")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Oxcarbazepine-loaded polymeric nanoparticles: development and permeability studies across in vitro models of the blood–brain barrier and human placental trophoblast
Authors Lopalco A, Ali H, Denora N, Rytting E
Received 14 November 2014
Accepted for publication 18 January 2015
Published 11 March 2015 Volume 2015:10(1) Pages 1985—1996
DOI https://doi.org/10.2147/IJN.S77498
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster

Antonio Lopalco,1–3,* Hazem Ali,1,* Nunzio Denora,3 Erik Rytting1,4,5
1Department of Obstretrics and Gynecology, University of Texas Medical Branch, Galveston, TX, USA; 2Department of Pharmaceutical Chemistry, University of Kansas, Lawrence, KS, USA; 3Department of Pharmacy – Drug Sciences, University of Bari Aldo Moro, Bari, Italy; 4Center for Biomedical Engineering, University of Texas Medical Branch, Galveston, TX, USA; 5Department of Pharmacology and Toxicology, University of Texas Medical Branch, Galveston, TX, USA
*These authors contributed equally to this work
Abstract: Encapsulation of antiepileptic drugs (AEDs) into nanoparticles may offer promise for treating pregnant women with epilepsy by improving brain delivery and limiting the transplacental permeability of AEDs to avoid fetal exposure and its consequent undesirable adverse effects. Oxcarbazepine-loaded nanoparticles were prepared by a modified solvent displacement method from biocompatible polymers (poly(lactic-co-glycolic acid) [PLGA] with or without surfactant and PEGylated PLGA [Resomer® RGPd5055]). The physical properties of the developed nanoparticles were determined with subsequent evaluation of their permeability across in vitro models of the blood–brain barrier (hCMEC/D3 cells) and human placental trophoblast cells (BeWo b30 cells). Oxcarbazepine-loaded nanoparticles with encapsulation efficiency above 69% were prepared with sizes ranging from 140–170 nm, polydispersity indices below 0.3, and zeta potential values below −34 mV. Differential scanning calorimetry and X-ray diffraction studies confirmed the amorphous state of the nanoencapsulated drug. The apparent permeability (Pe) values of the free and nanoencapsulated oxcarbazepine were comparable across both cell types, likely due to rapid drug release kinetics. Transport studies using fluorescently-labeled nanoparticles (loaded with coumarin-6) demonstrated increased permeability of surfactant-coated nanoparticles. Future developments in enzyme-prodrug therapy and targeted delivery are expected to provide improved options for pregnant patients with epilepsy.
Keywords: nanoparticles, epilepsy, PLGA, BeWo cells, coumarin-6, hCMEC/D3 cells
Introduction
Pregnancy is especially challenging for women with epilepsy since uncontrolled seizures may be hazardous to both the pregnant woman and her fetus. During the first trimester, uncontrolled seizures cause fetal developmental delay. On the other hand, the use of anticonvulsants for controlling seizures carries the risk of potential teratogenicity.1,2 Infants who have been exposed to antiepileptic drugs (AEDs) in utero run an increased risk of congenital malformations in a dose-dependent manner.3–7 In a review of the Medical Birth Registry of Norway from 1999 to 2011, the odds ratio for major congenital malformations in children exposed to any AED in utero was 1.27 (95% confidence interval: 1.02–1.59).8 Epileptic therapy during pregnancy, therefore, requires balance between the benefits to pregnant women and the associated risks of AEDs upon the developing fetus.
Oxcarbazepine, like other AEDs (eg, valproate and carbamazepine) crosses human placenta. A study published in 1993 revealed no congenital malformations among nine children born to patients exposed to oxcarbazepine during the first trimester, but one congenital malformation was reported out of 57 children exposed in utero to oxcarbazepine monotherapy.9 Another study reported three spontaneous abortions among twelve pregnant women.10 Therefore, there is a need for a drug delivery platform that can provide beneficial therapeutic outcomes for pregnant patients by improving the transport of medications across the blood–brain barrier (BBB) and simultaneously limit their transplacental passage.
The BBB is formed by capillary endothelial cells, which are connected by tight junctions, adherens junctions, and macula adherens, also known as desmosomes (Figure 1).11,12 This elaborated network of proteins, the absence of fenestrations, low pinocytic activity, and efflux transporters (P-glycoprotein, breast cancer resistance protein, and multidrug resistance proteins) may restrict the movement of compounds from the blood into the brain parenchyma.
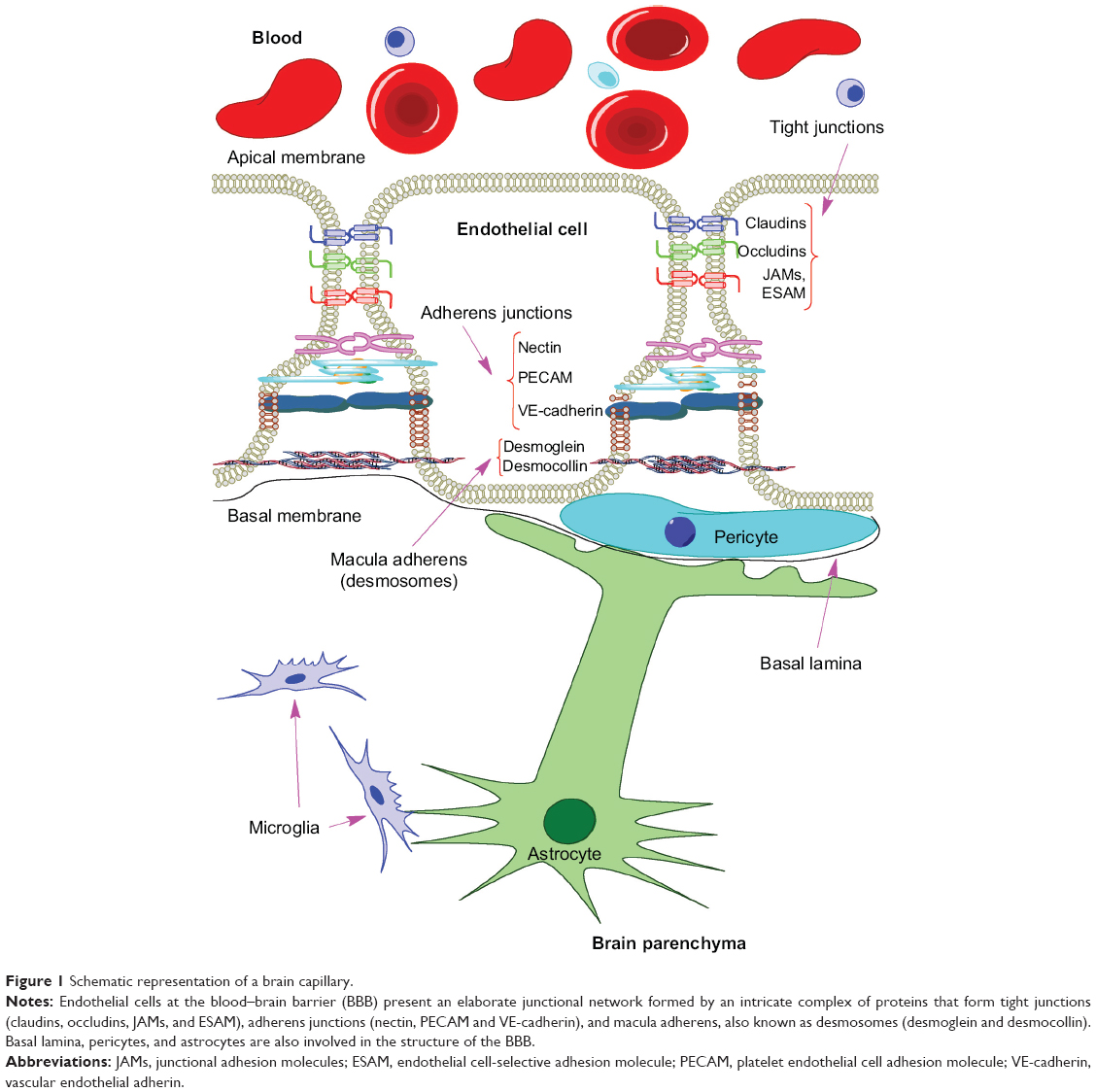 | Figure 1 Schematic representation of a brain capillary. |
The placental barrier (Figure 2) is composed of three layers: multinucleated syncytiotrophoblast cells, connective tissue, and the fetal vascular endothelium.13 Unlike the capillaries in the brain, the fetal capillaries within the placenta allow for more movement of molecules. The rate-limiting barrier for permeation across the human placenta is the syncytiotrophoblast layer that is characterized by complex tight junctions.14,15 Transport of molecules across the placental barrier, therefore, depends upon physicochemical properties such as molecular weight, charge, and lipophilicity.16
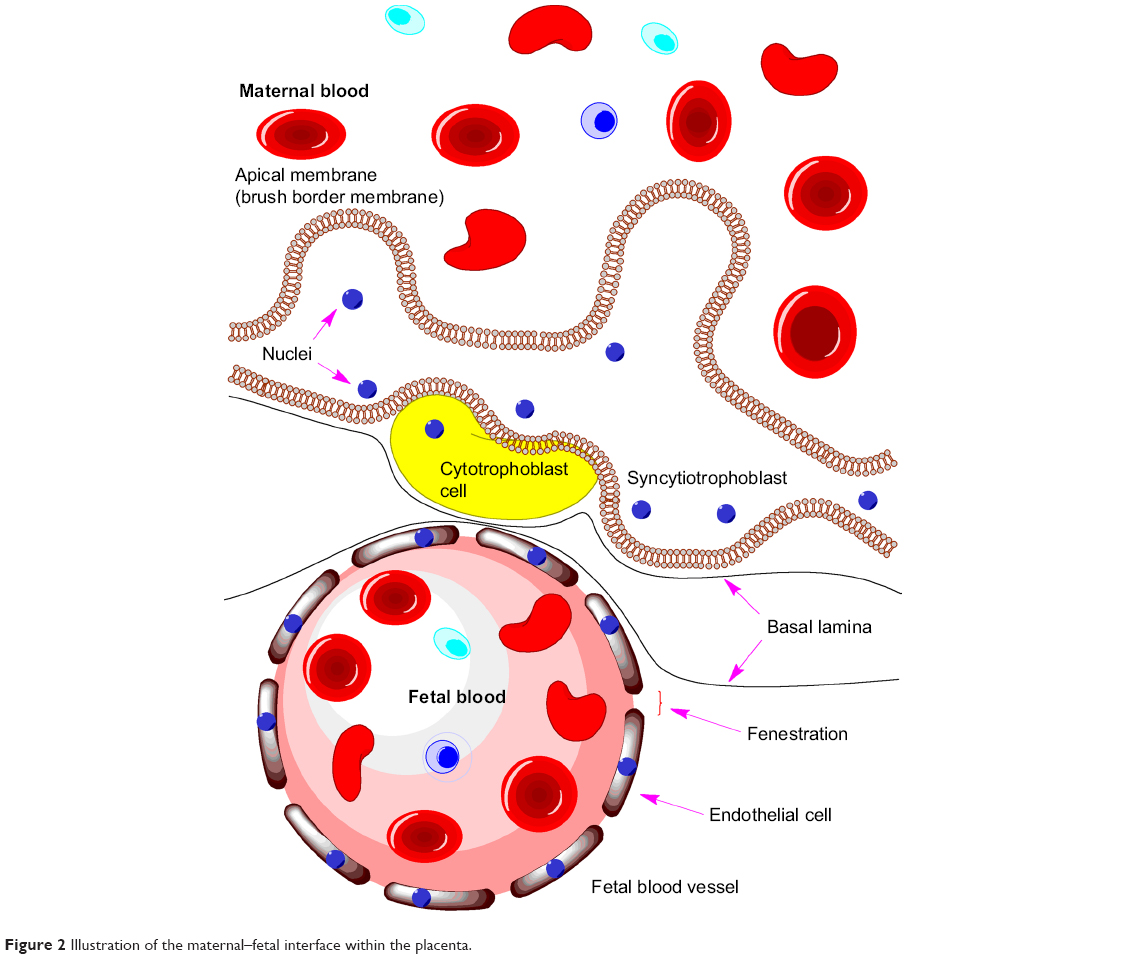 | Figure 2 Illustration of the maternal–fetal interface within the placenta. |
Nanoparticle delivery vehicles can target medications to the site of interest upon modifying their surfaces with ligands that recognize specific cell surface receptors. Liposomes, polymeric nanoparticles, solid lipid nanoparticles, polymeric micelles, and dendrimers are well-known nanoparticles that provide various beneficial therapeutic outcomes.17 They can be optimized in terms of their chemical composition, size, uptake properties, plasma half-life/clearance and immunogenicity for enhancing transcellular permeability of therapeutic agents and biomacromolecules across biological barriers.18 Nanoparticles have been used for targeted delivery to the brain,19,20 and it has been hypothesized that nanoparticles can improve antiepileptic therapy.21 Furthermore, nanoparticles can be employed in enzyme-prodrug therapeutic approaches to achieve pharmacological activity in target tissues.22,23 In the case of AED therapy during pregnancy, targeted nanomedicine strategies could potentially increase the proportion of a dose reaching the target tissue, subsequently resulting in reduced transplacental transfer of the AEDs. It is anticipated that this could lead to a reduced risk of birth defects in children exposed to AEDs in utero.24,25
Pharmaceutical development of therapeutic nanoparticles involves several steps, starting from their design with optimal drug encapsulation and drug release characteristics to production of nanoformulations with desired permeability across biological barriers and shelf-life stability. In this work, we have encapsulated oxcarbazepine into biodegradable and biocompatible nanoparticles composed of carboxylate end group 50:50 poly(lactic-co-glycolic acid) (PLGA), PEGylated PLGA (Resomer® RGPd5055), and PLGA nanoparticles coated with a surfactant (α-tocopherol polyethylene glycol-1000-succinate, TPGS). The nanoparticles were prepared by a modified solvent displacement method26 and characterized for morphology, particle size, zeta potential, and encapsulation efficiency. The second aim in this report was to investigate the transport of the oxcarbazepine-loaded nanoformulations and fluorescently-labeled nanoparticles (loaded with coumarin-6) across in vitro models of human placenta (BeWo b30 cells) and the BBB (hCMEC/D3 cells) using well-established protocols.27–29
Methods
Materials
Ascorbic acid was purchased from Sigma (St Louis, MO, USA); acetone and HPLC grade acetonitrile from Acros Organics (Fair Lawn, NJ, USA); carboxylate end group 50:50 poly(D,L-lactide-co-glycolide) (PLGA, inherent viscosity, 0.15–0.25 dL/g) from Durect Corporation (Pelham, AL, USA); chemically defined lipid concentrate from Invitrogen (Carlsbad, CA); dimethylsulfoxide from Sigma; Dulbecco’s Modified Eagle’s Medium (DMEM) from Gibco® (Grand Island, NY, USA); Dulbecco’s Phosphate Buffered Saline (without calcium and magnesium) from Cellgro Mediatech, Inc. (Manassa, VA, USA); EBM-2 basal medium from Lonza (Walkersville, MD, USA). Hanks’ buffered saline solution (HBSS) with calcium and magnesium and HEPES buffer from Cellgro Mediatech, Inc.; human Basic Fibroblast Growth Factor (bFGF) from Cell Signaling Technology (Danvers, MA, USA); human placental collagen (type IV) from Sigma-Aldrich; HPLC grade methanol from Ricca Chemical (Arlington, TX, USA); hydrocortisone from Fisher Scientific (Pittsburg, PA, USA); Minimum Essential Medium (MEM) from Gibco®; fetal bovine serum (FBS), MEM amino acid solution, and MEM vitamin solution from Hyclone (Logan, UT, USA); nonessential amino acids from Sigma-Aldrich; oxcarbazepine from TCI-America Inc. (Portland, OR, USA); penicillin/streptomycin and L-glutamine from Cellgro Mediatech, Inc.; Type I rat tail collagen from Fisher Scientific; Resomer® RGPd5055 (diblock PEGylated copolymer of PLGA containing 5% polyethylene glycol [PEG5000] from Boehringer Ingelheim [Ingelheim am Rhein, Germany]); α-Tocopherol polyethylene glycol-1000-succinate (TPGS) and coumarin-6 from Sigma; ultrapure water was obtained from a water purification system.
Preparation of oxcarbazepine- and coumarin-6-loaded nanoparticles
Polymeric nanoparticles (Table 1) were prepared at room temperature (22°C–23°C) by a modified solvent displacement method as described previously.26 Briefly, acetone was used to dissolve oxcarbazepine (5% w/w) or coumarin-6 (0.1% w/w) and 15 to 20 mg of the polymer. An aqueous solution of α-tocopherol polyethylene glycol-1000-succinate (TPGS, at 0%–0.03% (w/v)) was used as a surfactant. One milliliter of the organic phase was injected in 5 mL of water in the presence or absence of surfactant under magnetic stirring (450 rpm), at room temperature, using an electronically adjustable peristaltic pump (Traceable® Calibration Control Company, Friendswood, TX, USA) at a constant flow rate of 6 mL min−1. The resulting mixture was stirred (850 rpm) in the fume hood for 4–5 hours to allow for complete evaporation of the acetone. Coumarin-6-loaded nanoparticles were protected from light until further analysis.
 | Table 1 Composition of oxcarbazepine-loaded nanoparticles, coumarin-6-labeled nanoparticles, and unloaded nanoparticles |
Measurement of particle size, size distribution, and zeta potential (ζ)
Samples were diluted to 10% with purified water, and their corresponding Z-average particle size and polydispersity index (PDI) were measured by dynamic light scattering at 25°C with backscatter detection (173°) using a High Performance Particle Sizer (Malvern Instruments, Malvern, UK). The PDI describes the deviation of the measured autocorrelation function during particle size analysis from that of monodisperse spheres having the same diameter.30 Zeta potential was measured at 25°C by laser Doppler velocimetry using a Zetasizer 2000 (Malvern Instruments). Analyses were performed in triplicate unless otherwise specified.
Determination of nanoparticle entrapment efficiency
To determine the amount of oxcarbazepine incorporated into the nanoparticles, a 500-μL aliquot of the nanoparticle suspension was placed in the upper chamber of Vivaspin® 6 centrifugal concentrator (100 kDa molecular weight cutoff, Sartorius Stedim Biotech GmbH, Göttingen, Germany). The assembly was then centrifuged at 3,000 rpm for 15 minutes at 25°C using an Eppendorf® 5810R centrifuge (Hamburg, Germany). The nanoparticles – including the encapsulated oxcarbazepine – remained in the upper chamber, while the aqueous filtrate contained the free, unencapsulated oxcarbazepine. After centrifugation, the concentration of free oxcarbazepine in the filtrate was quantified by HPLC, as described in the next section. Encapsulation efficiency was measured in triplicate and calculated as:
|
|
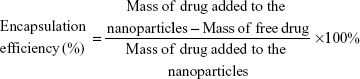
Instrumentation and chromatographic conditions
The HPLC analysis was carried out by a Waters apparatus equipped with a 1525 binary HPLC pump, 2707 autosampler unit, and 2998 photodiode array (PDA) detector (Waters® Corporation, Milford, MA, USA), and an Onyx® monolithic C18 column (4.6×100 mm, Phenomenex, Torrance, CA, USA). Data acquisition was performed using Empower™ software, version 2.0 (Waters® Corporation). An isocratic elution of 0.7 mL min−1 was performed using water:acetonitrile at a 64:36 ratio. The mobile phase was allowed to run for 30–45 minutes prior to analysis of each sample in order to wash the column. The detection of oxcarbazepine was carried out at λ =255 nm. An injection volume of 10 μL was used in all experiments, and the elution was carried out at room temperature.
In vitro release studies
In vitro release of oxcarbazepine from the nanoformulations was studied at pH 7.4 in phosphate buffered saline (0.8 g NaCl, 0.02 g KCl, 0.3 g Na2HPO4·7H2O, and 0.02 g KH2PO4 in 100 mL of deionized water). Several vials were prepared and shaken with an orbital shaker (50 rpm) in a hot box at 37°C. Nanoparticle suspensions were diluted 1:20 (v/v) in the buffer solution. Samples (500 μL) were taken at 0.5, 1, 2, 4, and 24 hours. The amount of oxcarbazepine released was determined by HPLC analysis of the concentration of free drug in the aqueous filtrate following centrifugal filtration, as previously described for the determination of encapsulation efficiency. The amount of drug released was determined in triplicate and expressed as a percentage of the initial amount of drug loaded in the nanoparticles.
Cryo-electron microscopy
Cryo-electron microscopy (cryo-EM) was utilized to examine oxcarbazepine-loaded nanoparticles and to ensure the absence of any free oxcarbazepine crystals that might crystallize out during the fabrication process. Oxcarbazepine-loaded nanoparticles were vitrified on holey carbon film grids (C-flat,™ Protochips, Raleigh, NC, USA) to minimize any potential morphological changes during specimen preparation and imaging.31,32 In brief, nanoparticle dispersions were applied to the holey films in a volume of ~2 μL, blotted with filter paper, and plunged into liquid ethane cooled in a liquid nitrogen bath. Frozen grids were stored under liquid nitrogen and transferred to a cryo-specimen 626 holder (Gatan, Inc., Pleasanton, CA, USA) under liquid nitrogen before loading them into a JEOL 2200 electron microscope, with a field emission gun operating at 200 keV. Grids were maintained at near-liquid nitrogen temperature (−172°C to −180°C) during imaging. Nanoparticles were imaged at 25,000× indicated microscope magnification with a 4 k ×4 k slow-scan CCD camera (UltraScan 895, Gatan, Inc.) using a low-dose imaging procedure.
X-ray diffraction and differential scanning calorimetry measurements
Prior to X-ray diffraction and differential scanning calorimetric (DSC) studies, nanoparticle dispersions were frozen at −80°C. Samples were then lyophilized for 24 hours using a FreeZone 2.5 L benchtop freeze dry system (Labconco Corporation, Kansas City, MO, USA) at a temperature of −50°C and a vacuum of 0.055 mbar.
The crystallinity of the oxcarbazepine-loaded nanoparticles was examined at room temperature with a DIP2030 X-ray diffractometer (MAC Science Inc., Yokohama, Japan). X-ray diffraction patterns were obtained by wide-angle X-ray scattering (WAXS, 2θ =5°–50°, step size =0.04) using a MacScience DIP2030H-VLM dual 30 cm diameter imaging plate detector with an M06XHF22, 100 μm ultra-fine-focus high-brilliancy X-ray generator and Rigaku Blue focusing multilayer optics. Data were recorded at a detector distance of 100 mm using Cu radiation (λ =1.5418 Å) at an anode voltage of 40 kV and a current of 30 mA. The freeze-dried nanoparticle samples were mounted in MiTeGen MicroRT capillaries and rotated during exposure. A blank, empty capillary exposure was used for background subtraction. The generated data were processed through FIT2D software (provided by Dr Joseph Reibenspies, Texas A&M University, College Station, TX) to convert the images into plots of intensity versus diffraction angle (2θ).
Thermal analysis was performed to complement the data generated by X-ray diffraction, to confirm the presence of oxcarbazepine in an amorphous state in the nanoparticle matrices. DSC analyses were performed using a Q200 DSC (TA Instruments, New Castle, DE, USA). Samples containing 2–3 mg of lyophilized nanoparticles were accurately weighed and hermetically sealed in aluminum pans. DSC scans were first recorded at a heating rate of 10°C min−1 from 20°C to 200°C and then allowed to cool to 10°C to observe the exothermic phase. During thermal scans, the crucible was purged with nitrogen. The generated data were analyzed with Universal Analysis 2000 software (TA Instruments).
Transport experiments
The transport of nanoparticles across in vitro models of human placental trophoblast and the BBB was investigated using BeWo cells (clone b30) and hCMEC/D3 cells, respectively. The BeWo cells were obtained from Dr LE Knudsen, University of Copenhagen, Denmark. Cell culture was carried out according to previously described protocols.33,34 Briefly, cells were cultured in DMEM with 10% FBS supplemented with penicillin/streptomycin, L-glutamine, and nonessential amino acids and maintained at 37°C, 5% CO2, and 95% relative humidity. Cells were passed every 5–7 days; cell passage numbers 24–28 were used in this study. Before transport experiments, cells were seeded onto polyester Transwell® inserts (Corning Inc., Corning, NY, USA) (pore size, 3 μm; 1.12 cm2 growth area; apical volume, 0.5 mL; basolateral volume, 1.5 mL), and the cell seeding density was 100,000 cells cm−2. Cell culture medium was changed daily until the cell monolayer became confluent (after 5–7 days). Formation of a single monolayer with tight junctions was verified by measuring the transepithelial electrical resistance (TEER) values in growth media (DMEM) at room temperature using an EndOhm-12 apparatus (World Precision Instruments, Sarasota, FL, USA). The actual TEER values corresponding to the cell barrier function were calculated by subtracting the resistance of an empty Transwell® insert (blank without cells) from an insert with cells. Only the inserts with cell monolayers displaying TEER values of at least 30±2 Ω·cm2 were used in the nanoparticle transport studies,35 and values were as high as 71±2 Ω·cm2.
The in vitro BBB model, hCMEC/D3 cells, were obtained from Dr U Bickel, Texas Tech University Health Sciences Center, Amarillo, Texas, USA (with permission from Dr PO Couraud, Inserm, Paris, France). Cells were cultured in EBM-2 with 5% FBS supplemented with penicillin/streptomycin, hydrocortisone (1.4 μM), ascorbic acid (5.0 μg/mL), chemically defined lipid concentrate (1.0%), HEPES (10.0 mM), and bFGF (1.0 ng/mL) and maintained at 37°C, 5% CO2, and 95% relative humidity. Cells were passed every 3–5 days; cell passage numbers 25–30 were used in this study. Before transport experiments, cells were seeded onto the aforementioned polyester Transwell® inserts at a density of 50,000 cells cm−2, and the cell culture medium was changed every 3 days until the cell monolayer became confluent (after 5–7 days). The formation of cell monolayer and verification of tight junctions was evaluated by measuring transendothelial electrical resistance values, which were in the range of 65–75 Ω·cm2.
At time zero, nanoparticle dispersions (or free oxcarbazepine) were vortexed in transport medium and added to the apical chamber, and the transport experiment was carried out under cell culture conditions (37°C, 5% CO2, 95% humidity) with constant stirring. At the specified time intervals, 100 μL samples were withdrawan from the basolateral chamber and replaced with 100 μL of fresh transport medium. The transport of oxcarbazepine-loaded nanoparticles and free oxcarbazepine was investigated at a theoretical concentration of 50 μg mL−1 oxcarbazepine. The collected samples were mixed with an equal volume of acetonitrile:methanol (50:50) to dissolve the drug with subsequent centrifugation to precipitate the serum protein. The supernatant was then analyzed by HPLC to determine the amount of oxcarbazepine that had crossed the cell monolayer, as described in the “Instrumentation and chromatographic conditions” section. Experiments were carried out in quadruplicate in parallel with transport studies across blank Transwell® inserts with no cells (for the determination of apparent permeability values, as described by the equations below).
Coumarin-6 transport studies were carried out in the same manner described earlier, except that the sample volumes were 200 μL. The cumulative amount of coumarin-6 transported was determined by measuring the fluorescence of the samples in phenol red-free DMEM at λex =485 nm and λem=528 nm using an FLX800 microplate reader (BioTek Instruments, Inc., Winooski, VT, USA). Data acquisition was performed using Gen5™ software (BioTek Instruments, Inc.). The concentration of coumarin-6 in the basal chamber was then calculated from calibration standards prepared by serial dilution of the coumarin-6-loaded nanoparticles.
At each time point t = tn, the mass transported (ΔQn) was determined and corrected for the mass removed during the previous sampling periods using the following equation:
|
|
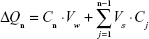
where Cn is the concentration of the sample measured at time tn, Vw is the volume of the well sampled (1.5 mL from the basolateral chamber), Vs is the sampling volume (100 μL for the oxcarbazepine transport studies and 200 μL for the transport of coumarin-6-loaded nanoparticles), and the term 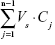 represents the correction for the cumulative mass removed by sampling during all sampling periods (from t = t1 until t = tn−1).
represents the correction for the cumulative mass removed by sampling during all sampling periods (from t = t1 until t = tn−1).
Permeability values (P) were calculated using the following equation:
|
|
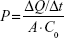
where ΔQ/Δt is the flux across the cell monolayer (mass s−1), A is the monolayer surface area (cm2), and C0 is the initial concentration of the oxcarbazepine on the apical chamber (in mass cm−3). The apparent permeability (Pe) of oxcarbazepine across the cell monolayer alone was calculated from the permeability across blank Transwell® filters membranes without cells (Pm), and the permeability across the Transwell® inserts containing cells (Pt) with the following equation:
|
|
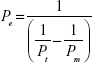
Statistical analysis
Data collected in this study were analyzed by one-way analysis of variance (ANOVA). Results were deemed significant if their corresponding P-values were less than 0.05.
Results and discussion
Synthesis of nanoparticles, particle size, size distribution, zeta potential, and encapsulation efficiency
In the present work, novel oxcarbazepine-loaded nanoparticles were prepared by a modified solvent displacement method to investigate the possibility of these nanoformulations crossing an in vitro model of the BBB (hCMEC/D3 cells) and an in vitro model of human placental trophoblast (BeWo cells) for potential future use for epileptic therapy during pregnancy.
Formulation development of oxcarbazepine-loaded nanoparticles included variations in polymer type, polymer concentration, and the use of a surfactant in the formulation to improve the nanoparticle stability. We selected these oxcarbazepine-loaded nanoparticle formulations based on results of a series of experiments in which we changed the polymer type using three different polymers, namely carboxylate end group 50:50 poly(lactic-co-glycolic acid) (PLGA), ester terminated 85:15 poly(lactic-co-glycolic acid), and poly(lactic acid) (PLA). The sizes of nanoparticles prepared from the last two polymers (at 20 mg/mL in acetone) were near 200 nm (data not shown), and when the nanoparticles were prepared at concentrations below 20 mg/mL in acetone using these two polymers, oxcarbazepine encapsulation efficiency diminished to below 60%. Since the objective was to develop nanoparticles with sizes near 150 nm, PLGA was pursued as the nanomaterial of choice.
To ensure monodisperse nanoformulations, it was desired that the nanoparticles have PDI values below 0.3. The mean size and PDI of the nanoparticles suspensions were measured by photon correlation spectroscopy after suitable dilution of bulk suspensions in deionized water. As shown in Table 2, nanoparticles had average diameters between 139 and 169 nm and a negative surface with zeta potential (ζ) values between −43 and −51 mV for PLGA nanoparticles and −34 and −38 mV for the Resomer® RGPd5055 nanoparticles. The presence of the PEG chains in Resomer® RGPd5055 nanoparticles reduced the absolute value of the negative charge. However, the decrease in zeta potential absolute value did not compromise the stability of these nanoparticles when they were stored in the refrigerator (5°C–8°C) for 30 days (data not shown). The encapsulation efficiency (EE%) values of the oxcarbazepine-loaded nanoparticles ranged from 69% to 72%, the highest EE% achieved at the designated polymer concentrations. From these results, it can be inferred that polymer type and the presence/absence of surfactant did not have a significant influence on encapsulation efficiency.
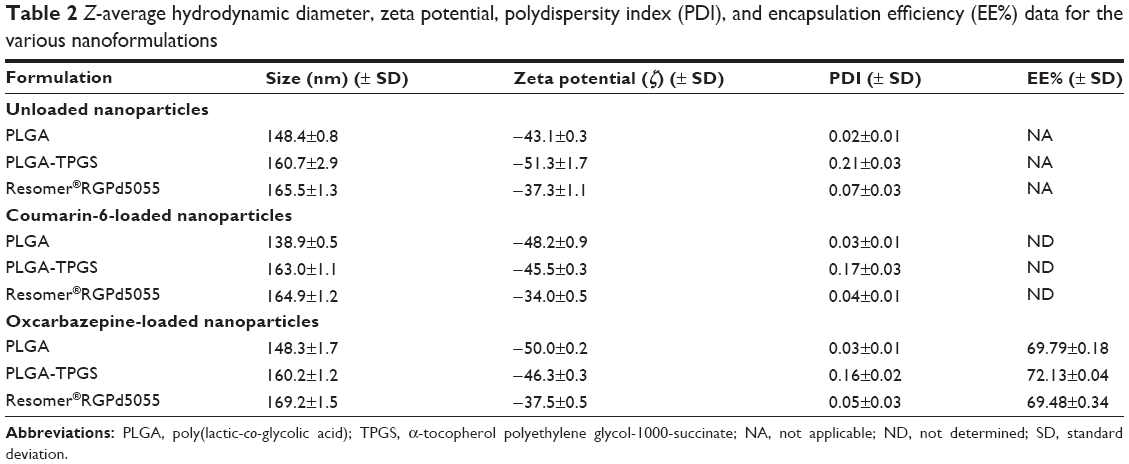 | Table 2 Z-average hydrodynamic diameter, zeta potential, polydispersity index (PDI), and encapsulation efficiency (EE%) data for the various nanoformulations |
X-ray diffraction and differential scanning calorimetry measurements
Nanoparticles may have limits in their ability to encapsulate certain drugs, and sometimes during nanoparticle preparation, the drug may crystallize out, depending on several factors such as aqueous solubility of the drug molecule, theoretical drug loading, polymer hydrophilicity and concentration, volume of organic solvent used, and the affinity of the drug molecule to the nanoparticle matrix. Since the particle size and monodispersity are critical quality attributes, nanoparticles were analyzed by wide-angle X-ray diffraction to study the physical state of oxcarbazepine upon nanoencapsulation.
X-ray diffraction is an established technique that has been used to characterize solid dispersions and evaluate the degree of crystallinity and polymorphism of drugs. Figure 3A and B present X-ray images and the diffraction patterns of freeze-dried oxcarbazepine-loaded nanoparticles. As shown, oxcarbazepine-loaded nanoparticles displayed smooth multiple colored rings (Figure 3A, a). The physical mixture of oxcarbazepine and unloaded nanoparticles resulted in a rougher image, because of the presence of oxcarbazepine crystals (Figure 3A, b). The image of oxcarbazepine-loaded nanoparticles was smooth and characterized by the absence of oxcarbazepine crystals, which confirmed the amorphous state of oxcarbazepine in the polymer matrices (Figure 3A, a). The X-ray diffraction pattern for oxcarbazepine powder showed major peaks at 2θ =5.3, 9.9, 10.2, 12.6, 15.7, 16.1, 18.4, 21.7, 23.9, and 26.2 (Figure 3B). The physical mixture of oxcarbazepine and unloaded nanoparticles (PLGA and TPGS), on the other hand, revealed the distinct oxcarbazepine peaks with low intensity (2θ =10.2, 15.7, 16.1, 18.4, and 23.9), indicating the existence of oxcarbazepine in crystalline form (Figure 3B). In contrast, freeze-dried dispersions of unloaded as well as oxcarbazepine-loaded nanoparticles displayed only the PLGA-derived broad peak at 2θ =10°–25°. The absence of peaks distinctive to the oxcarbazepine diffraction pattern suggested its amorphous state and its presence as a molecular dispersion in the PLGA matrices. Similar results were obtained when Resomer® RGPd5055 was used for nanoparticle synthesis (Figure 3B).
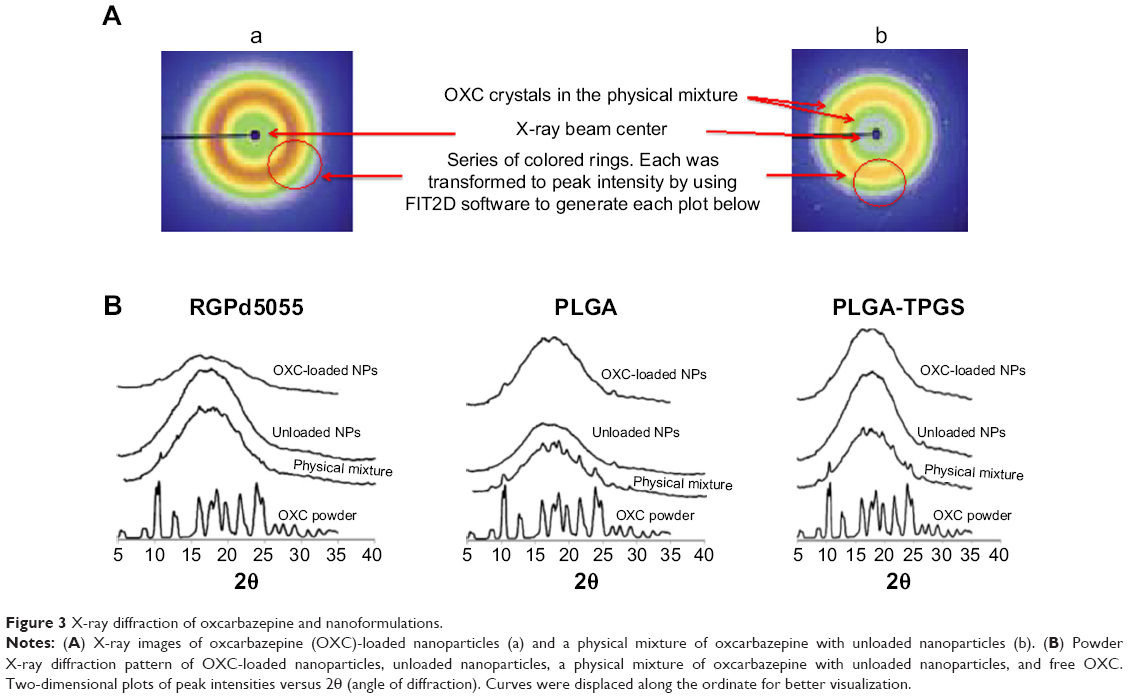 | Figure 3 X-ray diffraction of oxcarbazepine and nanoformulations. |
DSC analysis was carried out to confirm the amorphous state of encapsulated oxcarbazepine in the polymeric matrices of the nanoparticles (see Figure 4). The copolymers PLGA and Resomer RGPd5055 are amorphous and exhibit glass transition temperatures around 45°C–52°C. Oxcarbazepine (as free drug) has a melting point between 216°C and 230°C. Analysis was carried out to compare the thermal characteristics of oxcarbazepine-loaded nanoparticles to those of a binary mixture of unloaded nanoparticles with the free drug. DSC analysis of these physical mixtures displayed broad melting peaks at 216°C–220°C, which correspond to the melting range of free oxcarbazepine (see Figure 4, insets at right). No melting endotherm was observed in the DSC curves of the oxcarbazepine-loaded nanoparticles, however, indicating that the nanoencapsulated drug was present in a noncrystalline state.
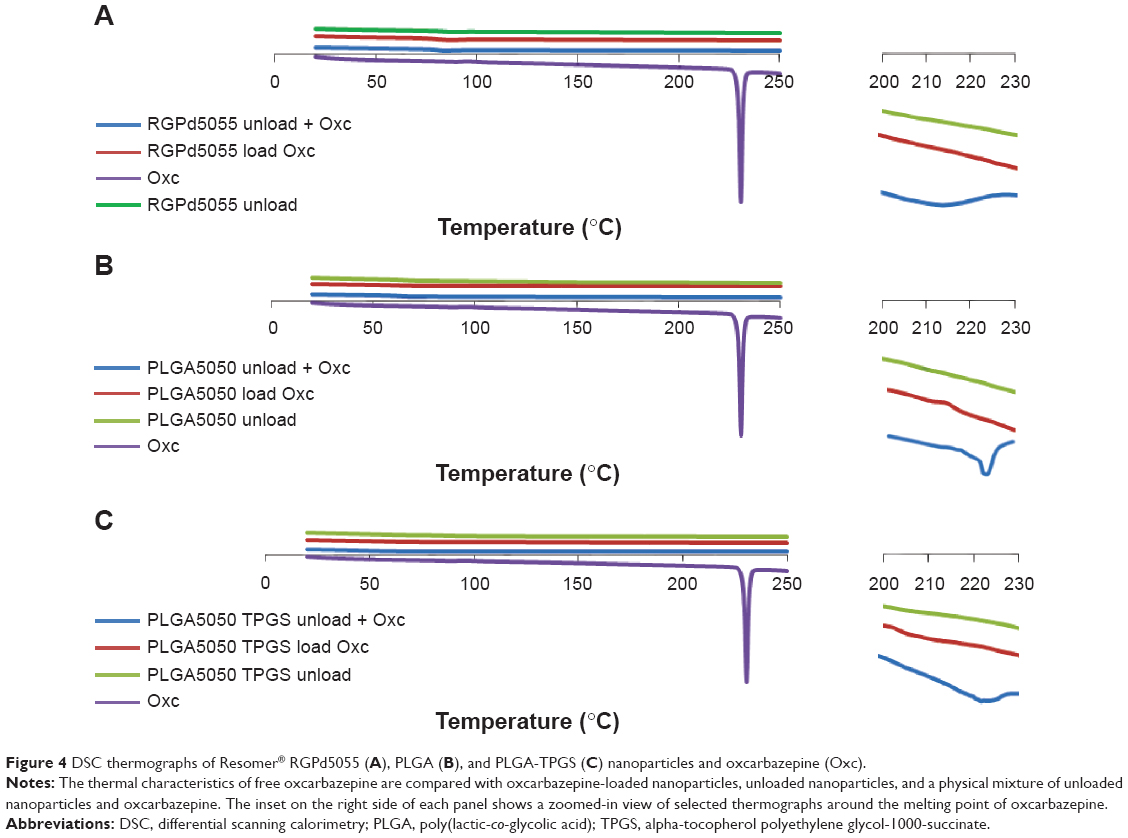 | Figure 4 DSC thermographs of Resomer® RGPd5055 (A), PLGA (B), and PLGA-TPGS (C) nanoparticles and oxcarbazepine (Oxc). |
Cryo-electron microscopy (cryo-EM)
Cryo-electron microscopy allows the preservation of the native state of the samples by extremely rapid freezing of suspensions by so-called water vitrification.31 In this process, the fast cooling rates (>105–106°C s−1) allow water to harden like glass, leaving the specimens embedded in a solid matrix to maintain the native conformations of biological macromolecules.32 Cryo-EM images were collected to investigate the shape of oxcarbazepine-loaded nanoparticles. As shown in the representative image (Figure 5), the nanoparticles are spherical. No oxcarbazepine crystals were visible, which confirmed the amorphous states depicted by the X-ray diffraction and the DSC results. The size of the nanoparticles was measured using DigitalMicrograph software (Gatan, Inc., Pleasanton, CA, USA), and these values agreed with the particle size data obtained by photon correlation spectroscopy as reported in Table 2.
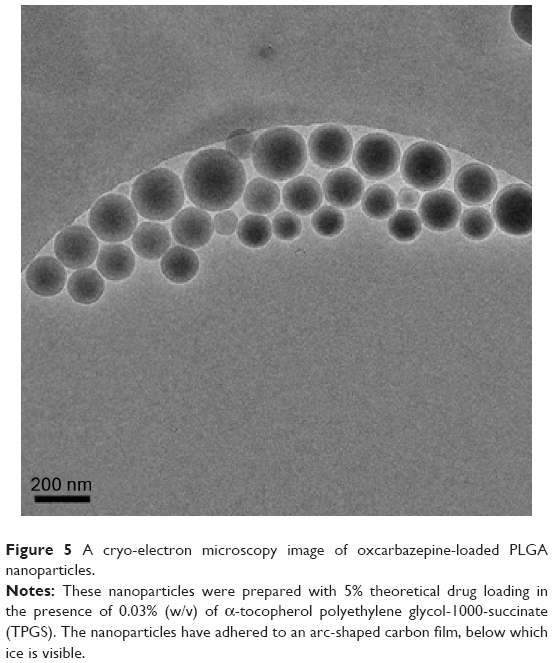 | Figure 5 A cryo-electron microscopy image of oxcarbazepine-loaded PLGA nanoparticles. |
In vitro transport experiments
Transport of the nanoformulations and free oxcarbazepine across in vitro models of human placenta (BeWo cells) and the BBB (hCMEC/D3 cells) was investigated at 37°C under cell culture conditions (5% CO2, 95% relative humidity). The results presented in Table 3 indicate that the apparent permeability values (Pe) of the free drug and nanoencapsulated drug were similar for each nanoformulation, presumably because of rapid release of the free drug molecules that are likely near the surface of the nanoparticles rather than completely encapsulated within the nanoparticle matrices. These results are in agreement with the drug release kinetics of these nanoformulations. Figure 6 shows that 60%–68% of the drug was released from the nanoparticles within two hours.
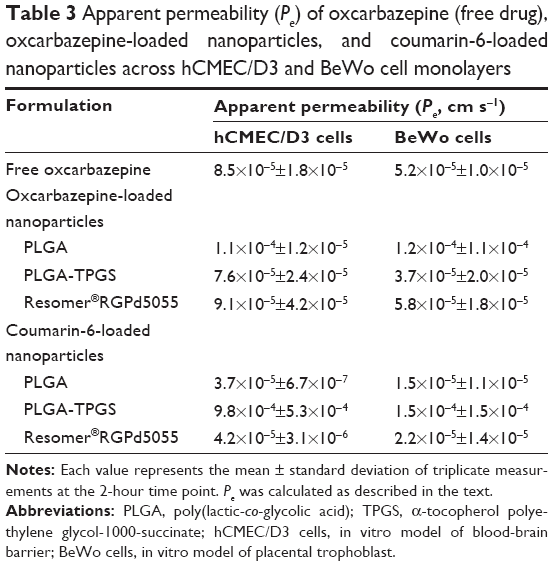 | Table 3 Apparent permeability (Pe) of oxcarbazepine (free drug), oxcarbazepine-loaded nanoparticles, and coumarin-6-loaded nanoparticles across hCMEC/D3 and BeWo cell monolayers |
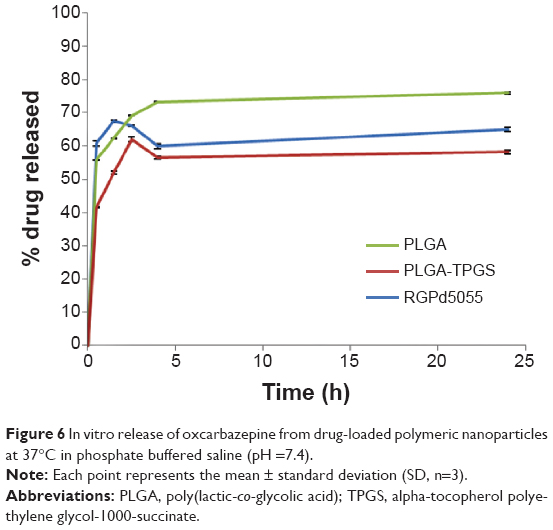 | Figure 6 In vitro release of oxcarbazepine from drug-loaded polymeric nanoparticles at 37°C in phosphate buffered saline (pH =7.4). |
Transport studies with the coumarin-6-labeled nanoparticles demonstrated that TPGS had a significant influence on nanoparticle transport, as the surfactant enhanced nanoparticle permeability across the hCMEC/D3 cells. We had previously observed increased permeability of PLGA nanoparticles coated with the surfactant sodium taurocholate across BeWo cells.36
Conclusion
With the long-term goal of developing a nanoparticle-based drug targeting strategy to improve epileptic therapy in pregnant women, we first investigated the encapsulation of oxcarbazepine into polymeric nanoparticles composed of the biocompatible and FDA-approved polymer PLGA.37 Monodisperse nanoformulations were prepared with average particle sizes between 139 and 169 nm and polydispersity indices below 0.3. Fair encapsulation efficiency values around 70% were obtained, and both X-ray diffraction and DSC measurements demonstrated the amorphous form of the drug in the nanoformulations. Owing to the rapid drug release kinetics, little difference was observed upon comparing the transport of free oxcarbazepine with that of the nanoencapsulated drug across in vitro models of the BBB (hCMEC/D3 cells) and human placental trophoblast (BeWo cells). Nevertheless, these studies demonstrate the utility of these two models for determining the permeability of drug-loaded nanoparticles. In order to improve epileptic therapy during pregnancy, additional advancements are necessary. These include steps to reduce burst drug release, such as encapsulation in another type of nanomaterial, controlling the encapsulation process, or the use of other AEDs with more favorable drug loading and drug release characteristics. Upon optimization of the nanoencapsulation approach, drug delivery strategies employing targeting ligands or enzyme-prodrug approaches can be pursued.
Acknowledgments
The authors would like to thank Andrea Latrofa, Associate Professor at the University of Bari Aldo Moro, for his scientific contributions and professional discussions. Mark White and Michael Sherman from the Department of Biochemistry and Molecular Biology and the Sealy Center for Structural Biology and Molecular Biophysics at the University of Texas Medical Branch are thanked for their assistance with the X-ray diffraction and cryo-TEM experiments, respectively. This research was supported in part by the William and Mary McGanity Research Fund in Obstetrics and Gynecology and by a research career development award (K12HD052023: Building Interdisciplinary Research Careers in Women’s Health Program, BIRCWH) from the National Institute of Allergy and Infectious Diseases (NIAID), the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD), and the Office of the Director (OD), National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIAID, NICHD, OD, or the National Institutes of Health.
Disclosure
The authors report no conflicts of interest.
References
Tomson T, Danielsson BR, Winbladh B. Epilepsi och graviditet. En balansgång mellan risker för mor och barn. [Epilepsy and pregnancy. Balancing between risks to the mother and child]. Läkartidningen 1997;94:2827–2835. Swedish. | ||
Tomson T, Battino D. Teratogenic effects of antiepileptic drugs. Lancet Neurol. 2012;11:803–813. | ||
Reimers A, Brodtkorb E. Second-generation antiepileptic drugs and pregnancy: a guide for clinicians. Expert Rev Neurother. 2012;12:707–717. | ||
Rolnitsky A, Merlob P, Klinger G. In utero oxcarbazepine and a withdrawal syndrome, anomalies, and hyponatremia. Pediatr Neurol. 2013;48:466–468. | ||
Wlodarczyk BJ, Palacios AM, George TM, Finnell RH. Antiepileptic drugs and pregnancy outcomes. Am J Med Genet A. 2012;158A:2071–2090. | ||
Molgaard-Nielsen D, Hviid A. Newer-generation antiepileptic drugs and the risk of major birth defects. JAMA. 2011;305:1996–2002. | ||
Battino D, Tomson T. Management of epilepsy during pregnancy. Drugs. 2007;67:2727–2746. | ||
Veiby G, Daltveit AK, Engelsen BA, Gilhus NE. Fetal growth restriction and birth defects with newer and older antiepileptic drugs during pregnancy. J Neurol. 2014;261:579–588. | ||
Myllynen P, Pienimaki P, Jouppila P, Vahakangas K. Transplacental passage of oxcarbazepine and its metabolites in vivo. Epilepsia. 2001;42:1482–1485. | ||
Friis ML, Kristensen O, Boas J, et al. Therapeutic experiences with 947 epileptic out-patients in oxcarbazepine treatment. Acta Neurol Scand. 1993;87:224–227. | ||
Brightman MW, Reese TS. Junctions between intimately apposed cell membranes in the vertebrate brain. J Cell Biol. 1969;40:648–677. | ||
Vorbrodt AW, Dobrogowska DH. Molecular anatomy of intercellular junctions in brain endothelial and epithelial barriers: electron microscopist’s view. Brain Res Brain Res Rev. 2003;42:221–242. | ||
Sastry BV. Techniques to study human placental transport. Adv Drug Deliv Rev. 1999;38:17–39. | ||
Dilworth MR, Sibley CP. Review: transport across the placenta of mice and women. Placenta. 2013;34(Suppl):S34–S39. | ||
Enders AC, Blankenship TN. Comparative placental structure. Adv Drug Deliv Rev. 1999;38:3–15. | ||
Audus KL. Controlling drug delivery across the placenta. Eur J Pharm Sci. 1999;8:161–165. | ||
Denora N, Laquintana V, Lopalco A, et al. In vitro targeting and imaging the translocator protein TSPO 18-kDa through G(4)-PAMAM-FITC labeled dendrimer. J Control Release. 2013;172:1111–1125. | ||
Denora N, Trapani A, Laquintana V, Lopedota A, Trapani G. Recent advances in medicinal chemistry and pharmaceutical technology – strategies for drug delivery to the brain. Curr Top Med Chem. 2009;9:182–196. | ||
Liu Y, Guo Y, An S, et al. Correction: targeting caspase-3 as dual therapeutic benefits by RNAi facilitating brain-targeted nanoparticles in a rat model of Parkinson’s disease. PLoS One. 2013;8:e62905. | ||
Li J, Feng L, Fan L, et al. Targeting the brain with PEG-PLGA nanoparticles modified with phage-displayed peptides. Biomaterials. 2011;32:4943–4950. | ||
Bennewitz MF, Saltzman WM. Nanotechnology for delivery of drugs to the brain for epilepsy. Neurotherapeutics. 2009;6:323–336. | ||
Chen Z, Penet MF, Nimmagadda S, et al. PSMA-targeted theranostic nanoplex for prostate cancer therapy. ACS Nano. 2012;6:7752–7762. | ||
Gwenin VV, Gwenin CD, Kalaji M. Colloidal gold modified with a genetically engineered nitroreductase: toward a novel enzyme delivery system for cancer prodrug therapy. Langmuir. 2011;27:14300–14307. | ||
Saunders M. Transplacental transport of nanomaterials. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2009;1:671–684. | ||
Mawer G, Briggs M, Baker GA, et al. Pregnancy with epilepsy: obstetric and neonatal outcome of a controlled study. Seizure. 2010;19:112–119. | ||
Beck-Broichsitter M, Rytting E, Lebhardt T, Wang X, Kissel T. Preparation of nanoparticles by solvent displacement for drug delivery: a shift in the “ouzo region” upon drug loading. Eur J Pharm Sci. 2010;41:244–253. | ||
Poulsen MS, Rytting E, Mose T, Knudsen LE. Modeling placental transport: correlation of in vitro BeWo cell permeability and ex vivo human placental perfusion. Toxicol In Vitro. 2009;23:1380–1386. | ||
Dauchy S, Miller F, Couraud PO, et al. Expression and transcriptional regulation of ABC transporters and cytochromes P450 in hCMEC/D3 human cerebral microvascular endothelial cells. Biochem Pharmacol. 2009;77:897–909. | ||
Ragnaill MN, Brown M, Ye D, et al. Internal benchmarking of a human blood–brain barrier cell model for screening of nanoparticle uptake and transcytosis. Eur J Pharm Biopharm. 2011;77:360–367. | ||
Jores K, Mehnert W, Drechsler M, Bunjes H, Johann C, Mader K. Investigations on the structure of solid lipid nanoparticles (SLN) and oil-loaded solid lipid nanoparticles by photon correlation spectroscopy, field-flow fractionation and transmission electron microscopy. J Control Release. 2004;95:217–227. | ||
Sherman MB, Weaver SC. Structure of the recombinant alphavirus Western equine encephalitis virus revealed by cryoelectron microscopy. J Virol. 2010;84:9775–9782. | ||
Lepault J, Booy FP, Dubochet J. Electron microscopy of frozen biological suspensions. J Microsc. 1983;129:89–102. | ||
Bode CJ, Jin H, Rytting E, Silverstein PS, Young AM, Audus KL. In vitro models for studying trophoblast transcellular transport. Methods Mol Med. 2006;122:225–239. | ||
Cartwright L, Poulsen MS, Nielsen HM, et al. In vitro placental model optimization for nanoparticle transport studies. Int J Nanomedicine. 2012;7:497–510. | ||
Morck TJ, Sorda G, Bechi N, et al. Placental transport and in vitro effects of Bisphenol A. Reprod Toxicol. 2010;30:131–137. | ||
Ali H, Kalashnikova I, White MA, Sherman M, Rytting E. Preparation, characterization, and transport of dexamethasone-loaded polymeric nanoparticles across a human placental in vitro model. Int J Pharm. 2013;454:149–157. | ||
Lu JM, Wang X, Marin-Muller C, et al. Current advances in research and clinical applications of PLGA-based nanotechnology. Expert Rev Mol Diagn. 2009;9:325–341. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.