")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 10
Profile of minocycline and its potential in the treatment of schizophrenia
Received 17 March 2014
Accepted for publication 22 April 2014
Published 17 June 2014 Volume 2014:10 Pages 1103—1111
DOI https://doi.org/10.2147/NDT.S64236
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Lulu Zhang,1,2 Jingping Zhao1
1Mental Health Institute of the Second Xiangya Hospital, Key Laboratory of Psychiatry and Mental Health of Hunan Province, Central South University, Changsha, Hunan, 2Department of Psychology, Guangzhou First People’s Hospital, Guangzhou, Guangdong, People’s Republic of China
Abstract: Accumulating evidence suggests that neuroinflammation affecting microglia plays an important role in the etiology of schizophrenia, and appropriate control of microglial activation may be a promising therapeutic strategy for schizophrenia. Minocycline, a second-generation tetracycline that inhibits microglial activation, has been shown to have a neuroprotective effect in various models of neurodegenerative disease, including anti-inflammatory, antioxidant, and antiapoptotic properties, and an ability to modulate glutamate-induced excitotoxicity. Given that these mechanisms overlap with neuropathologic pathways, minocycline may have a potential role in the adjuvant treatment of schizophrenia, and improve its negative symptoms. Here, we review the relevant studies of minocycline, ranging from preclinical research to human clinical trials.
Keywords: schizophrenia, minocycline, microglia, neuroinflammation
Introduction
Schizophrenia is a chronic and often debilitating illness affecting approximately 1% of the world population.1 It is a complex disorder characterized by profound disturbance of perception, cognition, emotion, and social function. The onset of schizophrenia is typically in late adolescence or early adulthood, and includes distinctive symptoms, commonly referred to as positive, negative, and cognitive. To date, effective treatments for schizophrenia have been limited to medications with antidopaminergic activity, which alleviate symptoms by augmenting dysfunctional neurotransmitter systems.2 While antipsychotics are most effective for positive symptoms, negative and cognitive symptoms are less well addressed.3 Treatment of patients with minimal or no response to adequate doses of antipsychotics represents an enormous challenge for clinicians. These antipsychotic-resistant patients constitute up to 25%–30% of all patients suffering from schizophrenia.2,4,5 One of the main reasons for this is our lack of understanding of the etiology of schizophrenia.
Although the exact mechanism of schizophrenia remains to be elucidated, several hypotheses have been proposed, including disruption of neurotransmitter systems,6–10 genetic factors,11 and neurodevelopmental and neurotoxic mechanisms.12,13 In recent years, however, growing evidence has supported the idea that neuroinflammation, in particular that focused on the microglia, plays an important role in the etiology of schizophrenia, so appropriate control of microglial activation may be a promising strategy in the treatment of the disease.1,14–16 Minocycline, a second-generation tetracycline, has a distinct neuroprotective profile independent of its antibacterial activity.17 Minocycline is almost completely absorbed when taken orally and shows excellent penetration of brain tissue. These properties, as well as its beneficial effect in animal models of neurologic disorders, has led to investigation of its potential use in the treatment of schizophrenia.18,19 Recent reports have demonstrated a possible antipsychotic effect for minocycline, which is a potent inhibitor of microglial activation. In these studies, use of minocycline as an adjuvant to antipsychotics was reported to be beneficial in patients with schizophrenia.20–23 Here we review the existing literature focusing on the preclinical and clinical potential of minocycline in the treatment of schizophrenia and propose directions for future research.
Overview of minocycline: howto confer neuroprotection?
Microglial hypothesis for schizophrenia
Although dopaminergic neurotransmission is involved in the pathophysiology of schizophrenia, the exact mechanism leading to dopaminergic dysfunction remains unclear. Accumulating evidence indicates the significance of neuroinflammation involving microglia in schizophrenia.16,24 Maternal inflammation during critical stages of gestation has been shown to form the basis of the link between prenatal infection and schizophrenia.25,26 Longitudinal studies in animal models further indicate that infection-induced developmental neuroinflammation may be pathologically relevant beyond the antenatal and neonatal periods, and may contribute to progression of disease, associated with gradual development of full-blown schizophrenia.27 Arion et al carried out a DNA microarray study that showed increased expression of genes related to immune and chaperone function in the prefrontal cortex in schizophrenia.28 Narayan et al profiled genome-wide expression patterns in the prefrontal cortex in subjects with schizophrenia at different stages of the illness. Their study demonstrated that the molecular basis for schizophrenia changes between the early to chronic stages, providing evidence that the nature of schizophrenia changes with disease progression, with the long-term illness possibly being associated with inflammation, stimulus response, and immune function.29
Microglial cells are the primary reservoirs of proinflammatory cytokines, such as interleukin-6, tumor necrosis factor-alpha (TNF-α), and interferon-gamma (IFN-γ), and act as the main antigen-presenting cells in the central nervous system (CNS).30 Microglia are important for the cross-talk between the immune system and glutamatergic neurotransmission.31 The cells gradually become present in the brain and participate in various aspects of brain development, including cell death, axonal remodeling, synaptogenesis, and synaptic pruning.32–36 Prolonged microglial hyperactivity may lead to neuronal apoptosis and brain damage, which are commonly seen in neurodegenerative disorders such as Parkinson’s disease and Alzheimer’s disease.37,38 A neurodegenerative and neurodevelopmental process is indicated in the course of schizophrenia and may be associated with microglial activation.39,40 Animal studies, autopsy studies, and positron emission tomography (PET) studies have demonstrated that the neuropathology of schizophrenia is associated with microglial activation.41–49 Bayer et al found that there were more activated microglia in the frontal cortex and hippocampus in patients with schizophrenia post mortem than in controls.45 Their findings were strengthened further by positive results from other autopsy studies in patients with schizophrenia.43,44,46,47 The evidence from autopsy studies suggests that schizophrenia is associated with an increased number of activated microglia cells. However, autopsy studies cannot reflect dynamic changes in microglial activation over time. PET, a noninvasive brain imaging technique, provides the opportunity to study the presence of microglial activationin vivo. Van Berckel et al found overactive microglia in patients with schizophrenia of recent onset.48 Doorduin et al also reported finding more activated microglia in the hippocampus of patients with schizophrenia than in healthy volunteers, and suggested that focal neuroinflammation may play an important role in schizophrenia.49 As such, there has been growing evidence of a “microglial hypothesis of schizophrenia” (Figure 1) whereby stressful events, damage-associated molecular patterns, and pathogen-associated molecular patterns activate microglia in the CNS via immunologic or inflammatory activators. Proinflammatory cytokines and free radicals released by overactivated microglia can cause neuronal degeneration, abnormalities in white matter, and decreased neurogenesis. The damage done to the neuron by overactivated microglia may thus lead eventually to the development of schizophrenia.1,16 If microglial pathology proves to be an important causative factor in schizophrenia, the processes involving modulators of microglial activation may represent a novel therapeutic strategy.50,51
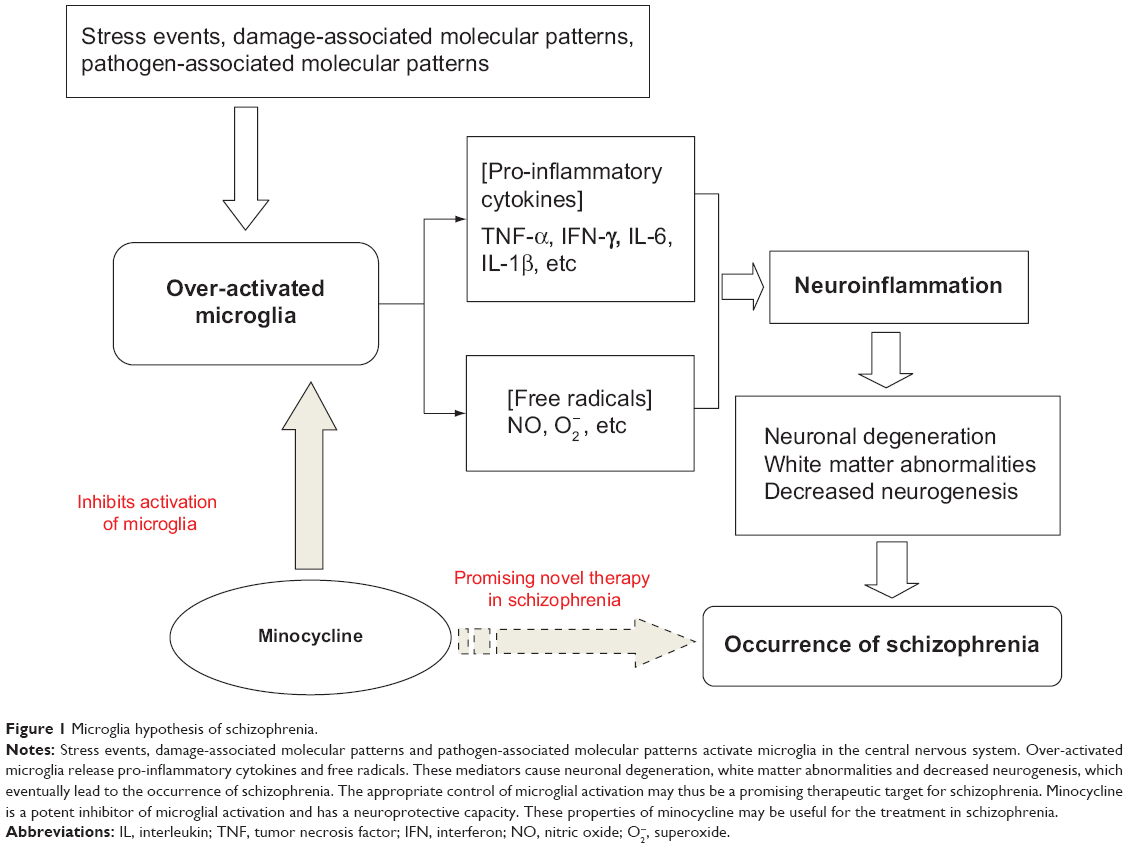 | Figure 1 Microglia hypothesis of schizophrenia. |
Targeting microglia
Inflammation of the CNS is detrimental to neurogenesis in the adult hippocampus.52,53 The negative effects of inflammation on differentiation and survival of neuronal cells are due to microglia-derived proinflammatory cytokines.52,54 Interleukin-1β and TNF-α have been reported to inhibit neurogenesis in vivo.55,56 However, neurogenesis can be restored by anti-inflammatory compounds such as minocycline, indomethacin, and cyclo-oxygenase-2 inhibitors (eg, celecoxib) which inhibit microglial activation.52,53,57,58 Other reports have shown that minocycline is able to inhibit microglial activation via anti-inflammatory and antioxidant properties which are independent of its antimicrobial action.59–67 Yrjanheikki et al showed that minocycline is able to provide neuroprotection against global ischemia in gerbils and focal brain ischemia in rats.59,60 This neuroprotection was associated with reduced activation of microglia and inhibition of interleukin-1β-converting enzyme, suggesting that minocycline may function by reducing the cytotoxic effects of the microglia. Minocycline can also reduce the proliferation and activation of resting microglia.61 Tikka et al investigated whether minocycline was able to reduce excitotoxicity in a primary neuronal culture, and found that glutamate-induced microglial activation occurred via the p38 mitogen-activated protein kinase pathway and that minocycline inhibited activation of this pathway in microglia, providing neuroprotection against excitotoxicity.62 Their findings were strengthened further by positive results in other studies using animal models of ischemic injury to the brain and neurodegenerative diseases.63–65 In addition, it was found that interleukin-1β levels following traumatic brain injury can be reduced in mice by a high dose of minocycline, and that increased TNF-α and nitric oxide levels in rats treated with 3-nitropropioic acid (an inhibitor of succinate dehydrogenase, an electron transport enzyme) can be attenuated by treatment with minocycline.66,67 Minocycline has also been reported to reduce superoxide production by granular neurons in the rat following treatment with N-methyl-D-aspartate, a glutamate receptor agonist.68 Overall, the above studies suggest that minocycline targets overactive microglia and removesproinflammatory cytokines and oxygen radicals.59–67
Attenuation of apoptosis
Increasing evidence indicates that the neurodegenerative course of schizophrenia involves an increased susceptibility to apoptotic death, and activation of the apoptotic process results in rapid neuronal death.69–73 The mechanisms of cell death in neurodegenerative diseases include oxidative stress, excitotoxicity, mitochondrial dysfunction, and inflammation.74 Proinflammatory cytokines, including TNF-α, have been well characterized as mediators of oxidative stress and induce apoptosis in human cortical neurons as well as oligodendrocytes.75,76 Apoptosis is indirectly suppressed by minocycline by inhibition of proinflammatory cytokines and nitric oxide.77
Moreover, minocycline has been shown to be an inhibitor of poly-(ADP-ribose), polymerase-1, and matrix metalloproteinases.78,79 The antiapoptotic profile of minocycline has been attributed to upregulation of the anti-apoptotic protein Bcl-2, reduced expression of caspases, and inhibition of release of proapoptotic proteins from the mitochondria.80–84 Animal studies have shown that minocycline is able to increase expression of antiapoptotic proteins and decrease expression of these proteins after treatment with transforming growth factor beta-2. Hydrogen peroxide can also be reversed by minocycline, which leads to inhibition of apoptosis.85,86 Minocycline may directly activate the kinase G pathway by activation of kinase G1, leading to increased expression of Bcl-2.19,87 Minocycline is able to block the mitochondrial permeability transition pore (a large-conductance mega channel in the inner mitochondrial membrane), inhibiting release of proapoptotic proteins from the mitochondria.88–91
Minocycline has also been shown to reduce glutamate excitotoxicity in the glutamatergic pathways.92–99 It has been reported that minocycline-treated rat cortical neurons show increased cell viability following glutamate administration, which indicates that minocycline is able to reduce glutamate-induced neurotoxicity.92,93 GluR1 receptors, which are critical for the cognitive processes that are impaired in schizophrenia, has been shown to be involved in the pathobiology of schizophrenia.94,95 Minocycline affects a subtype of glutamate receptors known as GluR1 alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptors in neurons in vitro and in the CNS in vivo. Increased membrane localization of the GluR1 AMPA receptor improves glutamatergic activity and modulates neuroplasticity.96 Overall, these findings suggest that minocycline has an antiapoptotic profile and is involved in several pathways known to be disturbed in schizophrenia.
Minocycline in schizophrenia: preclinical studies
Minocycline has been reported to reduce inflammation and provide neuroprotection in a variety of experimental models, including ischemic stroke, brain and spinal cord injury, multiple sclerosis, and Parkinson’s disease, Alzheimer’s disease, and Huntington’s disease.17 Du et alhave reported on the neuroprotective effects of minocycline in animal models of stroke/ischemic injury and Huntington’s disease, and shown that minocycline can prevent nigrostriatal dopaminergic neurodegeneration in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse model of Parkinson’s disease.97 Minocycline has been shown to play an important role in the treatment of disorders involving neuronal damage.17,97 However, in a rat model of intracerebral hemorrhage, Wasserman et al found that minocycline did not reduce neuronal loss outside the hematoma or striatal tissue, despite reducing the number of neutrophils and activated microglia. These authors suggested that minocycline did not appear to target the mechanisms responsible for cell death in this model of intracerebral hemorrhage.98 However, the complex behavioral tests needed to assess functional recovery in this model of intracerebral hemorrhage were not used, and it was not known whether rescuing the band of damaged neurons at the edge of the hematoma would have lasting benefits.
The neurotherapeutic potential of minocycline in the treatment of mental disorders has also been explored.19,99–101 It has been reported that minocycline is able to ameliorate behavioral changes as well as neurotoxicity at dopaminergic terminals after administration of methamphetamine, a compound that causes long-term cognitive deficits and psychiatric signs such as hallucination and delusions.99 Hashimoto et al did a PET study in conscious monkeys, and found that minocycline attenuated the reduction of dopamine transporters in monkeys treated with methamphetamine. They suggested that minocycline protects against methamphetamine-induced neurotoxicity in the monkey brain.102 Levkovitz et al100 compared the effects of minocycline and haloperidol in an animal model of schizophrenia (N-methyl-D-aspartate antagonist, dizocilpine maleate, MK801). After 3 days of treatment with minocycline 35 mg/kg/day, the rats were injected with MK801 and assessed using behavioral tests. The findings showed that MK801 caused cognitive visuospatial memory deficits and changes in sensorimotor gating, similar to those evident in schizophrenia. Minocycline reversed the cognitive effects of MK801, and the effect was similar to that of haloperidol. Levkovitz et al suggested that minocycline may be able to protect against the disturbed cognitive processes seen in the MK801 animal model of schizophrenia.100 Zhang et al also indicated that minocycline attenuates the behavioral changes (such as acute hyperlocomotion and prepulse inhibition deficits) occurring in mice after administration of dizocilpine, and suggested that minocycline could be a potential therapeutic compound for schizophrenia.103 Monte et al reported the prevention and reversal of ketamine-induced schizophrenia-related behavior by minocycline.101 Moreover, minocycline has been shown to have beneficial effects on cognition in animal models of schizophrenia.In a study by Fujita et al saline 10 mL/kg/day or phencyclidine 10 mg/kg/day were subcutaneously administered in mice for 10 days. Three days after the final dose of saline or phencyclidine, vehicle (10 mL/kg/day, physiologic saline) or minocycline (4.0 or 40 mg/kg/day) was administered intraperitoneally for 14 consecutive days. One day after the final injection, a novel object recognition test was performed, and it was found that phencyclidine-induced cognitive deficits in mice were significantly improved by subsequent subchronic administration of minocycline (40 mg/kg), suggesting minocycline could be a potential therapeutic compound for cognitive deficits in schizophrenic patients.104 Improvement by minocycline of impaired recognition memory in methamphetamine-treated mice was also reported by Mizoguchi et al.105
In summary, these findings indicate that minocycline may be useful in the treatment in schizophrenia. However, regardless of how encouraging the preclinical studies have been, clinical trials are required to verify these effects of minocycline.
Minocycline in schizophrenia: clinical studies
Miyaoka et al reported two cases of minocycline being used as adjunctive treatment. In these cases, minocycline produced a remarkable improvement in individuals with acute schizophrenia who had predominantly catatonic symptoms.106 These observations are supported by a case report of a patient with schizophrenia given minocycline 200 mg/day for 8 weeks in addition to a stable dose of olanzapine 20 mg/day.107 The combination of minocycline and antipsychotic treatment significantly reduced positive symptoms, with no negative symptoms seen in the baseline assessment. Significantly decreased hyperperfusion in the posterior cingulate gyrus along with a simultaneous improvement of positive symptoms was observed after treatment with minocycline. Based on these case reports, Miyaoka et al conducted an open-label study in 22 patients with schizophrenia, in whom minocycline was administered for 4 weeks as an adjunct to antipsychotic medication. Minocycline was initiated as 100 mg orally twice daily for the first week, and 150 mg orally three times daily from weeks 2 through 4. The patients showed statistically significant and robust clinical improvements on the Positive and Negative Syndrome Scale, and these improvements were maintained at the follow-up evaluation 4 weeks after the end of treatment with minocycline, with few adverse events.108 However, this was an open-label study without a control group, and the effects of the adjunctive medication cannot be ruled out as an explanation for the observed improvement.
Interestingly, some studies indicate that minocycline may have specific effects on negative symptoms and impaired cognitive function in patients with schizophrenia, which contribute to their poor social and occupational functioning. Case reports indicate the effective role of add on minocycline in treating negative symptoms in schizophrenia.109,110 Recently, Levkovitz et al carried out a double-blind, randomized, placebo-controlled study demonstrating that add on treatment with minocycline 200 mg/day for 6 months had a beneficial effect on negative symptoms and general outcomes in patients with early-phase schizophrenia. A similar pattern was seen for cognitive functioning, mainly of the executive type, ie, working memory, cognitive shifting, and cognitive planning.111 Seventy patients with early-phase schizophrenia were recruited for the study; however, of 54 patients randomly allocated in a 2:1 ratio to the minocycline group, only 13 completed the 6-month trial. This high dropout rate limits the reliability of the results of this study. Another randomized, double-blind, placebo-controlled clinical trial also suggested that addition of minocycline (200 mg/day for one year) to treatment as usual in patients with earlypsychosis could reduce negative symptoms without a detectable effect on cognition.112 The investigators recruited 144 patients with psychosis (including schizophrenia, schizoaffective disorder, psychosis not otherwise specified, or schizophreniform disorder) within 5 years of first onset in Brazil and Pakistan, and 94 completed the trial. Although the dropout rates were not high when compared with other trials, the validity and reliability of cognitive measures in two different racial groups and cultures could have increased the variability of the results. In addition, neither the type nor dose of antipsychotic medication was controlled, which may have further increased the variability and obscured the effects of the medication. Sofuoglu et al showed that minocycline attenuated dextroamphetamine-induced subjective reward effects and improved reaction times on a Go No-Go task in healthy volunteers.113
Although case reports should to be interpreted with caution and large-scale studies are needed to determine the effectiveness of minocycline and related compounds in clinical use, the results of the above studies suggest that minocycline may be a safe and effective adjunct to antipsychotic medication for the treatment of schizophrenia.
Conclusion
Although the etiology of schizophrenia remains unclear, there has been growing evidence suggesting that neuroinflammation, characterized by overactive microglia, plays an important role in the neuropathology of schizophrenia. The microglial hypothesis of schizophrenia may shed new light on a therapeutic strategy for the disease.1,16 According to this hypothesis and the results of previous related studies, we suggest that overactivation of microglia can lead to neuronal degeneration and abnormal development in the brain, and that this neuropathologic process may be more likely to result in negative symptoms and cognitive impairment in schizophrenia.1,16,21–23,48,49 Neuroinflammation could constitute the basic framework for further study in the neuropathology of negative schizophrenia.
As an inhibitor of microglial activation, minocycline has anti-inflammatory, antioxidant, and antiapoptotic properties, and can modulate glutamate-induced excitotoxicity. The therapeutic potential of minocycline has been demonstrated in studies using animal models of schizophrenia.19,99–105 In addition to the evidence presented here, recent clinical trials have demonstrated that minocycline can be used as an add on therapy in schizophrenia, especially for amelioration of negative symptoms.106–112
Because of the limited and preliminary nature of the studies mentioned above, more detailed studies are required to determine whether minocycline is effective as an adjunctive treatment for symptoms in patients with schizophrenia who derive limited benefit from standard antipsychotic treatment. Moreover, considering the complicated etiology and heterogeneity of schizophrenia, it will be necessary to categorize the disease phenotype in further studies, including patient status and the severity of negative symptoms, which possibly need to be used as inclusion/exclusion criteria or stratifying variables. Finally, given the mechanism of action of minocycline and the data on the role of overactivated microglia in schizophrenia, future clinical studies should include inflammatory markers in blood or cerebrospinal fluid, and functional imaging such as PET to verify the hypothesis that subgroups of schizophrenic patients with neuroinflammation are the most appropriate candidates for augmentation therapy with minocycline.
Acknowledgment
This study was supported by the National Natural Science Foundation of China (81071093).
Disclosure
The authors report no conflicts of interest in this work.
References
Monji A, Kato T, Kanba S. Cytokines and schizophrenia: microglia hypothesis of schizophrenia. Psychiatry Clin Neurosci. 2009;63(3):257–265. | ||
Kane JM, Correll CU. Past and present progress in the pharmacologic treatment of schizophrenia. J Clin Psychiatry. 2010;71(9):1115–1124. | ||
Leucht S, Arbter D, Engel RR, Kissling W, Davis JM. How effective are second-generation antipsychotic drugs? A meta-analysis of placebo-controlled trials. Mol Psychiatry. 2009;14(4):429–447. | ||
Essali A, Al Haj HN, Li C, Rathbone J. Clozapine versus typical neuroleptic medication for schizophrenia. Cochrane Database Syst Rev. 2009;1:CD000059. | ||
Asenjo LC, Komossa K, Rummel-Kluge C, et al. Clozapine versus other atypical antipsychotics for schizophrenia. Cochrane Database Syst Rev. 2010;11:CD006633. | ||
Gilmour G, Dix S, Fellini L, et al. NMDA receptors, cognition and schizophrenia – testing the validity of the NMDA receptor hypofunction hypothesis. Neuropharmacology. 2012;62(3):1401–1412. | ||
Terry AV, Jr. Role of the central cholinergic system in the therapeutics of schizophrenia. Curr Neuropharmacol. 2008;6(3):286–292. | ||
Sander K, Kottke T, Stark H. Histamine H3 receptor antagonists go to clinics. Biol Pharm Bull. 2008;31(12):2163–2181. | ||
Meltzer HY. Serotonergic mechanisms as targets for existing and novel antipsychotics. Handb Exp Pharmacol. 2012;(212):87–124. | ||
Ginovart N, Kapur S. Role of dopamine D(2) receptors for antipsychotic activity. Handb Exp Pharmacol. 2012;212:27–52. | ||
Mulle JG. Schizophrenia genetics: progress, at last. Curr Opin Genet Dev. 2012;22(3):238–244. | ||
Gupta S, Kulhara P. What is schizophrenia: a neurodevelopmental or neurodegenerative disorder or a combination of both? A critical analysis. Indian J Psychiatry. 2010;52(1):21–27. | ||
Rapoport JL, Giedd JN, Gogtay N. Neurodevelopmental model of schizophrenia: update 2012. Mol Psychiatry. 2012;17(12):1228–1238. | ||
Dean B. Understanding the role of inflammatory-related pathways in the pathophysiology and treatment of psychiatric disorders: evidence from human peripheral studies and CNS studies. Int J Neuropsychopharmacol. 2011;14(7):997–1012. | ||
Meyer U. Anti-inflammatory signaling in schizophrenia. Brain Behav Immun. 2011;25(8):1507–1518. | ||
Monji A, Kato TA, Mizoguchi Y, et al. Neuroinflammation in schizophrenia especially focused on the role of microglia. Prog Neuropsychopharmacol Biol Psychiatry. 2013;42(5):115–121. | ||
Yong VW, Wells J, Giuliani F, Casha S, Power C, Metz LM. The promise of minocycline in neurology. Lancet Neurol. 2004;3(12):744–751. | ||
Kim HS, Suh YH. Minocycline and neurodegenerative diseases. Behav Brain Res. 2009;196(2):168–179. | ||
Dean OM, Data-Franco J, Giorlando F, Berk M. Minocycline: therapeutic potential in psychiatry. CNS Drugs. 2012;26(5):391–401. | ||
Keller WR, Kum LM, Wehring HJ, Koola MM, Buchanan RW, Kelly DL. A review of anti-inflammatory agents for symptoms of schizophrenia. J Psychopharmacol. 2013;27(4):337–342. | ||
Levkovitz Y, Levi U, Braw Y, Cohen H. Minocycline, a second-generation tetracycline, as a neuroprotective agent in an animal model of schizophrenia. Brain Res. 2007;1154:154–162. | ||
Levkovitz Y, Mendlovich S, Riwkes S, et al. A double-blind, randomized study of minocycline for the treatment of negative and cognitive symptoms in early-phase schizophrenia. J Clin Psychiatry. 2010;71(2):138–149. | ||
Chaudhry IB, Hallak J, Husain N, et al. Minocycline benefits negative symptoms in early schizophrenia: a randomized double-blind placebo-controlled clinical trial in patients on standard treatment.J Psychopharmacol. 2012;26(9):1185–1193. | ||
Muller N, Myint AM, Schwarz MJ. Inflammation in schizophrenia. Adv Protein Chem Struct Biol. 2012;88:49–68. | ||
Ashdown H, Dumont Y, Ng M, Poole S, Boksa P, Luheshi GN. The role of cytokines in mediating effects of prenatal infection on the fetus: implications for schizophrenia. Mol Psychiatry. 2006;11(1):47–55. | ||
Shen Q, Li ZQ, Sun Y, et al. The role of pro-inflammatory factors in mediating the effects on the fetus of prenatal undernutrition: implications for schizophrenia. Schizophr Res. 2008;99(1–3):48–55. | ||
Meyer U. Developmental neuroinflammation and schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2013;42:20–34. | ||
Arion D, Unger T, Lewis DA, Levitt P, Mirnics K. Molecular evidence for increased expression of genes related to immune and chaperone function in the prefrontal cortex in schizophrenia. Biol Psychiatry. 2007;62(7):711–721. | ||
Narayan S, Tang B, Head SR, et al. Molecular profiles of schizophrenia in the CNS at different stages of illness. Brain Res. 2008;1239:235–248. | ||
Bessis A, Bechade C, Bernard D, Roumier A. Microglial control of neuronal death and synaptic properties. Glia. 2007;55(3):233–238. | ||
Steiner J, Bogerts B, Sarnyai Z, et al. Bridging the gap between the immune and glutamate hypotheses of schizophrenia and major depression: potential role of glial NMDA receptor modulators and impaired blood-brain barrier integrity. World J Biol Psychiatry. 2012;13(7):482–492. | ||
Alliot F, Godin I, Pessac B. Microglia derive from progenitors, originating from the yolk sac, and which proliferate in the brain. Brain Res Dev Brain Res. 1999;117(2):145–152. | ||
Pont-Lezica L, Bechade C, Belarif-Cantaut Y, Pascual O, Bessis A. Physiological roles of microglia during development. J Neurochem. 2011;119(5):901–908. | ||
Schlegelmilch T, Henke K, Peri F. Microglia in the developing brain: from immunity to behaviour. Curr Opin Neurobiol. 2011;21(1):5–10. | ||
Tremblay ME, Majewska AK. A role for microglia in synaptic plasticity? Commun Integr Biol. 2011;4(2):220–222. | ||
Tremblay ME, Stevens B, Sierra A, Wake H, Bessis A, Nimmerjahn A.The role of microglia in the healthy brain. J Neurosci. 2011;31(45):16064–16069. | ||
Nurun BN, Tanaka T, Kamino K, et al. Toll-like receptor 3 mediated hyperphosphorylation of tau in human SH-SY5Y neuroblastoma cells. Psychiatry Clin Neurosci. 2006;60 Suppl 1:S27–S33. | ||
Block ML, Hong JS. Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol. 2005;76(2):77–98. | ||
Lieberman JA. Is schizophrenia a neurodegenerative disorder? A clinical and neurobiological perspective. Biol Psychiatry. 1999;46(6):729–739. | ||
Perez-Neri I, Ramirez-Bermudez J, Montes S, Rios C. Possible mechanisms of neurodegeneration in schizophrenia. Neurochem Res. 2006;31(10):1279–1294. | ||
Nakki R, Koistinaho J, Sharp FR, Sagar SM. Cerebellar toxicity of phencyclidine. J Neurosci. 1995;15(3 Pt 2):2097–2108. | ||
Nakki R, Nickolenko J, Chang J, Sagar SM, Sharp FR. Haloperidol prevents ketamine- and phencyclidine-induced HSP70 protein expression but not microglial activation. Exp Neurol. 1996;137(2):234–241. | ||
Steiner J, Mawrin C, Ziegeler A, et al. Distribution of HLA-DR-positive microglia in schizophrenia reflects impaired cerebral lateralization. Acta Neuropathol. 2006;112(3):305–316. | ||
Steiner J, Bielau H, Brisch R, et al. Immunological aspects in the neurobiology of suicide: elevated microglial density in schizophrenia and depression is associated with suicide. J Psychiatr Res. 2008;42(2):151–157. | ||
Bayer TA, Buslei R, Havas L, Falkai P. Evidence for activation of microglia in patients with psychiatric illnesses. Neurosci Lett. 1999;271(2):126–128. | ||
Radewicz K, Garey LJ, Gentleman SM, Reynolds R. Increase in HLA-DR immunoreactive microglia in frontal and temporal cortex of chronic schizophrenics. J Neuropathol Exp Neurol. 2000;59(2):137–150. | ||
Wierzba-Bobrowicz T, Lewandowska E, Lechowicz W, Stepień T, Pasennik E. Quantitative analysis of activated microglia, ramified and damage of processes in the frontal and temporal lobes of chronic schizophrenics. Folia Neuropathol. 2005;43(2):81–89. | ||
Van Berckel BN, Bossong MG, Boellaard R, et al. Microglia activation in recent-onset schizophrenia: a quantitative (R)-[11C]PK11195 positron emission tomography study. Biol Psychiatry. 2008;64(9):820–822. | ||
Doorduin J, de Vries EF, Willemsen AT, de Groot JC, Dierckx RA, Klein HC. Neuroinflammation in schizophrenia-related psychosis:a PET study. J Nucl Med. 2009;50(11):1801–1807. | ||
Takahashi N, Sakurai T. Roles of glial cells in schizophrenia: possible targets for therapeutic approaches. Neurobiol Dis. 2013;53:49–60. | ||
Frick LR, Williams K, Pittenger C. Microglial dysregulation in psychiatric disease. Clin Dev Immunol. 2013;2013:608654. | ||
Monje ML, Toda H, Palmer TD. Inflammatory blockade restores adult hippocampal neurogenesis. Science. 2003;302(5651):1760–1765. | ||
Ekdahl CT, Claasen JH, Bonde S, Kokaia Z, Lindvall O. Inflammation is detrimental for neurogenesis in adult brain. Proc Natl Acad Sci U S A.2003;100(23):13632–13637. | ||
Cacci E, Claasen JH, Kokaia Z. Microglia-derived tumor necrosis factor-alpha exaggerates death of newborn hippocampal progenitor cells in vitro. J Neurosci Res. 2005;80(6):789–797. | ||
Iosif RE, Ekdahl CT, Ahlenius H, et al. Tumor necrosis factor receptor 1is a negative regulator of progenitor proliferation in adult hippocampal neurogenesis. J Neurosci. 2006;26(38):9703–9712. | ||
Kaneko N, Kudo K, Mabuchi T, et al. Suppression of cell proliferation by interferon-alpha through interleukin-1 production in adult rat dentate gyrus. Neuropsychopharmacology. 2006;31(12):2619–2626. | ||
Muller N, Krause D, Dehning S, et al. Celecoxib treatment in an early stage of schizophrenia: results of a randomized, double-blind, placebo-controlled trial of celecoxib augmentation of amisulpride treatment. Schizophr Res. 2010;121(1–3):118–124. | ||
Muller N, Riedel M, Scheppach C, et al. Beneficial antipsychotic effects of celecoxib add-on therapy compared to risperidone alone in schizophrenia. Am J Psychiatry. 2002;159(6):1029–1034. | ||
Yrjänheikki J, Keinänen R, Pellikka M, Hökfelt T, Koistinaho J.Tetracyclines inhibit microglial activation and are neuroprotective in global brain ischemia. Proc Natl Acad Sci U S A. 1998;95(26):15769–15774. | ||
Yrjänheikki J, Tikka T, Keinänen R, Goldsteins G, Chan PH, Koistinaho J.A tetracycline derivative, minocycline, reduces inflammation and protects against focal cerebral ischemia with a wide therapeutic window. Proc Natl Acad Sci U S A. 1999;96(23):13496–13500. | ||
Dommeguess MA, Plaisant F, Vermey C, Gressens P. Early microglial activation following neonatal excitotoxic brain damage in mice: a potential target for neuroprotection. Neuroscience. 2003;121(3):619–628. | ||
Tikka T, Fiebich BL, Goldsteins G, Keinanen R, Koistinaho J. Minocycline, a tetracycline derivative, is neuroprotective against excitotoxicity by inhibiting activation and proliferation of microglia. J Neurosci. 2001;21(8):2580–2588. | ||
Arvin KL, Han BH, Du Y, Lin SZ, Paul SM, Holtzman DM. Minocycline markedly protects the neonatal brain against hypoxic-ischemic injury. Ann Neurol. 2002;52(1):54–61. | ||
Chen M, Ona VO, Li M, et al. Minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat Med. 2000;6(7):797–801. | ||
Wu DC, Jackson-Lewis V, Vila M, et al. Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J Neurosci. 2002;22(5):1763–1771. | ||
Homsi S, Federico F, Croci N, et al. Minocycline effects on cerebral edema: relations with inflammatory and oxidative stress markers following traumatic brain injury in mice. Brain Res. 2009;1291:122–132. | ||
Ahuja M, Bishnoi M, Chopra K. Protective effect of minocycline, a semi-synthetic second-generation tetracycline against 3-nitropropionic acid (3-NP)-induced neurotoxicity. Toxicology. 2008;244(2–3):111–122. | ||
Garcia-Martinez EM, Sanz-Blasco S, Karachitos A, et al. Mitochondria and calcium flux as targets of neuroprotection caused by minocycline in cerebellar granule cells. Biochem Pharmacol. 2010;79(2):239–250. | ||
Davis KL, Stewart DG, Friedman JI, et al. White matter changes in schizophrenia: evidence for myelin-related dysfunction. Arch Gen Psychiatry. 2003;60(5):443–456. | ||
Kumra S, Ashtari M, Cervellione KL, et al. White matter abnormalities in early-onset schizophrenia: a voxel-based diffusion tensor imaging study. J Am Acad Child Adolesc Psychiatry. 2005;44(9):934–941. | ||
Salisbury DF, Kuroki N, Kasai K, Shenton ME, McCarley RW. Progressive and interrelated functional and structural evidence of post-onset brain reduction in schizophrenia. Arch Gen Psychiatry. 2007;64(5):521–529. | ||
Hulshoff Pol HE, Kahn RS. What happens after the first episode? A review of progressive brain changes in chronically ill patients with schizophrenia. Schizophr Bull. 2008;34(2):354–366. | ||
Glantz LA, Gilmore JH, Lieberman JA, Jarskog LF. Apoptotic mechanisms and the synaptic pathology of schizophrenia. Schizophr Res. 2006;81(1):47–63. | ||
Olanow CW, Jenner P, Brooks D. Dopamine agonists and neuroprotection in Parkinson’s disease. Ann Neurol. 1998;44(3 Suppl 1):S167–S174. | ||
Medina S, Martinez M, Hernanz A. Antioxidants inhibit the human cortical neuron apoptosis induced by hydrogen peroxide, tumor necrosis factor alpha, dopamine and beta-amyloid peptide 1-42. Free Radic Res. 2002;36(11):1179–1184. | ||
Buntinx M, Moreels M, Vandenabeele F, et al. Cytokine-induced cell death in human oligodendroglial cell lines: I. Synergistic effects of IFN-gamma and TNF-alpha on apoptosis. J Neurosci Res. 2004;76(6):834–845. | ||
Thomas M, Le WD. Minocycline: neuroprotective mechanisms in Parkinson’s disease. Curr Pharm Des. 2004;10(6):679–686. | ||
Alano CC, Kauppinen TM, Valls AV, Swanson RA. Minocycline inhibits poly(ADP-ribose) polymerase-1 at nanomolar concentrations. Proc Natl Acad Sci U S A. 2006;103(25):9685–9690. | ||
Brundula V, Rewcastle NB, Metz LM, Bernard CC, Yong VW. Targeting leukocyte MMPs and transmigration: minocycline as a potential therapy for multiple sclerosis. Brain. 2002;125 Pt 6:1297–1308. | ||
Wang J, Wei Q, Wang CY, Hill WD, Hess DC, Dong Z. Minocycline up-regulates Bcl-2 and protects against cell death in mitochondria.J Biol Chem. 2004;279(19):19948–19954. | ||
Castanares M, Vera Y, Erkkila K, et al. Minocycline up-regulates BCL-2 levels in mitochondria and attenuates male germ cell apoptosis. Biochem Biophys Res Commun. 2005;337(2):663–669. | ||
Scarabelli TM, Stephanou A, Pasini E, et al. Minocycline inhibits caspase activation and reactivation, increases the ratio of XIAP to SMAC/DIABLO, and reduces the mitochondrial leakage of cytochrome C and smac/DIABLO. J Am Coll Cardiol. 2004;43(5):865–874. | ||
Chen M, Ona VO, Li M, et al. Minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat Med. 2000;6(7):797–801. | ||
Teng YD, Choi H, Onario RC, et al. Minocycline inhibits contusion-triggered mitochondrial cytochrome c release and mitigates functional deficits after spinal cord injury. Proc Natl Acad Sci U S A. 2004;101(9):3071–3076. | ||
Yang D, Liu X, Zhang R, et al. Increased apoptosis and different regulation of pro-apoptosis protein bax and antiapoptosis protein bcl-2 in the olfactory bulb of a rat model of depression. Neurosci Lett. 2011;504(1):18–22. | ||
Kernt M, Neubauer AS, Eibl KH, et al. Minocycline is cytoprotective in human trabecular meshwork cells and optic nerve head astrocytes by increasing expression of XIAP, survivin, and Bcl-2. Clin Ophthalmol. 2010;4:591–604. | ||
Tang CM, Hwang CS, Chen SD, Yang DI. Neuroprotective mechanisms of minocycline against sphingomyelinase/ceramide toxicity: roles of Bcl-2 and thioredoxin. Free Radic Biol Med. 2011;50(6):710–721. | ||
Zhu S, Stavrovskaya IG, Drozda M, et al. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature. 2002;417(6884):74–78. | ||
Wang X, Zhu S, Drozda M, et al. Minocycline inhibits caspase- independent and dependent mitochondrial cell death pathways in models of Huntington’s disease. Proc Natl Acad Sci U S A. 2003;100(18):10483–10487. | ||
Fuks B, Talaga P, Huart C, et al. In vitro properties of 5-(benzylsulfonyl)-4-bromo-2- methyl- 3(2H)-pyridazinone: a novel permeability transition pore inhibitor. Eur J Pharmacol. 2005;519(1–2):24–30. | ||
Leung AW, Halestrap AP. Recent progress in elucidating the molecular mechanism of the mitochondrial permeability transition pore. Biochim Biophys Acta. 2008;1777(7–8):946–952. | ||
Morimoto N, Shimazawa M, Yamashima T, Nagai H, Hara H. Minocycline inhibits oxidative stress and decreases in vitro and in vivo ischemic neuronal damage. Brain Res. 2005;1044(1):8–15. | ||
Kraus RL, Pasieczny R, Lariosa-Willingham K, Turner MS, Jiang A, Trauger JW. Antioxidant properties of minocycline: neuroprotection in an oxidative stress assay and direct radical-scavenging activity.J Neurochem. 2005;94(3):819–827. | ||
Wiedholz LM, Owens WA, Horton RE, et al. Mice lacking the AMPA GluR1 receptor exhibit striatal hyperdopaminergia and ‘schizophrenia-related’ behaviors. Mol Psychiatry. 2008;13(6):631–640. | ||
Woolley ML, Waters KA, Gartlon JE, et al. Evaluation of the pro-cognitive effects of the AMPA receptor positive modulator, 5-(1-piperidinylcarbonyl)-2,1,3-benzoxadiazole (CX691), in the rat. Psychopharmacology (Berl). 2009;202(1–3):343–354. | ||
Wang JQ, Arora A, Yang L, et al. Phosphorylation of AMPA receptors: mechanisms and synaptic plasticity. Mol Neurobiol. 2005;32(3):237–249. | ||
Du Y, Ma Z, Lin S, et al. Minocycline prevents nigrostriatal dopaminergic neurodegeneration in the MPTP model of Parkinson’s disease. Proc Natl Acad Sci U S A. 2001;98(25):14669–14674. | ||
Wasserman JK, Schlichter LC. Neuron death and inflammation in a rat model of intracerebral hemorrhage: effects of delayed minocycline treatment. Brain Res. 2007;1136(1):208–218. | ||
Zhang L, Kitaichi K, Fujimoto Y, et al. Protective effects of minocycline on behavioral changes and neurotoxicity in mice after administration of methamphetamine. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30(8):1381–1393. | ||
Levkovitz Y, Levi U, Braw Y, Cohen H. Minocycline, a second-generation tetracycline, as a neuroprotective agent in an animal model of schizophrenia. Brain Res. 2007;1154:154–162. | ||
Monte AS, de Souza GC, McIntyre RS, et al. Prevention and reversal of ketamine-induced schizophrenia related behavior by minocycline in mice: possible involvement of antioxidant and nitrergic pathways. J Psychopharmacol. 2013;27(11):1032–1043. | ||
Hashimoto K, Tsukada H, Nishiyama S, Fukumoto D, Kakiuchi T, Iyo M. Protective effects of minocycline on the reduction of dopamine transporters in the striatum after administration of methamphetamine: a positron emission tomography study in conscious monkeys. Biol Psychiatry. 2007;61(5):577–581. | ||
Zhang L, Shirayama Y, Iyo M, Hashimoto K. Minocycline attenuates hyperlocomotion and prepulse inhibition deficits in mice after administration of the NMDA receptor antagonist dizocilpine. Neuropsychopharmacology. 2007;32(9):2004–2010. | ||
Fujita Y, Ishima T, Kunitachi S, et al. Phencyclidine-induced cognitive deficits in mice are improved by subsequent subchronic administration of the antibiotic drug minocycline. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(2):336–339. | ||
Mizoguchi H, Takuma K, Fukakusa A, et al. Improvement by minocycline of methamphetamine-induced impairment of recognition memory in mice. Psychopharmacology (Berl). 2008;196(2):233–241. | ||
Miyaoka T, Yasukawa R, Yasuda H, Hayashida M, Inagaki T, Horiguchi J. Possible antipsychotic effects of minocycline in patients with schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31(1):304–307. | ||
Chaves C, de Marque CR, Wichert-Ana L, et al. Functional neuroimaging of minocycline’s effect in a patient with schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34(3):550–552. | ||
Miyaoka T, Yasukawa R, Yasuda H, Hayashida M, Inagaki T, Horiguchi J. Minocycline as adjunctive therapy for schizophrenia: an open-label study. Clin Neuropharmacol. 2008;31(5):287–292. | ||
Kelly DL, Vyas G, Richardson CM, et al. Adjunct minocycline to clozapine treated patients with persistent schizophrenia symptoms. Schizophr Res. 2011;133(1–3):257–258. | ||
Jhamnani K, Shivakumar V, Kalmady S, Rao NP, Venkatasubramanian G.Successful use of add-on minocycline for treatment of persistent negative symptoms in schizophrenia. J Neuropsychiatry Clin Neurosci. 2013;25(1):E06–E07. | ||
Levkovitz Y, Mendlovich S, Riwkes S, et al. A double-blind, randomized study of minocycline for the treatment of negative and cognitive symptoms in early-phase schizophrenia. J Clin Psychiatry. 2010;71(2):138–149. | ||
Chaudhry IB, Hallak J, Husain N, et al. Minocycline benefits negative symptoms in early schizophrenia: a randomized double-blind placebo-controlled clinical trial in patients on standard treatment.J Psychopharmacol. 2012;26(9):1185–1193. | ||
Sofuoglu M, Mooney M, Kosten T, Waters A, Hashimoto K. Minocycline attenuates subjective rewarding effects of dextroamphetamine in humans. Psychopharmacology (Berl). 2011;213(1):61–68. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.