")
Back to Journals » Drug Design, Development and Therapy » Volume 8
Posttranslational regulation of phosphatase and tensin homolog (PTEN) and its functional impact on cancer behaviors
Authors Xu W, Yang Z, Zhou S, Lu N
Received 12 July 2014
Accepted for publication 4 August 2014
Published 6 October 2014 Volume 2014:8 Pages 1745—1751
DOI https://doi.org/10.2147/DDDT.S71061
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Wei Duan
Wenting Xu,1 Zhen Yang,1 Shu-Feng Zhou,2 Nonghua Lu1
1Department of Gastroenterology, The First Affiliated Hospital of Nanchang University, Nanchang, Jiangxi, People’s Republic of China; 2Department of Pharmaceutical Sciences, College of Pharmacy, University of South Florida, Tampa, FL, USA
Abstract: The incidence of cancer is increasing worldwide, but the biochemical mechanisms for the occurrence of cancer is not fully understood, and there is no cure for advanced tumors. Defects of posttranslational modifications of proteins are linked to a number of important diseases, such as cancer. This review will update our knowledge on the critical role of posttranscriptional regulation of phosphatase and tensin homolog (PTEN) and its activities and the functional impact on cancer behaviors. PTEN is a tumor suppressor gene that occupies a key position in regulating cell growth, proliferation, apoptosis, mobility, signal transduction, and other crucial cellular processes. The activity and function of PTEN are regulated by coordinated epigenetic, transcriptional, posttranscriptional, and posttranslational modifications. In particular, PTEN is subject to phosphorylation, ubiquitylation, somoylation, acetylation, and active site oxidation. Posttranslational modifications of PTEN can dynamically change its activity and function. Deficiency in the posttranslational regulation of PTEN leads to abnormal cell proliferation, apoptosis, migration, and adhesion, which are associated with cancer initiation, progression, and metastasis. With increasing information on how PTEN is regulated by multiple mechanisms and networked proteins, its exact role in cancer initiation, growth, and metastasis will be revealed. PTEN and its functionally related proteins may represent useful targets for the discovery of new anticancer drugs, and gene therapy and the therapeutic potentials should be fully explored.
Keywords: phosphorylation, ubiquitination, acetylation, oxidation, PTEN
Introduction
It is well-known that the pathogenesis of human cancer is a complicated and progressive process, with the accumulation of multiple genetic and epigenetic alterations leading to the initiation, growth, development, and metastasis of cancer.1,2 Cancer is a collective disease with more than 200 types of tumors derived from different tissues and organs. Despite extensive research on cancer biology, the detailed mechanisms and pathways underlying the pathogenesis and progression of cancer are still elusive, with a number of oncogenes, tumor suppressor genes, and signaling proteins identified to play critical roles in carcinogenesis, growth and metastasis. Dysregulation of these molecules are often observed in tumors.
The phosphatase and tensin homolog (PTEN) is a tumor suppressor gene featuring dual-specificity phosphatase activities.3 It plays a critical role in maintaining normal cell activities and functions.3 On one hand, it can dephosphorylate focal adhesion kinase (FAK), to regulate cell adhesion, as well as Src-homologous collagen (Shc), to modulate cell migration.4,5 On the other hand, it is capable of antagonizing phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) activity by hydrolyzing 3-phosphate on phosphatidylinositol-3,4,5,-triphosphate (PIP3) to generate phosphatidylinositol-4,5,-bisphosphate (PIP2), thereby prohibiting downstream signaling molecule activation and finally, inhibiting cellular proliferation, growth, and survival.6 The latter feature of PTEN contributes greatly to its tumor suppressor role. The activity of PTEN is regulated by multiple internal and external factors. Somatic missense mutation or partial deletion of PTEN resulting in enzyme activity reduction or loss is prevalent in various tumors, most notably in the endometrium, brain, skin, colorectal, prostate, and breast cancers.7–11 There is enough evidence that the activity and function of PTEN are regulated by coordinated epigenetic, transcriptional, posttranscriptional, and posttranslational modifications. In particular, PTEN is subject to phosphorylation, somoylation, acetylation, and active site oxidation.12 Deficiency in posttranslational regulation can lead to the disorder of cell proliferation, apoptosis, migration, and adhesion, which are associated with cancer initiation, progression, and metastasis.
This review will update our knowledge on the critical role of posttranslational regulation of PTEN on its activity and the functional impact on cancer behaviors. A better understanding of how PTEN is regulated at the posttranslational level is essential for deepening our insights into cancer biology and identifying new therapeutic interventions for cancer.
Regulatory domain of PTEN
The PTEN protein consists of 403 amino acids. The crystal structure of PTEN has revealed that it contains five domains (Figure 1): a short N-terminal (PIP2)-binding domain; a N-terminal phosphatase domain that facilitates the hydrolysis of phospholipids; a C2 domain, which mediates the binding of signal related proteins to plasma membranes; a C-terminal tail domain containing PEST sequences which are enriched with proline(P), glutamic acid(E), serine(S), and threonine(T) as well as various phosphorylation sites; and a PDZ interaction motif which can bind to the lipid.13 Two naturally occurring mutations on the phosphatase domain make PTEN deficient in tumor suppression function: a C124S mutation, which can make the loss of both protein and lipid phosphatase activity of PTEN; and a G129E mutation resulting in only loss of the lipid phosphatase activity.14,15 Even though the N-terminal phosphatase domain is principally involved in PTEN’s physiological activity, the majority of PTEN mutations associated with carcinogenesis occur in the C2 domain and C-terminal tail, which suggests the critical role of the C-terminal sequence in maintaining PTEN function.16,17
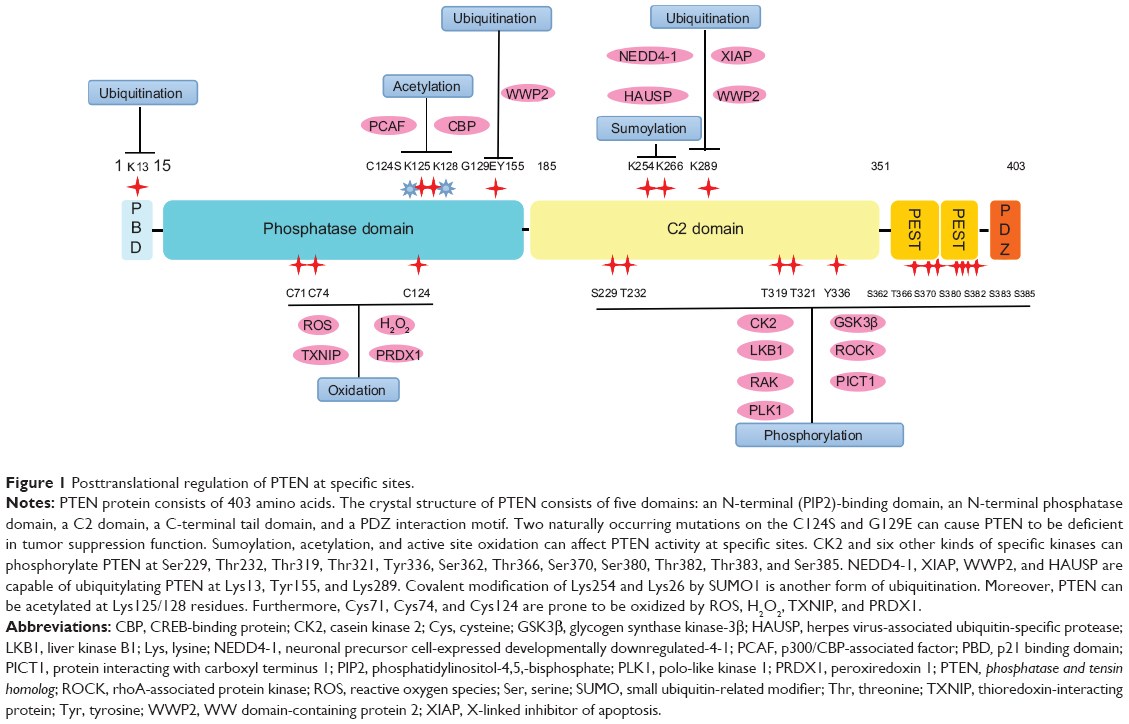 | Figure 1 Posttranslational regulation of PTEN at specific sites. |
Phosphorylation of PTEN
Phosphorylation of PTEN is a novel mechanism for PTEN inactivation, with great significance during carcinogenesis as shown in our previous work.18,19 Phosphorylated PTEN is still stable with a reduced activity. It can dephosphorylate PIP3 and thus activates PI3K/Akt signaling pathway. The phosphorylation sites of PTEN are mainly located in the C2 and the C-terminal domains.20,21 Nonphosphorylated PTEN can connect to the membrane at a faster rate and has a similar lifetime to the wild-type protein.22 Phosphorylated PTEN needs to be dephosphorylated before it can bind to membrane proteins to exert its full functionality.
Phosphorylation of PTEN can be mediated by multiple specific kinases (Table 1). As a ubiquitous Ser-/Thr-specific protein kinase required for viability and cell cycle progression, casein kinase 2 (CK2) can phosphorylate PTEN at Ser370, Ser380, Thr382, Thr383, and Ser385 (Figure 1), leading to a decrease of phosphatase activity and an increase of stability.23 Nonphosphorylated of PTEN will generate a closed and stable spatial structure at the C-terminal terminal domain, resulting in reduced membrane localization and phosphatase activity.21
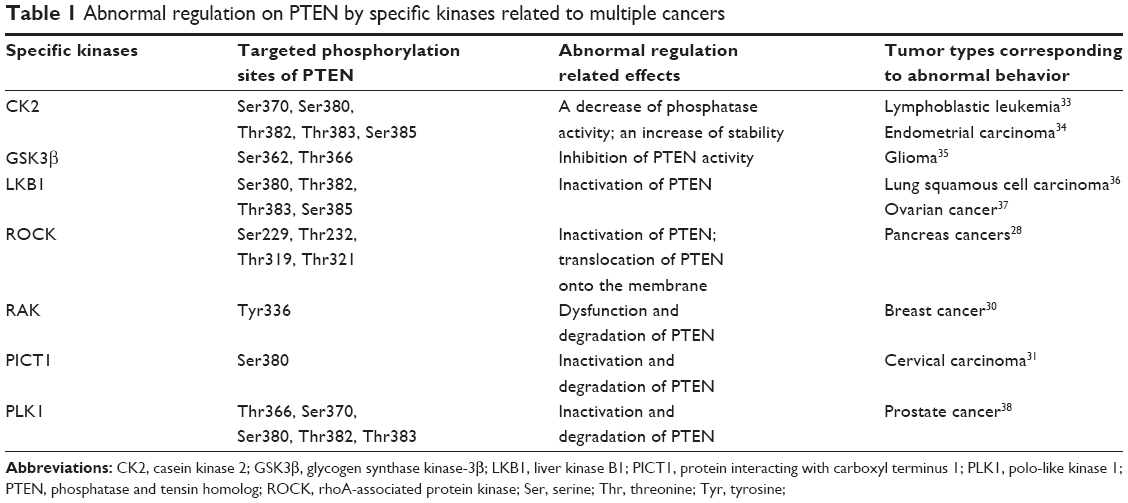 | Table 1 Abnormal regulation on PTEN by specific kinases related to multiple cancers |
Glycogen synthase kinase-3β (GSK3β) was also reported to phosphorylate PTEN at Ser362 and Thr366.24 Since GSK3β can be suppressed by insulin and some other activators of the PI3K signaling pathway, this intriguing finding suggests that GSK3β phosphorylation on PTEN is likely to be involved in the negative feedback loop of the PI3K signaling pathway.
Liver kinase B1 (LKB1) can phosphorylate Ser380, Thr382, Thr383, and Ser385 of PTEN.25 Phosphorylation of the above four residues can inactivate PTEN. Some argues that phosphorylated C-terminal interacts with C2 domain and PTEN phosphorylation domain, regarded as a pseudosubstrate and therefore induce auto-inhibition.26 According to this model, the Ser385 residue may be dephosphorylated followed with dephosphorylation of other neighboring regions, which possibly forms a more open structure and catalytic zone, causing an increase of the membrane affinity and PTEN activity.
Furthermore, rhoA-associated protein kinase (ROCK) can inactivate PTEN after phosphorylation on Ser229, Thr232, Thr319, and Thr321, and translocate it onto the membrane.27 The phenotype of PTEN and RhoA/ROCK pathway activation are closely linked.28 However, the p110 catalytic subunit of PI3K kinase is able to enhance tyrosine phosphorylation and decrease the activity of PTEN via the RhoA and ROCK pathway. The mechanism for this is to be determined.29
Moreover, a recent study has found that the RAK (a Russian word for cancer) nonreceptor tyrosine kinase is capable of enhancing PTEN tumor suppressor function, via phosphorylation of Tyr336, thus inhibiting its degradation by proteasome.30 This finding emphasizes the significance of accurate identification of specific tyrosine residues for phosphorylation in vivo, which may serve as indicators of cancer relapse or prognosis. Finally, protein interacting with carboxyl terminus 1 (PICT1) and polo-like kinase 1 (PLK1) have been reported to stabilize or activate PTEN.31,32
Ubiquitination and sumoylation of PTEN
With extensive studies on ubiquitination, which serves as an efficient approach for protein degradation, there is a great concern on ubiquitination of PTEN. The two PEST sequences on PTEN suggest the possibility of PTEN proteasomal degradation. The half-life of PTEN could be extended by inhibiting the proteasome, and ubiquitination of PTEN was shown to contribute to alterations of normal protein levels.39 A subsequent study confirmed that monoubiquitination of PTEN on the major conservative residues Lys13 and Lys289 made great sense in nuclear–cytoplasmic shuttling so that PTEN was able to inhibit cell growth independently of PI3K/Akt.40 Monoubiquitinated PTEN facilitates nuclear localization, while polyubiquitination causes proteasomal degradation in the cytosol, leading to loss of tumor suppressor activity of PTEN. Meanwhile, the conformational closure state of PTEN has difficulty in promoting ubiquitin-mediated proteasomal degradation. Consistently, a recent report has showed that the open conformational unphosphorylated PTEN is susceptible to ubiquitination, which promotes PTEN degradation and translocation into the nucleus.41
Neuronal precursor cell-expressed developmentally downregulated-4-1 (NEDD4-1) is the first identified E3 ligase for PTEN ubiquitination.42 NEDD4-1 is capable of monoubiquitylating PTEN, which is associated with nuclear shuttling, cell cycle arrest, and genomic stability. This explains why NEDD4-1 possesses tumor suppressive and oncogenic functions. But the mechanisms for NEDD4-1 in switching and balancing these two functions are yet to be determined. As NEDD4-1 is involved in the pathogenesis of non–small cell lung carcinoma and colorectal cancer, by inducing ubiquitination and degradation of PTEN,43,44 we propose that inhibition of NEDD4-1 has potential benefits in the treatment of cancer.
In addition, ubiquitination of PTEN is also considered to be mediated by X-linked inhibitor of apoptosis and E3 ubiquitin ligase WW domain-containing protein 2 (WWP2).45,46 It has been recently proved that phosphorylation at Tyr155 of PTEN is required for WWP2-associated PTEN ubiquitination.46 In order to maintain cell homeostasis, the deubiquitylating enzyme herpes virus-associated ubiquitin-specific protease (HAUSP) is thought to participate in the dynamic regulation of PTEN ubiquitination. This is supported by the fact that promyelocytic leukemia protein can affect PTEN activity through inhibition of HAUSP.48 In short, dynamic ubiquitinated modification of PTEN is important to its tumor suppressing function.
Intriguingly, SUMO family proteins were found to be able to regulate the activity of PTEN.48 Similar to ubiquitination, sumoylation displays function via covalent attachment to related proteins. After covalent modification by SUMO1, Lys254 and Lys266 in the C2 domain of PTEN bind to PIP3 through electrostatic interactions, thus downregulating the PI3K/Akt signaling pathway and inhibiting cellular growth and proliferation.49
Acetylation of PTEN
Acetylation and deacetylation of PTEN are available for normal exertion of its biological functions. Under certain circumstances, such as in response to growth factors, p300/calcium-binding PTEN-associated factor can acetylate Lys125/128 residues situated within the catalytic cleft of PTEN. The acetylation influences the substrate affinity of PTEN towards PIP3, thereby inhibiting its catalytic phosphatase activity.50 Acetylation of Lys402 located in PDZ-binding domain by CREB-binding protein can promote interactions between PTEN and proteins on the PDZ domain, hence affecting cellular functions.51 Conversely, the histone deacetylase sirtuin1 can modulate PTEN activity via deacetylation, thus contributing to the regulation of PTEN function.52
Oxidation of PTEN
PTEN is a member of the protein tyrosine phosphatase family, and its normal activity depends on the presence of a highly reactive cysteine residue. Interestingly, the cysteine located on the reactive site is prone to be oxidized; therefore, oxidative inactivation is vital to regulate the activity and function of PTEN.
Studies have shown that H2O2 causes oxidation of Cys124 in the catalytic center of PTEN so that it binds with Cys74 to form a disulfide bond, which leads to the decrease of PTEN phosphatase activity in time- and dose-dependent manners.53 Further, Lea et al found that both endogenous and exogenous H2O2 could cause oxidative inactivation of PTEN, followed by the increase of PIP3 concentration and Akt phosphorylation.54
However, reactive oxygen species (ROS) reduces PTEN activity through intramolecular disulfide bond formation between Cys124 and Cys71.55 Treatment of macrophages with phorbol ester, lipopolysaccharide, or the cellular mitochondrial electron transport chain inhibitors can provoke ROS production and raise the oxidized fraction of PTEN from 5% to 16%.56,57 ROS directly interacts with critical signaling molecules to initiate signaling in a broad variety of cellular processes, such as proliferation, survival, apoptosis, and autophagy. Notably, an increased level of ROS can inactivate PTEN by directly oxidizing its cysteine residue but also, by indirectly stimulating its phosphorylation, and thus gives rise to activation of the PI3K/Akt signaling pathway.58 Furthermore, 2,4,2-chloronitrobenzene, an inhibitor of thioredoxin reductase, has been shown to significantly suppress the reduction of oxidized PTEN, and conversely, overexpression of thioredoxin reductase can promote the deoxidation of PTEN and resumption of its normal tumor suppressing function.54
Additional studies have found that oxidation and reduction of PTEN can be influenced by thioredoxin-interacting protein and peroxiredoxin 1.58,59 This reversible inactivation of PTEN is commonly seen in cells treated with growth factors that stimulates peroxide production, implying that PTEN responds to mitogens through reduction-oxidation inactivation.60 Increase of H2O2 induced by disordered mitochondrial function results in oxidative inactivation of PTEN and PI3K signaling activity enhancement.61 Whether oxidized PTEN can regain its tumor suppressing function or not depends mainly on the ability of thioredoxin to decrease the inactive modus; however, the persistent abnormal reduction-oxidation status will no doubt cause a complete loss of PTEN function.62
Discussion and conclusion
Posttranslational modifications of proteins in eukaryotic cells can diversify and extend protein functions beyond what is dictated by the number of genes. These dynamic modifications reversibly or irreversibly change the structure, activity, and function of numerous important proteins through a variety of biochemical reactions, consequently regulating the signal integration, homeostasis, and physiological states. Abnormal and defective protein posttranslational modifications have been associated with a number of important human diseases, including cancer, diabetes, Alzheimer’s disease, and stroke. Alternation in PTEN level, activity, and function is one of the main factors that determine the occurrence and progression of tumor (Figure 2). A full understanding of the regulatory mechanisms of PTEN will help elucidate the full picture of tumor development, progression, and metastasis, and also provide a basis for discovery of new drug targets for cancer treatment. However, there are still a number of issues that need further exploration. For example, PTEN is a tumor suppressor gene, but PTEN2, its homologous gene, is a tumor testis antigen gene. Since both of them can be dephosphorylated, what on earth leads to the total opposite behavior and function? Additionally, how do phosphorylation, ubiquitylation, sumoylation, acetylation, and oxidation posttranslational regulations of PTEN cooperate with each other in maintaining the normal activity, and how do they play a joint role in tumorigenesis? What is more, although currently PTEN has been transfected into multiple cancer cell lines to confirm its growth inhibition function, so far, there is still no application of PTEN for gene therapy in human tissues.
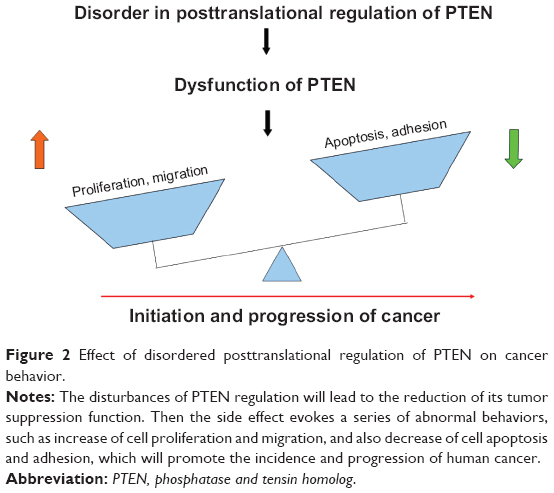 | Figure 2 Effect of disordered posttranslational regulation of PTEN on cancer behavior. |
Perspectives
As further studies on the mechanism of how PTEN and its related proteins display its specific role in tumor incidence, invasion, and metastasis, we conceive that biological gene therapy with PTEN is likely to be applied to cancer patients, especially those in the advanced stages. Other than providing the scientific basis for clinic surveillance and evaluation on cancer prognosis and drug efficacy, development of new anti-tumor drugs is equally important. In addition, excitingly, acute complete absence of PTEN, not only fails to promote tumorigenesis but also, causes cell senescence, which is different from the typical deletion. This aspect may offer a sound way for cancer treatment. Besides, PTEN is mainly found to be located in the nucleus of the original, differentiating, or resting cells; however, in cancer cells, which have a rapid proliferation, PTEN rarely enters the nucleus. Therefore, finding the mechanism of PTEN nuclear–cytoplasmic shuttling is also important for the discovery of new cures for cancer patients. With increasing evidence that PTEN is regulated by multiple mechanisms and networked proteins, we believe that its exact role in carcinogenesis, development, and metastasis will be further revealed. PTEN and its functionally related proteins may represent useful targets for the discovery of novel anticancer drugs and gene therapy, and the therapeutic potentials should be fully explored.
Acknowledgments
This research was supported by the National Science and Technology Major Projects for “Major New Drugs Innovation and Development” of the People’s Republic of China (grant number 2011ZX09302-007-03), the National Natural Science Foundation of the People’s Republic of China (grant numbers 81060038 and 81460377), the Natural Science Foundation of Jiangxi Province, People’s Republic of China (grant number 20142BAB215036), the Science and Technology Foundation of the Department of Education of Jiangxi Province, People’s Republic of China (grant number GJJ14169), and the “Talent 555 Project” of Jiangxi Province, People’s Republic of China.
Disclosure
The authors report no conflict of interest in this work.
References
Alexandrov LB, Nik-Zainal S, Wedge DC, et al; Australian Pancreatic Cancer Genome Initiative; ICGC Breast Cancer Consortium; ICGC MMML-Seq Consortium; ICGC PedBrain. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–421. | ||
Deng N, Goh LK, Wang H, et al. A comprehensive survey of genomic alterations in gastric cancer reveals systematic patterns of molecular exclusivity and co-occurrence among distinct therapeutic targets. Gut. 2012;61(5):673–684. | ||
Waite KA, Eng C. Protean PTEN: form and function. Am J Hum Genet. 2002;70(4):829–844. | ||
Zhang LL, Liu J, Lei S, Zhang J, Zhou W, Yu HG. PTEN inhibits the invasion and metastasis of gastric cancer via downregulation of FAK expression. Cell Signal. 2014;26(5):1011–1020. | ||
Schneider E, Keppler R, Prawitt D, et al. Migration of renal tumor cells depends on dephosphorylation of Shc by PTEN. Int J Oncol. 2011;38(3):823–831. | ||
Ye B, Jiang LL, Xu HT, Zhou DW, Li ZS. Expression of PI3K/AKT pathway in gastric cancer and its blockade suppresses tumor growth and metastasis. Int J Immunopathol Pharmacol. 2012;25(3):627–636. | ||
Baig RM, Mahjabeen I, Sabir M, et al. Genetic changes in the PTEN gene and their association with breast cancer in Pakistan. Asian Pac J Cancer Prev. 2011;12(10):2773–2778. | ||
Kandoth C, Schultz N, Cherniack AD, et al; Cancer Genome Atlas Research Network. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497(7447):67–73. | ||
Moon SH, Kim DK, Cha Y, Jeon I, Song J, Park KS. PI3K/Akt and Stat3 signaling regulated by PTEN control of the cancer stem cell population, proliferation and senescence in a glioblastoma cell line. Int J Oncol. 2013;42(3):921–928. | ||
Patel R, Gao M, Ahmad I, et al. Sprouty2, PTEN, and PP2A interact to regulate prostate cancer progression. J Clin Invest. 2013;123(3):1157–1175. | ||
Romano C, Schepis C. PTEN gene: a model for genetic diseases in dermatology. Scientific World Journal. 2012;2012:252457. | ||
Correia NC, Gírio A, Antunes I, Martins LR, Barata JT. The multiple layers of non-genetic regulation of PTEN tumour suppressor activity. Eur J Cancer. 2014;50(1):216–225. | ||
Lee JO, Yang H, Georgescu MM, et al. Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association. Cell. 1999;99(3):323–334. | ||
Papa A, Wan L, Bonora M, et al. Cancer-associated PTEN mutants act in a dominant-negative manner to suppress PTEN protein function. Cell. 2014;157(3):595–610. | ||
Liaw D, Marsh DJ, Li J, et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet. 1997;16(1):64–67. | ||
Shenoy SS, Nanda H, Lösche M. Membrane association of the PTEN tumor suppressor: electrostatic interaction with phosphatidylserine-containing bilayers and regulatory role of the C-terminal tail. J Struct Biol. 2012;180(3):394–408. | ||
Sun Z, Huang C, He J, et al. PTEN C-terminal deletion causes genomic instability and tumor development. Cell Rep. 2014;6(5):844–854. | ||
Yang Z, Yuan XG, Chen J, Luo SW, Luo ZJ, Lu NH. Reduced expression of PTEN and increased PTEN phosphorylation at residue Ser380 in gastric cancer tissues: a novel mechanism of PTEN inactivation. Clin Res Hepatol Gastroenterol. 2013;37(1):72–79. | ||
Xu WT, Yang Z, Lu NH. Roles of PTEN (Phosphatase and Tensin Homolog) in gastric cancer development and progression. Asian Pac J Cancer Prev. 2014;15(1):17–24. | ||
Cordier F, Chaffotte A, Terrien E, et al. Ordered phosphorylation events in two independent cascades of the PTEN C-tail revealed by NMR. J Am Chem Soc. 2012;134(50):20533–20543. | ||
Bolduc D, Rahdar M, Tu-Sekine B, et al. Phosphorylation-mediated PTEN conformational closure and deactivation revealed with protein semisynthesis. Elife. 2013;2:e00691. | ||
Rahdar M, Inoue T, Meyer T, Zhang J, Vazquez F, Devreotes PN. A phosphorylation-dependent intramolecular interaction regulates the membrane association and activity of the tumor suppressor PTEN. Proc Natl Acad Sci U S A. 2009;106(2):480–485. | ||
Liang K, Esteva FJ, Albarracin C, et al. Recombinant human erythropoietin antagonizes trastuzumab treatment of breast cancer cells via Jak2-mediated Src activation and PTEN inactivation. Cancer Cell. 2010;18(5):423–435. | ||
Jang HD, Noh JY, Shin JH, Lin JJ, Lee SY. PTEN regulation by the Akt/GSK-3β axis during RANKL signaling. Bone. 2013;55(1):126–131. | ||
Mehenni H, Lin-Marq N, Buchet-Poyau K, et al. LKB1 interacts with and phosphorylates PTEN: a functional link between two proteins involved in cancer predisposing syndromes. Hum Mol Genet. 2005;14(15):2209–2219. | ||
Odriozola L, Singh G, Hoang T, Chan, AM. Regulation of PTEN activity by its carboxyl-terminal autoinhibitory domain. J Biol Chem. 282(32);23306–23315. | ||
Vemula S, Shi J, Hanneman P, Wei L, Kapur R. ROCK1 functions as a suppressor of inflammatory cell migration by regulating PTEN phosphorylation and stability. Blood. 2010;115(9):1785–1796. | ||
Vo K, Amarasinghe B, Washington K, Gonzalez A, Berlin J, Dang TP. Targeting notch pathway enhances rapamycin antitumor activity in pancreas cancers through PTEN phosphorylation. Mol Cancer. 2011;10:138. | ||
Papakonstanti EA, Ridley AJ, Vanhaesebroeck B. The p110delta isoform of PI 3-kinase negatively controls RhoA and PTEN. EMBO J. 2007;26(13):3050–3061. | ||
Yim EK, Peng G, Dai H, et al. Rak functions as a tumor suppressor by regulating PTEN protein stability and function. Cancer Cell. 2009;15(4):304–314. | ||
Okahara F, Itoh K, Nakagawara A, Murakami M, Kanaho Y, Maehama T. Critical role of PICT-1, a tumor suppressor candidate, in phosphatidylinositol 3,4,5-trisphosphate signals and tumorigenic transformation. Mol Biol Cell. 2006;17(11):4888–4895. | ||
Choi BH, Pagano M, Dai W. Plk1 Protein Phosphorylates Phosphatase and Tensin Homolog (PTEN) and Regulates Its Mitotic Activity during the Cell Cycle. J Biol Chem. 2014;289(20):14066–14074. | ||
Gomes AM, Soares MV, Ribeiro P, et al. Adult B-cell acute lymphoblastic leukemia cells display decreased PTEN activity and constitutive hyperactivation of PI3K/Akt pathway despite high PTEN protein levels. Haematologica. 2014;99(6):1062–1068. | ||
Pallares J, Llobet D, Santacana M, et al. CK2beta is expressed in endometrial carcinoma and has a role in apoptosis resistance and cell proliferation. Am J Pathol. 2009;174(1):287–296. | ||
Lin C, Liang Y, Zhu H, Zhang J, Zhong X. R280T mutation of p53 gene promotes proliferation of human glioma cells through GSK-3β/PTEN pathway. Neurosci Lett. 2012;529(1):60–65. | ||
Xu C, Fillmore CM, Koyama S, et al. Loss of Lkb1 and Pten leads to lung squamous cell carcinoma with elevated PD-L1 expression. Cancer Cell. 2014;25(5):590–604. | ||
Tanwar PS, Mohapatra G, Chiang S, et al. Loss of LKB1 and PTEN tumor suppressor genes in the ovarian surface epithelium induces papillary serous ovarian cancer. Carcinogenesis. 2014;35(3):546–553. | ||
Liu XS, Song B, Elzey BD, et al. Polo-like kinase 1 facilitates loss of Pten tumor suppressor-induced prostate cancer formation. J Biol Chem. 2011;286(41):35795–35800. | ||
Torres J, Pulido R. The tumor suppressor PTEN is phosphorylated by the protein kinase CK2 at its C terminus. Implications for PTEN stability to proteasome-mediated degradation. J Biol Chem. 2001;276(2):993–998. | ||
Trotman LC, Wang X, Alimonti A, et al. Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell. 2007;128(1):141–156. | ||
Song MS, Carracedo A, Salmena L, et al. Nuclear PTEN regulates the APC-CDH1 tumor-suppressive complex in a phosphatase-independent manner. Cell. 2011;144(2):187–199. | ||
Wang X, Trotman LC, Koppie T, et al. NEDD4-1 is a proto-oncogenic ubiquitin ligase for PTEN. Cell. 2007;128(1):129–139. | ||
Amodio N, Scrima M, Palaia L, et al. Oncogenic role of the E3 ubiquitin ligase NEDD4-1, a PTEN negative regulator, in non-small-cell lung carcinomas. Am J Pathol. 2010;177(5):2622–2634. | ||
Eide PW, Cekaite L, Danielsen SA, et al. NEDD4 is overexpressed in colorectal cancer and promotes colonic cell growth independently of the PI3K/PTEN/AKT pathway. Cell Signal. 2013;25(1):12–18. | ||
Van Themsche C, Leblanc V, Parent S, Asselin E. X-linked inhibitor of apoptosis protein (XIAP) regulates PTEN ubiquitination, content, and compartmentalization. J Biol Chem. 2009;284(31):20462–20466. | ||
Maddika S, Kavela S, Rani N, et al. WWP2 is an E3 ubiquitin ligase for PTEN. Nat Cell Biol. 2011;13(6):728–733. | ||
Song MS, Salmena L, Carracedo A, et al. The deubiquitinylation and localization of PTEN are regulated by a HAUSP-PML network. Nature. 2008;455(7214):813–817. | ||
González-Santamaría J, Campagna M, Ortega-Molina A, et al. Regulation of the tumor suppressor PTEN by SUMO. Cell Death Dis. 2012;3:e393. | ||
Huang J, Yan J, Zhang J, et al. SUMO1 modification of PTEN regulates tumorigenesis by controlling its association with the plasma membrane. Nat Commun. 2012;3:911. | ||
Okumura K, Mendoza M, Bachoo RM, DePinho RA, Cavenee WK, Furnari FB. PCAF modulates PTEN activity. J Biol Chem. 2006;281(36):26562–26568. | ||
Ding L, Chen S, Liu P, et al. CBP loss cooperates with PTEN haploinsufficiency to drive prostate cancer: implications for epigenetic therapy. Cancer Res. 2014;74(7):2050–2061. | ||
Chae HD, Broxmeyer HE. SIRT1 deficiency downregulates PTEN/JNK/FOXO1 pathway to block reactive oxygen species-induced apoptosis in mouse embryonic stem cells. Stem Cells Dev. 2011;20(7):1277–1285. | ||
Cho SH, Lee CH, Ahn Y, et al. Redox regulation of PTEN and protein tyrosine phosphatases in H(2)O(2) mediated cell signaling. FEBS Lett. 2004;560(1–3):7–13. | ||
Lee SR, Yang KS, Kwon J, Lee C, Jeong W, Rhee SG. Reversible inactivation of the tumor suppressor PTEN by H2O2. J Biol Chem. 2002;277(23):20336–20342. | ||
Gebremedhin D, Terashvili M, Wickramasekera N, et al. Redox signaling via oxidative inactivation of PTEN modulates pressure-dependent myogenic tone in rat middle cerebral arteries. PLoS One. 2013;8(7):e68498. | ||
Leslie NR, Bennett D, Lindsay YE, Stewart H, Gray A, Downes CP. Redox regulation of PI 3-kinase signalling via inactivation of PTEN. EMBO J. 2003;22(20):5501–5510. | ||
Wu KL, Wu CA, Wu CW, Chan SH, Chang AY, Chan JY. Redox-sensitive oxidation and phosphorylation of PTEN contribute to enhanced activation of PI3K/Akt signaling in rostral ventrolateral medulla and neurogenic hypertension in spontaneously hypertensive rats. Antioxid Redox Signal. 2013;18(1):36–50. | ||
Wu Y, Zhou H, Wu K, Lee S, Li R, Liu X. PTEN phosphorylation and nuclear export mediate free fatty acid-induced oxidative stress. Antioxid Redox Signal. 2014;20(9):1382–1395. | ||
Cao J, Schulte J, Knight A, et al. Prdx1 inhibits tumorigenesis via regulating PTEN/AKT activity. EMBO J. 2009;28(10):1505–1517. | ||
Hui ST, Andres AM, Miller AK, et al. Txnip balances metabolic and growth signaling via PTEN disulfide reduction. Proc Natl Acad Sci U S A. 2008;105(10):3921–3926. | ||
Kwon J, Lee SR, Yang KS, et al. Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proc Natl Acad Sci U S A. 2004;101(47):16419–16424. | ||
Xu J, Tian W, Ma X, et al. The molecular mechanism underlying morphine-induced Akt activation: roles of protein phosphatases and reactive oxygen species. Cell Biochem Biophys. 2011;61(2):303–311. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.