")
Back to Journals » International Medical Case Reports Journal » Volume 7
Presymptomatic genetic analysis during pregnancy for vascular type Ehlers–Danlos syndrome
Authors Naing BT, Watanabe A, Tanigaki S, Ono M, Iwashita M, Shimada T
Received 30 December 2013
Accepted for publication 13 February 2014
Published 19 June 2014 Volume 2014:7 Pages 99—102
DOI https://doi.org/10.2147/IMCRJ.S59879
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Banyar Than Naing,1 Atsushi Watanabe,1,2 Shinji Tanigaki,3 Masae Ono,4 Mitsutoshi Iwashita,3 Takashi Shimada1,2
1Department of Biochemistry and Molecular Biology, Nippon Medical School, Tokyo, Japan; 2Division of Clinical Genetics, Nippon Medical School Hospital, Tokyo, Japan; 3Department of Obstetrics and Gynecology, Kyorin University School of Medicine, Tokyo, Japan; 4Department of Pediatrics, Kyorin University School of Medicine, Tokyo, Japan
Abstract: The vascular type of Ehlers–Danlos syndrome (EDS), EDS type IV (Online Mendelian Inheritance in Man [MIM] #130050) is characterized by thin, translucent skin, easy bruising, and arterial, intestinal, and/or uterine fragility during pregnancy, which may lead to sudden death. It is an autosomal dominant inherited disorder caused by type III procollagen gene (COL3A1: MIM #120180) mutations. Approximately 50% of the COL3A1 mutations are inherited from an affected parent, and 50% are de novo mutations. Each child of an affected individual has a 50% chance of inheriting the mutation and developing the disorder. Pregnant women with vascular EDS are at an increased risk of uterine and arterial rupture during the peripartum period, with high maternal morbidity and mortality rates. We report the first case of an asymptomatic 35-year-old woman at a risk of complications of vascular EDS who underwent presymptomatic evaluation during pregnancy. The sequencing results of both her brother and mother had a one-base-pair deletion, resulting in Glutamate at position 730 changing to Lysine and causing a frame shift and premature termination codon at 61 amino acids from the mutation position (p. Glu730Lysfs*61) on exon 32 of COL3A1. This deletion caused frameshift, leading to a premature termination codon (TAG) at 181 nucleotides downstream in exon 35, which could not be detected by previous total RNA (ribonucleic acid) method. Thus, she was at risk of complications of vascular EDS, and diagnostic testing was employed at 8 weeks of pregnancy to minimize the risk of developing vascular EDS-related complications. The negative presymptomatic diagnostic result allowed the patient to choose normal delivery at term. Vascular EDS is a serious disorder, with high mortality, especially in high-risk women with vascular EDS during pregnancy. The presymptomatic genetic testing of vascular EDS during pregnancy for a high-risk family can help with the early establishment of preventive measures.
Keywords: vascular EDS, type III procollagen gene (COL3A1), testing
Introduction
The vascular type of Ehlers–Danlos syndrome (EDS), EDS type IV (Online Mendelian Inheritance in Man [MIM] #130050)1,2 is an autosomal dominant inherited disorder. Its primary clinical features are rupture of the blood vessels or internal organs such as bowel and uterine rupture during pregnancy, which may lead to sudden death.2,3 Vascular EDS causes severe fragility of connective tissues, with arterial and intestinal ruptures and complications of surgical and radiological treatment, and is of particular importance to medical professionals of many specialties: surgeons, internists, radiologists, and obstetricians. An accurate diagnosis may help in the management of visceral complications. Vascular EDS is mainly caused by type III procollagen gene (COL3A1: MIM #120180) mutations.4 The majority of identified mutations are point mutations that result in single amino acid substitutions for glycine in the GLY-X-Y repeat of the triple helical region. Approximately 50% of the COL3A1 mutations are inherited from an affected parent, and 50% are de novo mutations.5 Most individuals with vascular EDS will experience a significant medical problem in their lifetime, at rates of 25% and 80% by the ages of 20 years and 40 years, respectively.3
In some patients, potentially life-threatening complications of vascular EDS appear only during pregnancy,6 with the maternal mortality rate in the range of 12%–25% because of peripartum arterial or uterine rupture.3,7,8 To reduce the risks of late pregnancy and delivery, pregnant women with the COL3A1 mutation of vascular EDS are treated as high risk and selected for an early delivery by elective cesarean section.6,8,9 Genetic testing is one option that can help those in a family with the COL3A1 mutation to make informed decisions on risk management. Here, we report an asymptomatic woman at risk of complications of vascular EDS who underwent presymptomatic genetic testing during pregnancy.
Case report
An asymptomatic 35-year-old woman (client) at a risk of complications of vascular EDS during pregnancy came to our clinic for genetic counseling. The elder brother (proband) was diagnosed with vascular EDS with pneumothorax and COL3A1 heterozygous mutation, resulting in Glutamate at position 730 changing to Lysine and causing a frame shift and premature termination codon at 61 amino acids from the mutation position (p. Glu730Lysfs*61) by using the GenBank genetic sequence database available at http://www.ncbi.nlm.nih.gov/nuccore/NM_000090.3 (GenBank ID: NM_000090.3) as the reference, as shown in Figure 1. This deletion existed within the triple helical domain of COL3A1, but it caused a frameshift, resulting in a premature termination codon at 181 nucleotides downstream in exon 35. This could not be detected by the previous total ribonucleic acid (RNA) method because of nonsense-mediated messenger RNA decay. Because there was no other family history of vascular EDS, we analyzed the COL3A1 mutation in both parents using polymerase chain reaction (PCR)-direct sequencing of the genomic deoxyribonucleic acid (DNA) to confirm whether or not the mutation occurred de novo. The mother had the same mutation, in spite of having no signs or symptoms of vascular EDS; therefore, the client had a 50% risk for the same mutation of COL3A1.
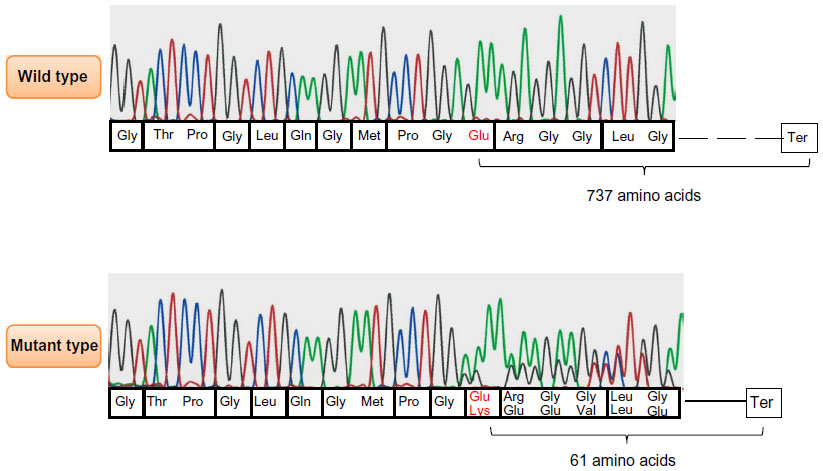 | Figure 1 COL3A1 results obtained by polymerase chain reaction direct sequencing method. |
Before pregnancy, genetic counseling was performed for the client based on these results: she did not want to take the genetic test. However, when the client became pregnant, the obstetrician told her that it was important to consider pregnancy- and peripartum-related risks of vascular EDS and the need for an early delivery via elective cesarean section, even though the echocardiogram was normal, including the size of the main aorta. Thus, the client underwent genetic counseling again at 8 weeks of gestation and determined that she wanted to undergo presymptomatic genetic testing for vascular EDS. With the approval of the ethical committee of Nippon Medical School and after obtaining informed consent from the client, the presymptomatic genetic testing was conducted.10
To detect the mutation, PCR-direct sequencing using genomic DNA was performed. We used forward (5′CAGCATTCCATTCACCTAGG 3′) and reverse primers (5′TCGATTACTTTCCAGGAGTG 3′) that covered the deletion position on exon 32. Capillary electrophoresis was performed using ABI Prism 3130 (Life Technologies, Carlsbad, CA, USA). We then undertook genotyping of common single nucleotide polymorphisms of COL3A1 around the mutation in each allele to confirm the mutation test in the family; these procedures have been previously reported.11 By combining the single nucleotide polymorphism genotyping results, we could prove which allele of each parent was derived to the proband and client (Figure 2). The client did not have the allele containing the COL3A1 mutation, allowing her to deliver normally at term. The healthy baby was born with no complications.
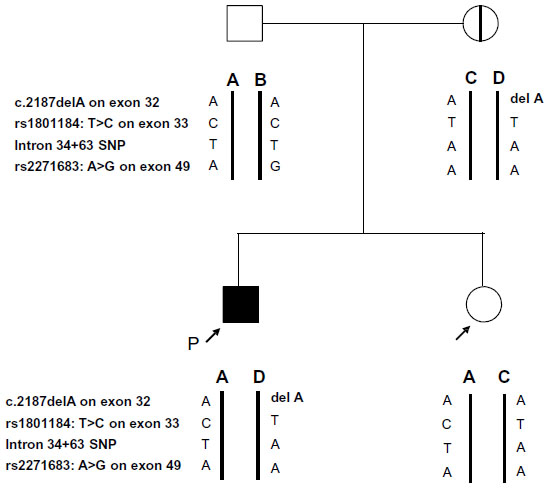 | Figure 2 The family pedigree with the result of the allele confirmation test. |
Discussion
Vascular EDS is usually caused by mutations of COL3A1 in single amino acid substitutions for glycine in the GLY-X-Y repeat of the triple helical region or in variant splice sites, causing a dominant-negative effect.12 Individuals with these mutations have severe symptoms because of high penetrance. COL3A1 haploinsufficiency, which has been found to be an alternative mechanism,13 results in a variety of vascular EDS with delayed onset of complications and longer life expectancy compared with other mutation types.14 The mother with a COL3A1 mutation in our report did not show any clinical symptoms, possibly because of haploinsufficiency caused by nonsense-mediated messenger RNA decay. Some vascular EDS patients with null mutations including nonsense-mediated messenger RNA decay also showed the same severe phenotypes, like that of patients with missense or splice-site mutations.14 Severities of vascular EDS were varied between the members of the family with the same mutation. In vascular EDS, genotype–phenotype correlations also add important information in predicting outcome and selecting the treatment. Determining the mutation type is important for the management of vascular EDS. Although de novo COL3A1 mutations occur in approximately 50% of cases, genetic testing of parents is important for evaluating the risk in other family members.
Management is also necessary for vascular EDS following presymptomatic diagnosis. In pregnant women with vascular EDS and COL3A1 mutation, it is prudent to follow the high-risk obstetric management protocols.6,9 It is essential to educate pregnant women with regard to the possible complications, and the requirement for close monitoring is recommended.7,15,16 During pregnancy, these patients are more susceptible to complications such as uterine and vascular rupture because of plasma volume expansion.17 Depending on the patient’s condition, one of the choices is to choose early delivery via elective cesarean section (32 weeks’ gestation)7 because the risk of complication is considered greatest during labor, delivery, and immediately postpartum.18 In order to be prepared in an emergency, from a surgical perspective, we also need to be aware that vessels are often very friable. This observation was made mainly during operations.19 Thus, a firm diagnosis of vascular EDS may help in the management of visceral complications, pregnancy, and genetic counseling.12
To the best of our knowledge, this is the first report on presymptomatic genetic testing in a pregnant woman with a family history of vascular EDS and known COL3A1 mutation. Presymptomatic genetic testing is an important choice for individuals at risk of complications of vascular EDS during pregnancy and facilitates informed decision-making and effective management of complications.
Acknowledgments
The authors thank the patient and her family members who participated in this study. This work was supported in part by a grant from Grants-in Aid for Scientific Research from the Ministry of Health, Education, Culture, Sports, Science, and Technology of Japan.
Disclosure
The authors report no conflicts of interest in this work.
References
Germain DP. Clinical and genetic features of vascular Ehlers–Danlos syndrome. Ann Vasc Surg. 2002;16(3):391–397. | |
Watanabe A, Kosho T, Wada T, et al. Genetic aspects of the vascular type of Ehlers–Danlos syndrome (vEDS, EDSIV) in Japan. Circ J. 2007;71(2):261–265. | |
Pepin M, Schwarze U, Superti-Furga A, et al. Clinical and genetic features of Ehlers–Danlos syndrome type IV, the vascular type. N Engl J Med. 2000;342(10):673–680. | |
Dalgleish R. The Human Collagen Mutation Database 1998. Nucleic Acids Res. 1998;26(1):253–255. | |
Germain DP. Ehlers–Danlos syndrome type IV. Orphanet J Rare Dis. 2007;2:32. | |
Hammond R, Oligbo N. Ehlers–Danlos syndrome type IV and pregnancy. Arch Gynecol Obstet. 2012;285(1):51–54. | |
Lurie S, Manor M, Hagay ZJ. The threat of type IV Ehlers–Danlos syndrome on maternal well-being during pregnancy: early delivery may make the difference. J Obstet Gynaecol. 1998;18(3):245–248. | |
Rudd NL, Nimrod C, Holbrook KA, et al. Pregnancy complications in type IV Ehlers–Danlos Syndrome. Lancet. 1983;1(8314–8315):50–53. | |
Peaceman AM, Cruikshank DP. Ehlers–Danlos syndrome and pregnancy: association of type IV disease with maternal death. Obstet Gynecol. 1987;69(3 Pt 2):428–431. | |
Skirton H, Goldsmith L, Jackson L, et al. Quality in genetic counselling for presymptomatic testing – clinical guidelines for practice across the range of genetic conditions. Eur J Hum Genet. 2013;21(3):256–260. | |
Naing BT, Watanabe A, Shimada T. A novel mutation screening system for Ehlers–Danlos syndrome, vascular type by high-resolution melting curve analysis in combination with small amplicon genotyping using genomic DNA. Biochem Biophys Res Commun. 2011;405(3):368–372. | |
Watanabe A, Shimada T. Vascular type of Ehlers–Danlos syndrome. J Nippon Med Sch. 2008;75(5):254–261. | |
Schwarze U, Schievink WI, Petty E, et al. Haploinsufficiency for one COL3A1 allele of type III procollagen results in a phenotype similar to the vascular form of Ehlers–Danlos syndrome, Ehlers–Danlos syndrome type IV. Am J Hum Genet. 2001;69(5):989–1001. | |
Leistritz DF, Pepin MG, Schwarze U, et al. COL3A1 haploinsufficiency results in a variety of Ehlers–Danlos syndrome type IV with delayed onset of complications and longer life expectancy. Genet Med. 2011;13(8):717–722. | |
Chetty SP, Shaffer BL, Norton ME. Management of pregnancy in women with genetic disorders, Part 1: Disorders of the connective tissue, muscle, vascular, and skeletal systems. Obstet Gynecol Surv. 2011;66(11):699–709. | |
Bergqvist D, Bjorck M, Wanhainen A. Treatment of vascular Ehlers–Danlos syndrome: a systematic review. Ann Surg. 2013;258(2):257–261. | |
Palmquist M, Pappas JG, Petrikovsky B, et al. Successful pregnancy outcome in Ehlers–Danlos syndrome, vascular type. J Matern Fetal Neonatal Med. 2009;22(10):924–927. | |
Erez Y, Ezra Y, Rojansky N. Ehlers–Danlos type IV in pregnancy. A case report and a literature review. Fetal Diagn Ther. 2008;23(1):7–9. | |
Kimura K, Sakai-Kimura M, Takahashi R, et al. Too friable to treat? Lancet. 2010;375(9725):1578. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.