin 1 protects protein kinase C-delta from down-regulation in mouse keratinocytes in parallel with its inhibition of phorbol ester-induced differentiation. Mol Pharmacol. 1994;46:840–850. | |
Thijsen SF, Schuurhuis GJ, van Oostveen JW, et al. Effects of bryostatin-1 on chronic myeloid leukaemia-derived haematopoietic progenitors. Br J Cancer. 1999;79:1406–1412. | |
Zhang K, Xu J, Huang X, et al. Trichosanthin down-regulated p210Bcr-Abl and enhanced imatinib-induced growth arrest in chronic myelogenous leukemia cell line K562. Cancer Chemother Pharmacol. 2007;60:581–587. | |
Bradeen HA, Eide CA, O’Hare T, et al. Comparison of imatinib mesylate, dasatinib (BMS-354825), and nilotinib (AMN107) in an N-athyl-N-nitrosourea (ENU)-based mutagenesis screen: high efficacy of drug combinations. Neoplasia. 2006;108:2332–2338. | |
Binato R, Mencalha A, Pizzatti L, Scholl V, Zalcberg I, Abdelhay E. RUNX1T1 is overexpressed in imatinib mesylate-resistant cells. Mol Med Rep. 2009;2:657–661. | |
Dufies M, Jacquel A, Belhacene N, et al. Mechanisms of AXL overexpression and function in imatinib-resistant chronic myeloid leukemia cells. Oncotarget. 2011;2:874–885. | |
Kalle AM, Sachchidanand S, Pallu R. Bcr-Abl-independent mechanism of resistance to imatinib in K562 cells: induction of cyclooxygenase-2 (COX-2) by histone deacetylases (HDACs). Leuk Res. 2010;34:1132–1138. | |
Jørgensen HG, Allan EK, Mountford JC, et al. Enhanced CML stem cell elimination in vitro by bryostatin priming with imatinib mesylate. Exp Hematol. 2005;33:1140–1146. | |
Lothstein L, Savranskaya L, Sweatman TW. N-benzyladriamycin-14-valerate (AD 198) cytotoxicity circumvents Bcr-Abl anti-apoptotic signaling in human leukemia cells and also potentiates imatinib cytotoxicity. Leuk Res. 2007;31:1085–1095. | |
Guzman ML, Li X, Corbett CA, et al. Rapid and selective death of leukemia stem and progenitor cells induced by the compound 4-benzyl, 2-methyl, 1,2,4-thiadiazolidine, 3,5 dione (TDZD-8). Blood. 2007;110:4436–4444. | |
Robert G, Ben Sahra I, Puissant A, et al. Acadesine kills chronic myelogenous leukemia (CML) cells through PKC-dependent induction of autophagic cell death. PloS One. 2009;18;4:e7889. | |
Pellicano F, Copland M, Jorgensen HG, Mountford J, Leber B, Holyoake TL. BMS-214662 induces mitochondrial apoptosis in chronic myeloid leukemia (CML) stem/progenitor cells, including CD34+38- cells, through activation of protein kinase Cβ. Blood. 2009;5;114:4186–4196. | |
Zhang B, Li M, McDonald T, et al. Microenvironmental protection of CML stem and progenitor cells from tyrosine kinase inhibitors through N-cadherin and Wnt-β-catenin signaling. Blood. 2013;121:1824–1838. | |
Higuchi S, Li M, Zhu P, Ashraf M. Delta-opioid receptor activation promotes mesenchymal stem cell survival via PKC/STAT3 signaling pathway. Circ J. 2012;76:204–212. | |
Song BW, Chang W, Hong BK, et al. Protein kinase C activation stimulates mesenchymal stem cell adhesion through activation of focal adhesion kinase. Cell Transplant. 2013;22:797–809. | |
Tsai TL, Manner PA, Li WJ. Regulation of mesenchymal stem cell chondrogenesis by glucose through protein kinase C/transforming growth factor signaling. Osteoarthritis Cartilage. 2013;21:368–376. | |
Kinehara M, Kawamura S, Tateyama D, et al. Protein kinase C regulates human pluripotent stem cell self-renewal. PLoS One. 2013;8:e54122. | |
Sengupta A, Duran A, Ishikawa E, et al. Atypical protein kinase C (aPKC¶ and aPKC») is dispensable for mammalian hematopoietic stem cell activity and blood formation. Proc Natl Acad Sci U S A. 2011;108:9957–9962. | |
Chen Z, Forman LW, Williams RM, Faller DV. Protein kinase C-delta inactivation inhibits the proliferation and survival of cancer stem cells in culture and in vivo. BMC Cancer. 2014;14:90. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.