")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 9 » Issue 1
Pulmonary adenocarcinoma mutation profile in smokers with smoking-related interstitial fibrosis
Authors Primiani A, Dias-Santagata D, Iafrate AJ, Kradin R
Received 5 February 2014
Accepted for publication 31 March 2014
Published 24 May 2014 Volume 2014:9(1) Pages 525—531
DOI https://doi.org/10.2147/COPD.S61932
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Andrea Primiani,1 Dora Dias-Santagata,1 A John Iafrate,1 Richard L Kradin1,2
1Pathology Service, 2Pulmonary Medicine/Critical Care Unit, Department of Medicine, Massachusetts General Hospital and Harvard Medical School, Boston, MA, USA
Abstract: Cigarette smoking is an established cause of lung cancer. However, pulmonary fibrosis is also an independent risk factor for the development of lung cancer. Smoking-related interstitial fibrosis (SRIF) has recently been reported. We hypothesized that adenocarcinomas in lungs with SRIF might show distinct molecular changes and examined the molecular phenotype of 168 resected lung adenocarcinomas in lungs with and without SRIF. The diagnosis of SRIF was determined by histological examination, based on the presence of alveolar septal thickening, due to pauci-inflamed, hyalinized, “ropy” collagen, in areas of lung greater than 1 cm away from the tumor. Tumors were concomitantly examined genotypically for mutations in genes frequently altered in cancer, including EGFR and KRAS, by SNaPshot and by fluorescence in situ hybridization for possible ALK rearrangements. Fluorescence in situ hybridization for ROS1 rearrangement (n=36) and/or MET amplification (n=31) were performed when no mutation was identified by either SNaPshot or ALK analysis. Sixty-five cases (38.7%) showed SRIF, which was distributed in all lobes of the lungs examined. No differences were observed in sex, average age, or smoking history in patients with and without SRIF. There was no difference in either the percent or types of adenocarcinoma genetic mutations in patients with SRIF versus those without. This data suggests that SRIF does not represent an independent risk factor for the development of the major known and targeted mutations seen in pulmonary adenocarcinoma. However, additional research is required to investigate the potential significance of SRIF in the pathogenesis of lung cancer.
Keywords: lung, cancer, smoking, SRIF
Introduction
Cigarette smoking is the leading cause of lung carcinoma. Lung cancer has also been linked to various fibrosing lung diseases, including idiopathic pulmonary fibrosis (IPF), systemic sclerosis, and dermatomyositis, and certain pneumoconioses (eg, asbestosis and silicosis) such that pulmonary fibrosis may represent an independent risk factor for developing lung cancer.1–4 However, the role of pulmonary fibrosis in pathogenesis of lung cancer has not been elucidated. Recently, investigators have described a distinct type of pulmonary interstitial fibrosis in smokers, smoking-related interstitial fibrosis (SRIF). SRIF is characterized by the presence of alveolar septal thickening by hyalinized collagen with little associated inflammation and is histologically distinguishable from other fibrosing lung diseases.5,6 Furthermore, pulmonary fibrosis associated with emphysema has been recognized as a pattern of lung abnormality in the radiographic imaging of smokers and has been associated with an increased risk of lung cancer in prior studies.7–10 Thus, we hypothesized that SRIF might be associated with lung cancer with specific genetic alterations. For this reason, we interrogated multiple genes for patterns of mutations in lung adenocarcinomas in patients with concomitant evidence of SRIF.
Materials and methods
Case selection
The files of the James Homer Wright Pathology Laboratory of the Massachusetts General Hospital were searched for lung specimens resected between 2008 and 2012 with the diagnosis of pulmonary adenocarcinoma. Only cases in which a substantial portion of lung parenchyma uninvolved by tumor was resected (ie, large wedge resections, multiple wedge resections, segmentectomies, lobectomies, or pneumonectomies) were included. Additionally, only cases with tumors on which genetic testing had been performed for clinical purposes were included. Specimens resected from nonsmokers and patients with a prior history of neoadjuvant radiation therapy were excluded. A smoking history was obtained from the patients’ medical records and based on self-reporting. If available, imaging studies were reviewed.
Histologic evaluation
Two pathologists (AP and RLK) retrospectively examined hematoxylin and eosin (H&E)-stained sections of lung parenchyma uninvolved by and distant from tumor in order to evaluate for emphysema and SRIF. The number of slides examined was between two and ten per case, depending on the size of the resection specimen and extent of tumor involvement. The criteria used for the diagnosis of SRIF were the presence of paucicellular, eosinophilic, collagenous thickening of alveolar septae in at least 30% of the examined lung parenchyma that was uninvolved by tumor (specifically, >1 cm away from the tumor), with no identified confounding cause. Respiratory bronchiolitis was defined by the presence of pigmented (smoker’s) macrophages within the respiratory bronchioles and/or surrounding peribronchiolar alveoli.
Mutational analysis
Tumors (n=168) were examined genotypically for 71 hot spot mutations in 14 genes frequently altered in cancers (including AKT1, APC, BRAF, B-catenin, EGFR, IDH1, KIT, KRAS, MAP2K1, NOTCH1, NRAS, PIK3CA, PTEN, and TP53) by SNaPshot (Applied Biosystems, Foster City, CA, USA), a previously described mutational analysis assay.11 Briefly, DNA from formalin-fixed, paraffin-embedded tissue was subjected to a multiplexed polymerase chain reaction (PCR) and single-base extension reaction platform in order to generate fluorescently labeled signals that interrogate hot-spot mutation sites. The SNaPshot products were then analyzed by capillary electrophoresis to determine selected hot-spot mutations. In addition, direct PCR sequencing techniques were used to assay EGFR exons 19 and 20 and ERBB2 (HER2) exon 20 for in-frame activating insertions or deletions.
Fluorescence in situ hybridization (FISH) was performed on deparaffinized, protease-treated, 5 μm thick sections cut from tumor material from most cases (n=162) in order to evaluate for the presence of ALK rearrangement. H&E-stained slides were reviewed to select regions of the tissue that contain mostly tumor cells to be used for hybridization. Dual-color break-apart probes, constructed from BAC clones, specific for the 3′ and 5′ sequences of the ALK gene were hybridized according to the manufacturer’s protocol using a Hybrite slide processor (Abbott Molecular, Des Plaines, IL, USA). A case was considered positive for ALK rearrangement if 15% of cells contained split fluorescent signals.
In tumors in which no mutation was identified by the SNaPshot assay and no ALK rearrangement was present, FISH for ROS1 rearrangement (n=36) and/or FISH for MET amplification (n=31) were performed. The presence of ROS1 rearrangement was evaluated using a protocol identical to that described for ALK FISH, with ROS1-specific 3′ and 5′ sequence probes. To analyze MET gene amplification status, deparaffinized, protease-treated, 5 μm thick sections cut from tumor material (as selected based on review of H&E-stained slides) were hybridized with dual-color probes for the MET locus and centromere on chromosome 7 according to the manufacturer’s protocol using a Hybrite slide processor (Abbott Molecular). MET amplification was considered present when the MET: centromere 7 signal ratio was 2.2 or greater.
Statistical analysis
The difference in age and smoking history in patients with and without SRIF was compared by means of an unpaired t-test. The Fisher’s exact test was used to compare the sex distribution, presence of emphysema, and distribution of examined parenchyma in cases with and without SRIF. The presence of genetic alterations in evaluated genes was compared between lung adenocarcinoma cases with and without background SRIF using the Fisher’s exact test. Two-tailed P-values less than 0.05 were considered statistically significant.
Results
One hundred and sixty-eight lung adenocarcinoma resections were included in the study; these included four wedge resections, four with multiple wedge resections, four segmentectomies, two partial lobectomies, 144 lobectomies, and ten pneumonectomies. The number of H&E-stained slides from lung parenchyma uninvolved by and at a distance greater than 1 cm from the pulmonary malignancy that were examined varied from two to ten per case. On histologic review, 65 cases (38.7%) showed SRIF in the lung uninvolved by tumor; 103 (61.3%) cases did not show SRIF. In cases with SRIF, alveolar septal fibrosis with dense, eosinophilic, paucicellular collagen bundles was present in the subpleural lung parenchyma with centrilobular peribronchiolar accentuation (Figure 1). The findings of SRIF were distributed in all lung lobes (Table 1). Among cases with SRIF, 61 (93.8%) had concomitant emphysema on histologic examination. In these cases, SRIF was seen both in and away from the emphysematous lung parenchyma. In contrast, 85 (82.5%) patients without SRIF had emphysema on histologic exam (P=0.04). Of note, all cases showed patchy evidence of respiratory bronchiolitis.
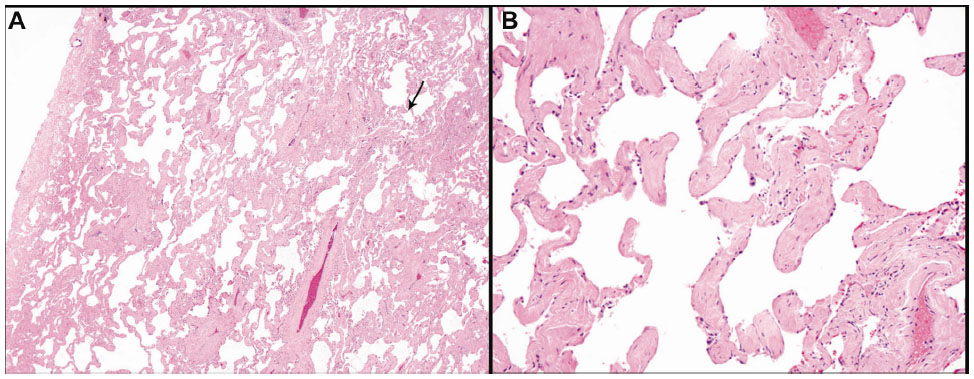 | Figure 1 (A) Smoking-related interstitial fibrosis involving subpleural lung parenchyma (pleural surface is at the left) with centrilobular accentuation (arrow); (B) thickening of the alveolar septa by dense, eosinophilic, paucicellular collagen bundles. |
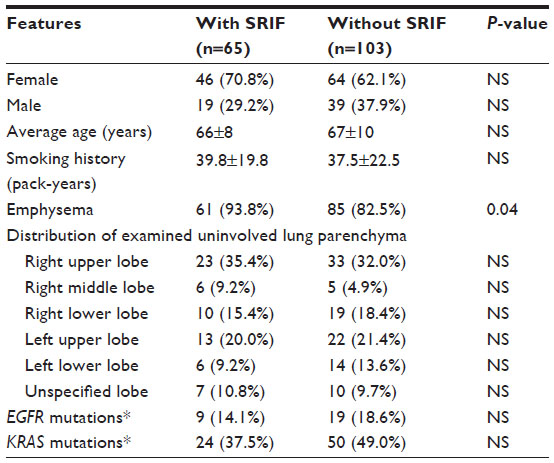 | Table 1 Clinical and pathologic features of pulmonary adenocarcinoma cases with and without SRIF |
A history of cigarette smoking was documented in all cases. The smoking history elicited from patients with and without SRIF was 39.8±19.8 pack years and 37.5±22.5 pack years, respectively (P=0.41) (Table 1). The average age at resection among patients with and without SRIF was 66±8 years and 67±10 years, respectively (P=0.85). There was no significant difference in the sex of patients with or without SRIF (70.8% vs 62.1%, respectively; P=0.32). Results of imaging studies were not consistently available for review; however, few patients had evidence of SRIF on computed tomography (CT) evaluation (Figure 2).
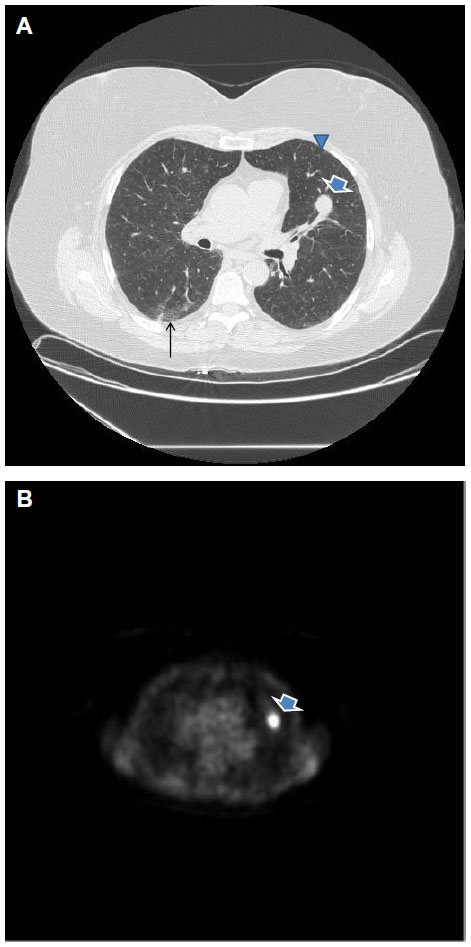 | Figure 2 Spiculated nodule in the left upper lobe (arrow in A) with associated 18-FDG uptake on positron emission tomography scan (B), consistent with lung cancer. |
Genetic analysis
One of three sequentially updated versions of SNaPshot and direct PCR sequencing assays were performed on 64 cases with SRIF and 102 cases without SRIF. ALK FISH, ROS FISH, and MET FISH were performed on 64, 16, and 13 cases with SRIF, respectively, and on 99, 20, and 18 cases without SRIF, respectively. There was no difference in the number of mutations present in each of the 14 interrogated genes or in the prevalence of individual hotspot mutations in cases with and without SRIF. Mutations in EGFR were observed in 9 (14.1%) cases with SRIF and 19 (18.6%) cases without SRIF (P=0.53); mutations in KRAS occurred in 24 (37.5%) cases with SRIF and in 50 (49.0%) cases without SRIF (P=0.15) (Table 1). Finally, there was no difference in the number of cases with and without SRIF in which no genetic mutation was identified (27 and 30, respectively; P=0.13).
Discussion
SRIF was present in the background lung parenchyma of 38.7% of lungs from smokers with pulmonary adenocarcinoma. This is comparable to other recently reported studies of SRIF.12–15 Katzenstein et al evaluated lobectomy specimens resected for a malignant neoplasm in 20 smokers and identified patchy widening of alveolar septa by acellular collagen deposition with minimal or focal associated inflammation in 45% of cases.12 SRIF was predominantly subpleural, but also occurred with or without associated emphysema in deep lung parenchyma in a centrilobular distribution, as in the present study. Similar lesions had previously been described in up to 50% of smokers, although authors have ascribed different terms to comparable histologic findings, including “respiratory bronchiolitis-associated interstitial lung disease with fibrosis” and “airspace enlargement with fibrosis”.13–15 These interstitial changes were absent in nonsmokers and must be distinguished from other fibrosing interstitial lung diseases, including IPF and nonspecific interstitial pneumonia, which have other important clinical implications.4,5,13–15 The distinction of SRIF from other chronic interstitial fibrosing lesions not only has prognostic implications but also affects therapeutic interventions.
While SRIF seems to be a common finding in smokers, clinical symptoms and radiologic findings associated with this pattern of interstitial lung disease are uncommon.12–14 However, the index group of patients with SRIF described by Yousem et al presented with signs and symptoms of chronic interstitial lung disease and, for this reason, underwent lung biopsy.13 Katzenstein et al identified mild to moderate reduction in diffusion capacity in four of nine (44%) patients with SRIF who underwent pulmonary function testing.12 Notably, several radiology studies have reviewed CT findings in smokers and found subtle interstitial changes in a subset of smokers. Lederer et al found ground glass and reticular opacities on CT scan in 2.2% of smokers, with increasing numbers of these areas in patients with increasing pack years.16 More recently, Washko et al and Sverzellati et al reported interstitial lung abnormalities in 8% and 22.8% of smokers, respectively.17,18 Thus, while SRIF is often an incidental finding recognized in lung parenchyma, these studies suggest that the increasing use of high-resolution CT may increase the detection of interstitial lung abnormalities in asymptomatic smokers and lung biopsies may be utilized to provide insight into the etiology of a patient’s pulmonary interstitial abnormalities.
It is possible that SRIF is part of the spectrum of changes seen in emphysema as, in this study, emphysema was more often present in cases with SRIF than in those without SRIF. Smokers with both pulmonary fibrosis and emphysema have been previously described.7,19–22 Cottin et al coined the term “combined pulmonary fibrosis and emphysema” (CPFE) in a retrospective study of 61 patients with upper lobe emphysema and lower lobe pulmonary fibrosis on high-resolution CT scan.7 Notably, CPFE has been associated with an increased risk of lung cancer. Kitaguchi et al reported a 46.8% prevalence of lung cancer in patients with CPFE as compared to 7.3% in patients with chronic obstructive pulmonary disease alone.8 More recently, Usui et al evaluated 1,143 consecutive patients with primary lung carcinoma and found that 8.9% had evidence of CPFE on CT scan at the time of cancer diagnosis.9 This association between CPFE and lung cancer may reflect the importance of cigarette smoking as a predisposing factor. However, the presence of interstitial fibrosis might be an independent factor in the development and progression of lung cancer.9,10
In 1939, Friedrich23 first proposed the association between pulmonary fibrosis and lung cancer. Subsequent studies have supported this relationship.2–4 IPF has been implicated as an independent risk factor for the development of lung cancer, although there is an increased incidence of IPF in smokers.1,24–30 Furthermore, molecular studies have demonstrated the presence of genetic alterations, especially in tumor suppressor genes such as p53, within lung tissue from patients with IPF.31–36 Thus, it is hypothesized that repeated inflammation, injury, and repair of the lung parenchyma may predispose to the development of lung cancer. More recently, inappropriate cytokine signaling has been implicated as a causative mechanism of IPF and may play a role in pulmonary carcinogenesis.37
Lung cancer in the setting of ILD is the most common malignancy to develop in patients with systemic sclerosis.38–42 Similarly to IPF, lung cancer in these patients may result from cellular injury and genetic damage caused by continued pulmonary inflammation and fibrosis. In addition, other rheumatologic diseases, such as dermatomyositis, rheumatoid arthritis, systemic lupus erythematosus, and sarcoidosis, and pneumoconioses, including asbestosis, silicosis, and berylliosis, have been associated with an increased risk of the development of lung cancer.2–4 Notably, although the literature is controversial, studies have suggested that, among patients with asbestosis, there is an increased prevalence of tumors within the lower lung lobes; ie, in the most fibrotic regions of the lung parenchyma.43,44
In light of the strong association of pulmonary fibrosis and lung cancer, we hypothesized that SRIF might be associated with a distinct molecular pathway in the development of lung cancer in smokers, despite the underlying relationship between both SRIF and cancer with cigarette smoking. Recent studies have showed an inverse relationship between the presence of interstitial lung disease and EGFR mutations in associated lung tumors.45,46 However, unlike our study, these studies did not specifically evaluate the role of SRIF in cancer progression, as both smokers and nonsmokers and patients with multiple subtypes of interstitial fibrosis were included in these studies. We found no difference in the number or types of genetic mutations in adenocarcinomas that develop in lungs with and without SRIF, which suggests that there is no difference in the molecular pathogenesis of adenocarcinomas that arise within smokers in the presence of SRIF. However, due to the limited sample size, additional research must be done to further investigate the potential clinical significance of SRIF and subsequent impact on cancer progression. In addition, the mechanism by which other fibrosing lung diseases, such as IPF, can lead to cancer development must be clarified.
This study has several limitations. Firstly, information regarding results of pulmonary function tests and imaging studies were not consistently available for review. A comparison of the mutations present in tumors from patients in our cohort with normal and abnormal lung function and those with and without evidence of interstitial fibrosis on imaging may have proved insightful. Secondly, this study was retrospective and nonrandomized. Most mutations, such as rearrangements in ALK and ROS, occurred in only small subsets of lung adenocarcinomas, so that it was not possible to make statistically relevant conclusions concerning these cases. A prospective randomized controlled study with systematic sampling of both subpleural and deep lung parenchyma away from the tumor would allow for more accurate assessment of the presence of SRIF. In addition, limited numbers of genes were included in the SNaPshot and FISH analysis that were performed, such that other mutations of possible relevance were not interrogated. Future studies adopting broad next-generation sequencing techniques may yield greater insight into the possible role of SRIF in the pathways of carcinogenesis. It is, of course, possible that the same carcinogens that lead to mutations in the lung epithelial genome are responsible for SRIF, but this remains to be investigated. However, despite the limitations of the present study, it is, to our knowledge, the first to explore the possible relationship between SRIF and the pathogenesis of lung cancer.
Acknowledgments
We thank Dr Subba Digumarthy from the Radiology Department at Massachusetts General Hospital for his help in selecting radiographic images, and Douglas Hayden from the Massachusetts General Hospital Biostatistics Center for his advice regarding the statistical analysis used in this study.
Disclosure
DDS and AJI serve as consultants for Bioreference Laboratories. The authors declare no further conflicts of interest in this work.
References
Aubry MC, Myers JL, Douglas WW, et al. Primary pulmonary carcinoma in patients with idiopathic pulmonary fibrosis. Mayo Clin Proc. 2002;77(8):763–770. | |
Artinian V, Kvale PA. Cancer and interstitial lung disease. Curr Opin Pulm Med. 2004;10(5):425–434. | |
Daniels CE, Jett JR. Does interstitial lung disease predispose to lung cancer? Curr Opin Pulm Med. 2005;11(5):431–437. | |
Archontogeorgis K, Steiropoulous P, Tzouvelekis EN, Bouros D. Lung cancer and interstitial lung disease: a systematic review. Pulm Med. 2012;2012:1–11. | |
Katzenstein AL. Smoking-related interstitial fibrosis (SRIF), pathogenesis and treatment of usual interstitial pneumonia (UIP), and transbronchial biopsy in UIP. Mod Pathol. 2012;25 Suppl 1:S68–S78. | |
Katzenstein AL. Smoking-related interstitial fibrosis (SRIF): pathologic findings and distinction from other chronic fibrosing lung diseases. J Clin Pathol. 2013;66(10):882–887. | |
Cottin V, Nunes H, Brillet PY, et al; Groupe d’Etude et de Recherche sur les Maladies Orphelines Pulmonaires (GERM O P). [Combined pulmonary fibrosis and emphysema: a distinct underrecognised entity]. Eur Respir J. 2005;26(4):586–593. French. | |
Kitaguchi Y, Fujimoto K, Hanaoka M, Kawakami S, Honda T, Kubo K. Clinical characteristics of combined pulmonary fibrosis and emphysema. Respirology. 2010;15(2):265–271. | |
Usui K, Tanai C, Tanaka Y, Noda H, Ishihara T. The prevalence of pulmonary fibrosis combined with emphysema in patients with lung cancer. Respirology. 2011;16(2):326–331. | |
Jankowich MD, Rounds SI. Combined pulmonary fibrosis and emphysema syndrome: a review. Chest. 2012;141(1):222–231. | |
Dias-Santagata D, Akhavanfard S, David SS, et al. Rapid targeted mutational analysis of human tumours: a clinical platform to guide personalized cancer medicine. EMBO Mol Med. 2010;2(5):146–158. | |
Katzenstein AL, Mukhopadhyay S, Zanardi C, Dexter E. Clinically occult interstitial fibrosis in smokers: classification and significance of a surprisingly common finding in lobectomy specimens. Hum Pathol. 2010;41(3):316–325. | |
Yousem SA. Respiratory bronchiolitis-associated interstitial lung disease with fibrosis is a lesion distinct from fibrotic nonspecific interstitial pneumonia: a proposal. Mod Pathol. 2006;19(11):1474–1479. | |
Fraig M, Shreesha U, Savici D, Katzenstein AL. Respiratory bronchiolitis: a clinicopathologic study in current smokers, ex-smokers, and never-smokers. Am J Surg Pathol. 2002;26(5):647–653. | |
Kawabata Y, Hoshi E, Murai K, et al. Smoking-related changes in the background lung of specimens resected for lung cancer: a semiquantitative study with correlation to postoperative course. Histopathology. 2008;53(6):707–714. | |
Lederer DJ, Enright PL, Kawut SM, et al. Cigarette smoking is associated with subclinical parenchymal lung disease: the Multi-Ethnic Study of Atherosclerosis (MESA)-lung study. Am J Respir Crit Care Med. 2009;180(5):407–414. | |
Washko GR, Hunninghake GM, Fernandez IE, et al; COPDGene Investigators. Lung volumes and emphysema in smokers with interstitial lung abnormalities. N Engl J Med. 2011;364(10):897–906. | |
Sverzellati N, Guerci L, Randi G, et al. Interstitial lung diseases in a lung cancer screening trial. Eur Respir J. 2011;38(2):392–400. | |
Wiggins J, Strickland B, Turner-Warwick M. Combined cryptogenic fibrosing alveolitis and emphysema: the value of high resolution computed tomography in assessment. Respir Med. 1990;84(5):365–369. | |
Jankowich MD, Polsky M, Klein M, Rounds S. Heterogeneity in combined pulmonary fibrosis and emphysema. Respiration. 2008;75(4):411–417. | |
Grubstein A, Bendayan D, Schactman I, Cohen M, Shitrit D, Kramer MR. Concomitant upper-lobe bullous emphysema, lower-lobe interstitial fibrosis and pulmonary hypertension in heavy smokers: report of eight cases and review of the literature. Respir Med. 2005;99(8):948–954. | |
Reddy TL, Mayo J, Churg A. Respiratory bronchiolitis with fibrosis. High-resolution computed tomography findings and correlation with pathology. Ann Am Thorac Soc. 2013;10(6):590–601. | |
Friedrich G. Periphere Lungenkrebse auf dem Bodem pleuranaher narben. Virchows Arch Pathol Anat Physiol. 1939;304(1–2):230–247. German. | |
Meyer EC, Liebow AA. Relationship of interstitial pneumonia honeycombing and atypical epithelial proliferation to cancer of the lung. Cancer. 1965;18:322–351. | |
Matsushita H, Tanaka S, Saiki Y, et al. Lung cancer associated with usual interstitial pneumonia. Pathol Int. 1995;45(12);925–932. | |
Hironaka M, Fukayama M. Pulmonary fibrosis and lung carcinoma: a comparative study of metaplastic epithelia in honeycombed areas of usual interstitial pneumonia with or without lung carcinoma. Pathol Int. 1999;49(12):1060–1066. | |
Hubbard R, Venn A, Lewis S, Britton J. Lung cancer and cryptogenic fibrosing alveolitis. A population-based cohort study. Am J Respir Crit Care Med. 2000;161(1):5–8. | |
Park J, Kim DS, Shim TS, et al. Lung cancer in patients with idiopathic pulmonary fibrosis. Eur Respir J. 2001;17(6):1216–1219. | |
Ozawa Y, Suda T, Naito T, et al. Cumulative incidence of and predictive factors for lung cancer in IPF. Respirology. 2009;14(5):723–728. | |
Harris JM, Johnston ID, Rudd R, Taylor AJ, Cullinan P. Cryptogenic fibrosing alveolitis and lung cancer: the BTS study. Thorax. 2010;65(1):70–76. | |
Yamanouchi H, Fujita J, Hojo S, et al. Neutrophil elastase: alpha-1 proteinase inhibitor complex in serum and bronchoalveolar lavage fluid in patients with pulmonary fibrosis. Eur Respir J. 1998;11(1):120–125. | |
Hojo S, Fujita J, Yamadori I, et al. Heterogeneous point mutations of the p53 gene in pulmonary fibrosis. Eur Respir J. 1998;12(6):1404–1408. | |
Vassilakis DA, Sourvinos G, Spandidos DA, Siafakas NM, Bouros D. Frequent genetic alterations at the microsatellite level in cytologic sputum samples of patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2000;162(3 Pt 1):1115–1119. | |
Uematsu K, Yoshimura A, Gemma A, et al. Aberrations in the fragile histidine triad (FHIT) gene in idiopathic pulmonary fibrosis. Cancer Res. 2001;61(23):8527–8533. | |
Takahashi T, Munakata M, Ohtsuka Y, et al. Expression and alteration of ras and p53 proteins in patients with lung carcinoma accompanied by idiopathic pulmonary fibrosis. Cancer. 2002;95(3):624–633. | |
Plataki M, Koutsopoulos AV, Darivianaki K, Delides G, Siafakas NM, Bouros D. Expression of apoptotic and antiapoptotic markers in epithelial cells in idiopathic pulmonary fibrosis. Chest. 2005;127(1):266–274. | |
Noble PW, Homer RJ. Idiopathic pulmonary fibrosis: new insights into pathogenesis. Clin Chest Med. 2004;25(4):749–758. | |
Roumm AD, Medsger TA. Cancer and systemic sclerosis: an epidemiologic study. Arthritis Rheum. 1985;28(12):1336–1340. | |
Abu-Shakra M, Guillemin F, Lee P. Cancer in systemic sclerosis. Arthritis Rheum. 1993;36(4):460–464. | |
Rosenthal AK, McLaughlin JK, Gridley G, Nyren O. Incidence of cancer among patients with systemic sclerosis. Cancer. 1995;76(5):910–914. | |
Hesselstrand R, Scheja A, Akesson A. Mortality and causes of death in a Swedish series of systemic sclerosis patients. Ann Rheum Dis. 1998;57(11):682–686. | |
Hill CL, Nguyen AM, Roder D, Roberts-Thomson P. Risk of cancer in patients with scleroderma: a population based cohort study. Ann Rheum Dis. 2003;62(8):728–731. | |
Selikoff IJ, Bader RA, Bader ME, Churg J, Hammond EC. Asbestosis and neoplasia. Am J Med. 1967;42(4):487–496. | |
Sluis-Cremer GK. The relationship between asbestosis and bronchial cancer. Chest. 1980;78(Suppl 2):380–381. | |
Usui K, Ushijima T, Tanaka Y, et al. The frequency of epidermal growth factor receptor mutation of nonsmall cell lung cancer according to the underlying pulmonary diseases. Pulm Med. 2011;2011:290132. | |
Fujimoto D, Tomii K, Otoshi T, et al. Preexisting interstitial lung disease is inversely correlated to tumor epidermal growth factor receptor mutation in patients with lung adenocarcinoma. Lung Cancer. 2013;80(2):159–164. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.