")
Back to Journals » Journal of Inflammation Research » Volume 8
Regulation of indoleamine 2,3-dioxygenase in primary human saphenous vein endothelial cells
Authors Mouratidis P, George AJT
Received 4 February 2015
Accepted for publication 13 March 2015
Published 21 May 2015 Volume 2015:8 Pages 97—106
DOI https://doi.org/10.2147/JIR.S82202
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Ning Quan
Petros XE Mouratidis,* Andrew JT George*
Department of Immunology, Imperial College London, London, UK
*These authors contributed equally to this work
Background: Indoleamine 2,3-dioxygenase (IDO) is an enzyme associated with the regulation of immune responses. Cytokines such as IFNγ induce its expression in endothelial cells originating from immune-privileged sites. In this study, we investigate regulators of IDO in primary endothelial cells from a non-immune-privileged site and determine whether IDO expression affects immune cell behavior.
Methods: IDO expression was determined using real-time quantitative polymerase chain reaction and immunoblotting. IDO activity was estimated using an IDO enzyme assay. Primary cells were transfected using microporation, and T-cell migration was determined using a cell transmigration assay.
Results: IDO is expressed in human saphenous vein endothelial cells after stimulation with IFNγ but not after treatment with TNFα, IL-1β, IL-2, IL-4, IL-6, or IL-10. VEGFβ and heparin negatively regulate IFNγ-driven increases in IDO. Overexpression of IDO in endothelial cells does not affect transmigration of T-cells.
Conclusion: IDO is expressed in human saphenous vein endothelial cells after stimulation with IFNγ. Heparin and angiogenesis stimulators such as VEGFβ negatively regulate its expression.
Keywords: IFNγ, VEGFβ, endothelium, inflammation, HSVEC
Introduction
The endothelial cell plays a central role in the control of inflammatory processes and can be involved in graft rejection, atherosclerosis, and vasculitides.1–3 In addition to the effect of endothelial cells in the recruitment of leukocytes into inflammation sites,4,5 they can also present antigens to white blood cells influencing their activation and transmigration.6–9
Indoleamine 2,3-dioxygenase (IDO) is an intracellular heme-containing enzyme that catalyzes the oxidative cleavage of the indole ring of l-tryptophan to produce kynurenine metabolites.10 Depletion of l-tryptophan from the local microenvironment together with the production of kynurenine can modulate inflammation responses.11,12 IDO activation has been associated with antimicrobial effects and the modulation of T-cell responses,13 at least partly by enhancement of proliferation of regulatory T-cells.14,15 Increased expression of IDO is related to a favorable environment for tumor growth and graft survival,16–19 the pathophysiology of depression,20,21 and protection against pulmonary hypertension.22 IDO activity may be affected not only at the transcriptional and translational level, but also by posttranslational modification and SOCS3-associated differential degradation.23–25
What is currently not known is whether immune-regulatory molecules such as IDO mediate graft rejection exhibited by endothelial cells. When human umbilical vein endothelial cells (HUVECs) were stimulated with IFNγ, IDO expression and activity were increased. In addition, it was shown that IDO activity inhibition enhanced the ability of HUVECs to stimulate allogeneic T-cell responses.26 Treatment of cells originating from non-immune-privileged sites such as human saphenous vein endothelial cells (HSVECs) with IFNγ did not upregulate IDO.26 In a non-vascular in vitro model of human corneal endothelial cells (HCECs) IFNγ-driven upregulation of IDO had no effect on T cell proliferation.27 In this study, we used primary cells isolated from a non-immune-privileged source, the saphenous vein, to investigate factors that affect IDO regulation and determined whether forced expression of IDO in these cells affected immune cell migration.
Materials and methods
Isolation of primary endothelial cells from human saphenous veins
Primary endothelial cells from human saphenous veins were isolated using the method described elsewhere.28 Briefly, both ends of the vein were sutured and the lumen was filled with 10 mL prewarmed 1 mg/mL collagenase II solution (Sigma-Aldrich Co, St Louis, MO, USA) and incubated at 37°C for 10 minutes. Then collagenase II solution was collected and vein was flushed with 10 mL trypsin–ethylenediaminetetraacetic acid and the effluent was collected again. Cells were pelleted and plated in a T25 flask. Endothelial cells were further purified with anti-CD105 microbeads using MS columns (Miltenyi Biotech, Auburn, CA, USA). Cell fractions showing high levels of staining for von Willebrand factor were used.
Cell culture and propagation
Cells were maintained in a subconfluent monolayer at 37°C in a humidified atmosphere containing 5% CO2. Endothelial medium was prepared by combining equal volumes of endothelial SFM (Thermo Fisher Scientific, Waltham, MA, USA) and endothelial EGM2 medium (Walkersville, MD, USA) and adding per 500 mL medium 5 mL of penicillin/streptomycin, 5 mL glutamine, and 5 mL of 5 mg/mL endothelial cell growth supplement purchased from BD Biosciences (San Jose, CA, USA). They were subcultured using trypsin, and screening for mycoplasma contamination was carried out on a regular basis. For experimental use, cells were treated with recombinant human IFNγ, recombinant human TNFα, recombinant human IL-2, recombinant human IL-4, recombinant human IL-6, recombinant human IL-10, and recombinant human VEGFβ, all purchased from Peprotech (Rocky Hill, NJ, USA).
IDO activity assay
IDO activity was determined in wells exhibiting cell confluency greater than 70% using a colorimetric assay based on detection of kynurenine as described previously.29,30 Briefly, l-kynurenine standards (0–200 μM) were prepared in endothelial medium. Kynurenine in control, test, and standard samples was precipitated by incubation with 30% trichloroacetic acid at 50°C for 30 minutes and centrifuged at 2,000× g for 10 minutes. Visualization of signal took place by adding equal volumes of Ehrlich reagent to the supernatant and incubation at 65°C for 15 minutes. Ehrlich reagent was prepared using 6.2 mL 1-propanol, 1.5 mL distilled water, 2.6 mL of 70% perchloric acid, and 1.5 g of 4-dimethylbenzinamide, all purchased from Sigma-Aldrich Co. Absorbance was measured at 492 nm in a colorimetric plate reader.
Cell homogenization and Western blotting
Control and treated cells were harvested from flasks, washed in phosphate-buffered saline, and lysed in SDS sample buffer (62.5 mM Tris-HCl, pH 6.8, 2% w/v SDS, 10% glycerol, 50 mM DTT, and 0.01% bromophenol blue) for 5 minutes on ice and sonicated for 10–15 seconds. Samples were then heated for 5 minutes at 90°C, micro-centrifuged briefly, and stored at −20°C. Antibodies against IDO and SOCS3 were purchased from Abcam (Cambridge, UK). Primary antibody of interest diluted in blocking buffer (1.5% milk in Tris-buffered saline/Tween 20, pH 7.6) containing 5% BSA was added to the membranes and left incubating overnight at 4°C. Membranes were then washed and incubated with secondary antibody. Signal was visualized on X-ray films using ECL Western blotting reagents (Amersham Biosciences, Little Chalfont, UK).
Real-time polymerase chain reaction
Cells were washed with phosphate-buffered saline, and mRNA was obtained using TRIzol. cDNA was obtained using the SuperScript First-Strand Synthesis System for RT-PCR. Detection of product was made using SYBR Green (Thermo Fisher Scientific). Relative quantification of mRNA transcription was performed by normalizing the Ct value of IDO to the Ct value of actin in control:
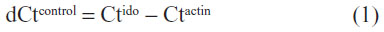
and treated cells:
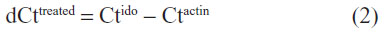
Then the ΔΔCt product was calculated by subtracting the dCt value of controls from the dCt value of treated cells:
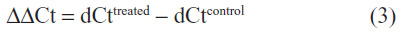
Using the formula 2−ΔΔCt, the relative enhancement ratio of IDO transcription was estimated. The following primer sequences were used: HPRT forward: 5′-GCAGACTTTGCTTTCCTTGGTC-3′; HPRT reverse: 5′-CTGGCTTATATCCAACACTTCGTG-3′; IDO forward: 5′-GGTCATGGAGATGTCCGTTAA-3′; IDO reverse: 5′-ACCAATAGAGAGACCAGGAAGAA-3′.
Transfection of endothelial cells using microporation
Microporation apparatus, tips, buffers and the MP-1096 kit were purchased from Labtech International (Ringmer, UK). The desired number of cells were resuspended in 24 μL of microporation suspension buffer and kept at 4°C for the duration of the experiment. One microgram DNA was added and cells were transfected using microporation. The vectors used were pcDNA3.1 (empty vector), pSMART2G (empty vector), pcDNA3.1-EGFP, and pSMART2G-IDO. Voltage was set at 1,350 mV, pulse number at 1, and duration at 30 ms, and p10 tips were used. After microporation, cell aliquots were put in endothelial medium in 24-well plates at 37°C and left to incubate overnight. For experiments, cells were used in the first 3 days after transfection.
Transmigration assay
HSVECs were plated on a 24-well Transwell chamber at full confluence and left to incubate for 1 day. PBMCs were obtained from human donors or buffy coat using density centrifugation with Ficoll-Paque. T-cells were obtained from PBMCs using MACS bead separation, employing negative selection with biotin-conjugated antibodies against CD14, CD16, CD19, CD36, CD56, and CD235. Fractions were stained with CD3 fluorescein isothiocyanate to determine purity and those staining positive were used for experiments. T-cell fractions containing 3×105 cells were applied in 1:10 dilution on the top of the endothelial cells in the Transwell chambers and co-cultured for a further day. T-cells transmigrating through the endothelial monolayer were collected on the bottom chamber and their number determined using a hemocytometer.
Statistical analysis of results
Results are presented as means + standard error of the mean. Statistical significance was carried out using a t-test assuming one-tailed distribution using two-sample unequal variance. P<0.05 was considered as a statistically significant value. Microsoft Excel data processing functions were used for data processing and analysis.
Results
IFNγ is the main regulator of IDO expression in primary HSVECs
Differential regulation of IDO in endothelial cells stimulated with IFNγ was evident in other studies depending on the endothelial cell origin.26,27 To demonstrate whether IDO expression in primary HSVECs was increased after IFNγ stimulation, these primary cells were treated with concentrations of IFNγ (0–120 ng/mL) for 2 days. IDO expression was assessed using immunoblotting, and IDO activity was assessed using an IDO activity assay. Results show that treatment of HSVECs with IFNγ (80 ng/mL) resulted in increased IDO expression and IDO activity (Figures 1 and S1).
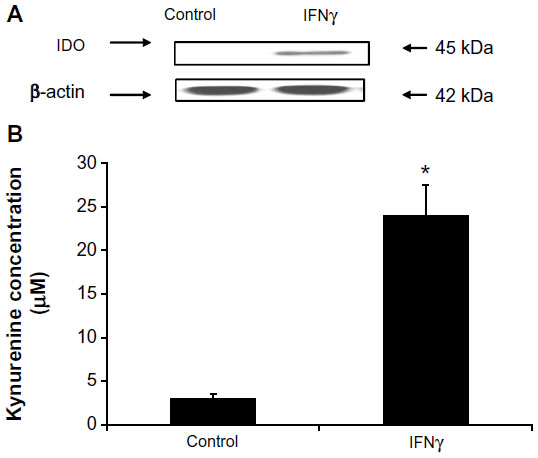 | Figure 1 IFNγ upregulates IDO in primary human saphenous vein endothelial cells (HSVECs). |
To investigate what other regulators drive expression of IDO, HSVECs were treated with IFNγ alone or in combination with cytokines in relevant concentrations such as TNFα (80 ng/mL), IL-1β (20 ng/mL), IL-2 (20 ng/mL), IL-4 (40 ng/mL), IL-6 (40 ng/mL), IL-10 (40 ng/mL), and TGFβ (100 ng/mL) for 2 days. IDO mRNA expression was assessed using real-time quantitative polymerase chain reaction. Results show that IDO mRNA was increased after treatment with IFNγ but not after treatment with the other cytokines (Figure 2A). In addition, co-treatment of HSVECs with IFNγ and these cytokines did not result in significant change in IFNγ-driven increases in IDO expression and activity, suggesting that IFNγ was the main regulator of IDO in HSVECs (Figures 2B and S2).
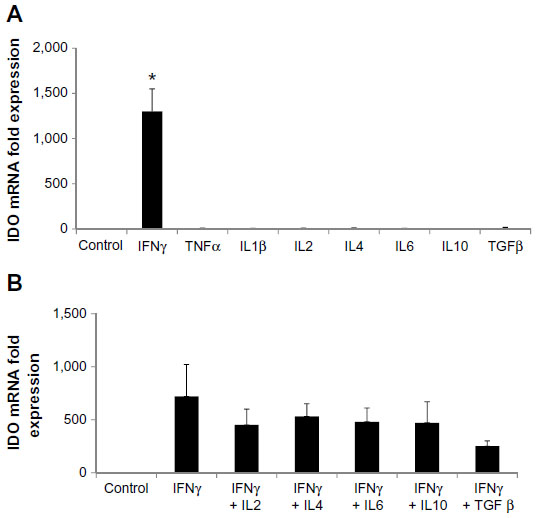 | Figure 2 IFNγ is the main regulator of IDO in human saphenous vein endothelial cells (HSVECs). |
VEGF, heparin, and nitric oxide are negative regulators of IDO in HSVECs
IFNγ has exhibited antiangiogenic effects in human cornea by downregulating expression of VEGFα.31 To identify whether crosstalk of IFNγ and VEGF pathways exists in HSVECs, these cells were stimulated with IFNγ (80 ng/mL) alone or in combination with VEGFβ (40 ng/mL) for 1 day and 2 days. Expression of IDO was determined using immunoblotting with a monoclonal antibody against human IDO. IDO was not expressed in unstimulated cells or cells treated with VEGFβ only (data not shown). When HSVECs were treated with IFNγ, then IDO protein levels were increased, whereas co-treatment of HSVECs with IFNγ and VEGFβ decreased IDO levels relative to stimulation with IFNγ only (Figure 3A). SOCS3 has been shown to target IDO for proteasomal degradation.25 To investigate the possibility that SOCS3 protein levels are regulated in a similar manner to IDO’s protein levels, its expression was investigated using immunoblotting. SOCS3 protein levels remained unchanged in cells treated with IFNγ only. However, when cells were co-treated with IFNγ and VEGFβ, SOCS3 protein levels increased after 24 hours and completely eliminated after 48 hours (Figure 3A).
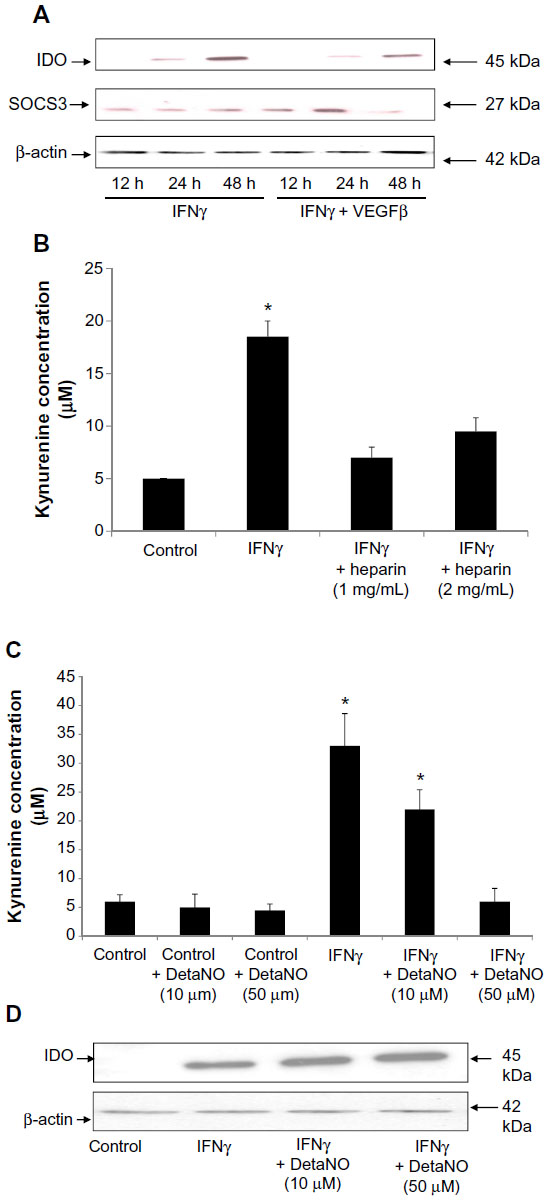 | Figure 3 Negative regulators of IDO expression in primary human saphenous vein endothelial cells (HSVECs). |
We have previously used heparin in the culture of endothelial cells and questioned whether it interferes with the induction of IDO.26 For this purpose, we measured the IDO response to IFNγ in the presence and absence of heparin. HSVECs were stimulated with IFNγ (80 ng/mL) with or without cell culture-relevant concentrations of heparin (1 mg/mL and 2 mg/mL). IDO activity was assessed using an IDO activity assay. This concentration of heparin was chosen because it is detected in endothelial cell media. Results show that heparin abrogated IFNγ-driven increases in IDO activity (Figure 3B).
Differential regulation of IDO activity by nitric oxide (NO) has been shown in monocyte-derived macrophages but not in microglial cells.32 To investigate whether NO has a regulatory effect on IDO activity in HSVECs, cells were stimulated with IFNγ (80 ng/mL) with or without the NO donor DetaNO (10 μM and 50 μM). IDO expression levels were assessed using immunoblotting and IDO activity was assessed using the IDO activity assay. Results show that NO decreased IFNγ-driven increases in IDO activity. However, IDO protein levels did not follow the same regulation pattern (Figure 3C and D).
Increased expression of IDO in HSVECs does not affect T-cell transmigration properties
To determine the effect of IDO overexpressed in endothelial cells independently of IFNγ stimulation, primary HSVECs were transiently transfected with EGFP using microporation. EGFP fluorescence was detected up to 6 days using fluorescence microscopy (Figure 4A). Then, HSVECs were transiently transfected with an IDO plasmid. Overexpression of IDO was confirmed using immunoblotting (Figure 4B). IDO-overexpressing HSVECs were plated on Transwell chambers and left to incubate for 1 day. T-cells were then added in the Transwell chambers and coincubated with HSVECs for 24 hours further. The number of cells detected in the lower compartment of the Transwell chamber was counted using a hemocytometer. Results show that IDO overexpression in HSVECs did not affect the number of T-cells transmigrating through the endothelial cell monolayer barrier (Figure 4C).
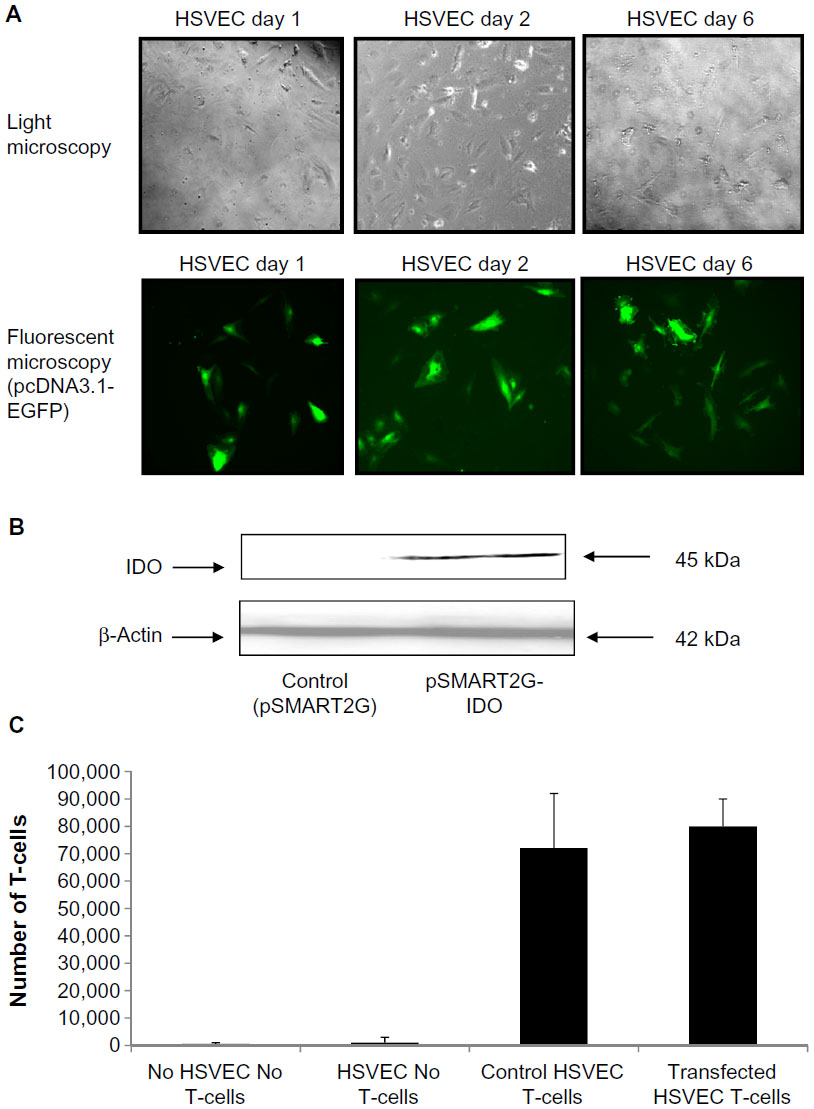 | Figure 4 IDO does not affect T-cell transmigration properties. |
Discussion
IDO is an enzyme that metabolizes tryptophan and is known to be central in the regulation of immune responses. IDO can be expressed in endothelial cells and alters T-cell responses to the endothelium. The aim of this study was to identify regulators of IDO expression in HSVECs and to determine whether IDO expression in these primary cells pooled together from different donors affects inflammation-related processes.
HSVECs were treated with proinflammatory and anti-inflammatory cytokines. IDO transcription and protein levels and activity were then assessed. We were not able to detect IDO expression in endothelial cells cultured under normal conditions. Stimulation of HSVECs with different concentrations of TNFα, IL-1β, IL-2, IL-4, IL-6, and IL-10 either alone or in combinations had a negligible effect on IDO expression that did not result in a significant functional effect as determined by production of kynurenines (data not shown). However, stimulation of cells with IFNγ resulted in an increase in IDO expression and activity. The high concentrations of IFNγ used to achieve upregulation of IDO in HSVECs did not result in detrimental effects to the cells. Co-stimulation of IFNγ-treated endothelial cells with cytokines such as TNFα, IL-1β, IL-2, IL-4, IL-6, and IL-10 did not affect IDO expression and activity relative to cells treated with IFNγ.
Conflicting reports on the ability of HSVECs to increase IDO expression after treatment with IFNγ have been reported, and we have described that IFNγ is unable to upregulate IDO activity in HSVECs while confirming upregulation in HUVECs.26 However, those previous experiments were performed with heparin as an additive to endothelial cell media to enhance cell proliferation and survival. To investigate whether heparin affects IFNγ-driven increases in IDO activity in HSVECs we replaced heparin with endothelial cell growth supplement factors, golden grade fetal calf serum and used specialized endothelial cell medium. By optimizing these cell culture conditions no significant loss of endothelial cell viability was observed. In addition HSVECs confluency was greater than 70% in experimental conditions. Using these experimental conditions we have shown that heparin, in concentrations routinely used in cell culture and experimental media, counteracts the ability of IFNγ to upregulate IDO in HSVECs. Heparin interferes with IFNγ activity by binding to the cytokine and reducing its ability to bind and activate the IFNγ receptor.33,34 Heparin counteracts IFNγ-induced increases of iNOS in rat endothelial cells,35 apoptosis of primary trophoblasts,36 and fractalkine expression in human endothelial cells.37 These results suggest that endothelial cells located in non-immune-privileged tissues can exhibit induction of IDO activity after IFNγ stimulation in a similar manner to the endothelial cells associated with immune-privileged tissue.
VEGFα has been associated with IDO expression upregulation in prostaglandin E2- and TNFα-treated dendritic cells.38 In addition, IFNγ has exhibited antiangiogenic effects in human cornea by downregulating expression of VEGFα.31 In this study, we show for the first time that angiogenesis stimulators such as VEGFβ have a negative effect on IFNγ-driven increases in IDO expression and also regulate SOCS3 protein levels. Orabona et al and Pallota et al have shown that SOCS3 mediates proteasome degradation of IDO in dendritic cells.25,39 It is tempting to hypothesize that the same mechanism takes place in primary endothelial cells co-treated with VEGFβ and IFNγ. However, further research is needed to prove a direct link between VEGFβ and SOCS3-mediated proteasome degradation of IDO. NO has been shown to affect IDO activity in monocyte-derived macrophages at the posttranslational levels but not in other biological models such as microglial cells.32 In our study, we confirm that NO has a negative effect on IDO activity at the posttranslational level. IDO catabolizes tryptophan by inserting oxygen to generate N-formyl-kynurenine. NO may inhibit this function by displacing bound tryptophan and oxygen after binding to IDO’s active heme site.40
The recruitment of inflammatory leukocytes from blood vessels is controlled by the expression of adhesion molecules on the endothelium as well as the secretion and presentation of chemokines.26 To determine whether IDO expression in HSVECs affected T-cell transmigration, IDO was overexpressed in these cells using microporation. This method resulted in transfection efficiency of approximately 50% that is stable for at least 6 days. Transfected HSVECs were allowed to attach onto Transwell chambers and form a monolayer. The number of T-cells transmigrating through the endothelial monolayer transfected with IDO versus the number of T-cells transmigrating through the endothelial cells expressing basal levels of IDO was then determined. Using this technique, we were not able to demonstrate a significant change in T-cell transmigration. It is possible that the number of non-transfected cells in the monolayer was high enough to allow passage of T-cells. However, controls using IFNγ-treated HSVECs did not result in a significant change in the number of transmigrated cells.
IDO regulates immune responses which otherwise could cause tissue damage. The endothelial cell is one of the first contacts of an activated lymphocyte, hence whether it has the mechanistic capability to increase IDO expression and regulate the immune response is highly significant. Max et al have demonstrated that IFNγ-induced IDO expression in Ewing sarcoma cells decreased T-cell proliferation in co-culture assays 3 days after treatment, but no effect on T-cell proliferation was evident 1 day after treatment.41 In contrast, Lahdou et al have shown that upregulation of IDO in corneal endothelial cells does not affect T-cell proliferation.27 In addition, they demonstrated that extraordinarily high levels of l-kynurenine close to 400 μM are necessary for a suppressive effect on T-cell proliferation to be detected. In our study, we achieved relatively low levels of l-kynurenine concentration up to 10 μM (transfected cells) (data not shown) and 40 μM (IFNγ-treated cells) when assessed in 24-well plates. However, the levels of IFNγ used in our study were higher than the physiological levels of IFNγ found in blood serum of patients treated for infections, although it cannot be excluded that higher levels of IFNγ accumulate in the target tissue’s microenvironment. So we believe that, with our chosen experimental procedures, we do not underestimate the IFNγ concentration and IDO expression the in vivo endothelial cells may be subjected to. In addition, it is important to distinguish between effects on cell proliferation and direct effects on transmigration properties of the cells. It is possible that the duration of lymphocyte exposure and the concentration of kynurenines the lymphocytes are subjected to, in the microenvironment of IDO-overexpressing cells, may dictate T-cell response. It is of interest to establish whether IDO may have additional roles in endothelium. Park et al have suggested that the role of IDO is different between target tissues and infiltrated immune cells while demonstrating that low endothelial IDO levels correlate with lower non-relapse mortality in patients with acute graft-versus-host disease after allogeneic hematopoietic stem cell transplantation.42 Wang et al have shown in mouse models of systemic inflammation that IDO expressed in vascular endothelial cells results in the production of kynurenines which regulate vascular tone and blood pressure regulation.43
The data presented in this study build on previous published observations and altogether provide an insight into the mechanism by which endothelial cells regulate immune responses and the role of endothelial IDO. Our results show that HSVECs have the capability to upregulate IDO after stimulation with IFNγ but, at relatively low kynurenine concentrations, do not directly affect T-cell transmigration properties. We also show for the first time that angiogenesis stimulators negatively regulate the IFNγ-driven increases in IDO expression. Given the interest that exists in IDO and graft survival, it is of interest to investigate whether angiogenesis regulators can promote or inhibit graft survival in vivo and the possibility that this might be an IDO-dependent effect.
Acknowledgment
This study was funded by the British Heart Foundation (BHF).
Disclosure
The authors report no conflicts of interest in this work.
References
Manduteanu I, Simionescu M. Inflammation in atherosclerosis: a cause or a result of vascular disorders? J Cell Mol Med. 2012;16(9):1978–1990. | |
Biasucci LM, Cardillo MT. Biomarkers of inflammation and endothelial function: the holy grail of experimental and clinical medicine? Vascul Pharmacol. 2012;56(1–2):26–28. | |
Sis B. Endothelial molecules decipher the mechanisms and functional pathways in antibody-mediated rejection. Hum Immunol. 2012;73(12):1218–1225. | |
Daniel AE, van Buul JD. Endothelial junction regulation: a prerequisite for leukocytes crossing the vessel wall. J Innate Immun. 2013;5(4):324–335. | |
Heemskerk N, van Rijssel J, van Buul JD. Rho-GTPase signaling in leukocyte extravasation: an endothelial point of view. Cell Adh Migr. 2014;8(2):67–75. | |
Charreau B. Molecular regulation of endothelial cell activation: novel mechanisms and emerging targets. Curr Opin Organ Transplant. 2011;16(2):207–213. | |
Mai J, Virtue A, Shen J, Wang H, Yang XF. An evolving new paradigm: endothelial cells – conditional innate immune cells. J Hematol Oncol. 2013;6:61. | |
Muller WA. Getting leukocytes to the site of inflammation. Vet Pathol. 2013;50(1):7–22. | |
Marelli-Berg FM, Frasca L, Weng L, Lombardi G, Lechler RI. Antigen recognition influences transendothelial migration of CD4+ T cells. J Immunol. 1999;162(2):696–703. | |
Fallarino F, Grohmann U, Puccetti P. Indoleamine 2,3-dioxygenase: from catalyst to signaling function. Eur J Immunol. 2012;42(8):1932–1937. | |
Munn DH, Zhou M, Attwood JT, et al. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science. 1998;281(5380):1191–1193. | |
Mellor AL, Sivakumar J, Chandler P, et al. Prevention of T cell-driven complement activation and inflammation by tryptophan catabolism during pregnancy. Nat Immunol. 2001;2(1):64–68. | |
Murakami Y, Hoshi M, Imamura Y, Arioka Y, Yamamoto Y, Saito K. Remarkable role of indoleamine 2,3-dioxygenase and tryptophan metabolites in infectious diseases: potential role in macrophage-mediated inflammatory diseases. Mediators Inflamm. 2013;2013:391984. | |
Schmetterer KG, Neunkirchner A, Pickl WF. Naturally occurring regulatory T cells: markers, mechanisms, and manipulation. FASEB J. 2012;26(6):2253–2276. | |
Sant AJ, Chaves FA, Leddon SA, Tung J. The control of the specificity of CD4 T cell responses: thresholds, breakpoints, and ceilings. Front Immunol. 2013;4:340. | |
Munn DH. Blocking IDO activity to enhance anti-tumor immunity. Front Biosci (Elite Ed). 2012;4:734–745. | |
Dürr S, Kindler V. Implication of indolamine 2,3 dioxygenase in the tolerance toward fetuses, tumors, and allografts. J Leukoc Biol. 2013; 93(5):681–687. | |
Platten M, Wick W, Van den Eynde BJ. Tryptophan catabolism in cancer: beyond IDO and tryptophan depletion. Cancer Res. 2012;72(21):5435–5440. | |
Johnson TS, Munn DH. Host indoleamine 2,3-dioxygenase: contribution to systemic acquired tumor tolerance. Immunol Invest. 2012;41(6–7):765–797. | |
Catena-Dell’Osso M, Rotella F, Dell’Osso A, Fagiolini A, Marazziti D. Inflammation, serotonin and major depression. Curr Drug Targets. 2013;14(5):571–577. | |
Mándi Y, Vécsei L. The kynurenine system and immunoregulation. J Neural Transm. 2012;119(2):197–209. | |
Xiao Y, Christou H, Liu L, et al. Endothelial indoleamine 2,3-dioxygenase protects against development of pulmonary hypertension. Am J Respir Crit Care Med. 2013;188(4):482–491. | |
Fujigaki S, Saito K, Takemura M, et al. L-tryptophan-L-kynurenine pathway metabolism accelerated by Toxoplasma gondii infection is abolished in gamma interferon-gene-deficient mice: cross-regulation between inducible nitric oxide synthase and indoleamine-2,3- dioxygenase. Infect Immun. 2002;70(2):779–786. | |
Oh GS, Pae HO, Choi BM, et al. 3-Hydroxyanthranilic acid, one of metabolites of tryptophan via indoleamine 2,3-dioxygenase pathway, suppresses inducible nitric oxide synthase expression by enhancing heme oxygenase-1 expression. Biochem Biophys Res Commun. 2004; 320(4):1156–1162. | |
Orabona C, Pallotta MT, Volpi C, et al. SOCS3 drives proteasomal degradation of indoleamine 2,3-dioxygenase (IDO) and antagonizes IDO-dependent tolerogenesis Proc Natl Acad Sci U S A. 2008;105(52):20828–20833. | |
Beutelspacher SC, Tan PH, McClure MO, Larkin DF, Lechler RI, George AJ. Expression of indoleamine 2,3-dioxygenase (IDO) by endothelial cells: implications for the control of alloresponses. Am J Transplant. 2006;6(6):1320–1330. | |
Lahdou I, Engler C, Mehrle S, et al. Role of human corneal endothelial cells in T-cell-mediated alloimmune attack in vitro. Invest Ophthalmol Vis Sci. 2014;55(3):1213–1221. | |
Mouratidis PX, George AJ. Transfection of indoleamine 2,3 dioxygenase in primary endothelial cells. Methods Mol Biol. 2010;616:149–159. | |
Takikawa O, Kuroiwa T, Yamazaki F, Kido R. Mechanism of interferon-gamma action. Characterization of indoleamine 2,3-dioxygenase in cultured human cells induced by interferon-gamma and evaluation of the enzyme-mediated tryptophan degradation in its anticellular activity. J Biol Chem. 1988;263(4):2041–2048. | |
Alegre E, López AS, González A. Tryptophan metabolites interfere with the Ehrlich reaction used for the measurement of kynurenine. Anal Biochem. 2005;339(1):188–189. | |
Kommineni VK, Nagineni CN, William A, Detrick B, Hooks JJ. IFN-gamma acts as anti-angiogenic cytokine in the human cornea by regulating the expression of VEGF-A and sVEGF-R1. Biochem Biophys Res Commun. 2008;374(3):479–484. | |
Alberati-Giani D, Malherbe P, Ricciardi-Castagnoli P, Köhler C, Denis-Donini S, Cesura AM. Differential regulation of indoleamine 2,3-dioxygenase expression by nitric oxide and inflammatory mediators in IFN-gamma-activated murine macrophages and microglial cells. J Immunol. 1997;159(1):419–426. | |
Sarrazin S, Bonnaffé D, Lubineau A, Lortat-Jacob H. Heparan sulfate mimicry: a synthetic glycoconjugate that recognizes the heparin binding domain of interferon-gamma inhibits the cytokine activity. J Biol Chem. 2005;280(45):37558–37564. | |
Fluhr H, Spratte J, Heidrich S, Ehrhardt J, Steinmüller F, Zygmunt M. Heparin inhibits interferon-γ signaling in human endometrial stromal cells by interference with the cellular binding of interferon-γ. Fertil Steril. 2011;95(4):1272–1277. | |
Bonmann E, Jüttler E, Krestel HE, Spranger M. Heparin inhibits induction of nitric oxide synthase by cytokines in rat brain microvascular endothelial cells. Neurosci Lett. 1998;253(2):95–98. | |
Hills FA, Abrahams VM, González-Timón B, et al. Heparin prevents programmed cell death in human trophoblast. Mol Hum Reprod. 2006;12(4):237–243. | |
Hatakeyama M, Imaizumi T, Tamo W, et al. Heparin inhibits IFN-gamma-induced fractalkine/CX3CL1 expression in human endothelial cells. Inflammation. 2004;28(1):7–13. | |
Marti LC, Pavon L, Severino P, Sibov T, Guilhen D, Moreira-Filho CA. Vascular endothelial growth factor-A enhances indoleamine 2,3-dioxygenase expression by dendritic cells and subsequently impacts lymphocyte proliferation. Mem Inst Oswaldo Cruz. 2013;109(1):70–79. | |
Pallotta MT, Orabona C, Volpi C, Grohmann U, Puccetti P, Fallarino F. Proteasomal degradation of indoleamine 2,3-dioxygenase in CD8 dendritic cells is mediated by suppressor of cytokine signaling 3 (SOCS3). Int J Tryptophan Res. 2010;3:91–97. | |
Thomas SR, Terentis AC, Cai H, et al. Post-translational regulation of human indoleamine 2,3-dioxygenase activity by nitric oxide. J Biol Chem. 2007;282(33):23778–23787. | |
Max D, Kühnöl CD, Burdach S, Niu L, Staege MS, Föll JL. Indoleamine- 2,3-dioxygenase in an immunotherapy model for Ewing sarcoma. Anticancer Res. 2014;34:6431–6441. | |
Park G, Choia YJ, Lee SE, et al. A paradoxical pattern of indoleamine 2,3-dioxygenase expression in the colon tissues of patients with acute graft-versus-host disease. Exp Hematol. 2014;42:734–740. | |
Wang Y, Liu H, McKenzie GK etal. Kynurenine is an endothelium-derived relaxing factor produced during inflammation. Nat Med. 2010;16(3):279–285. |
Supplementary materials
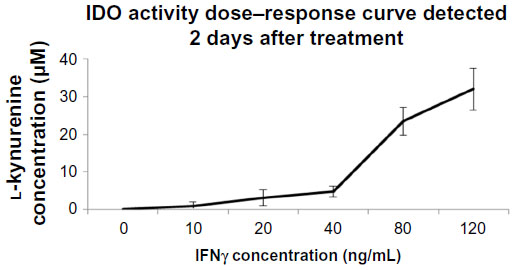 | Figure S1 IDO dose–response curve. |
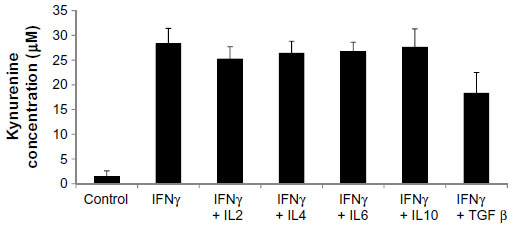 | Figure S2 IDO activity after treatment of human saphenous vein endothelial cells with cytokines for 2 days. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.