")
Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 8
Once-daily dose regimen of ribavirin is interchangeable with a twice-daily dose regimen: randomized open clinical trial
Authors Balk J, Haenen G, Koc, Peters R, Bast A, van der Vijgh W, Koek G
Received 12 February 2015
Accepted for publication 8 April 2015
Published 12 August 2015 Volume 2015:8 Pages 137—144
DOI https://doi.org/10.2147/PGPM.S82782
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Martin H Bluth
Jiska M Balk,1 Guido RMM Haenen,1 Özgür M Koc,2 Ron Peters,3 Aalt Bast,1 Wim JF van der Vijgh,1 Ger H Koek,4
1Department of Toxicology, NUTRIM School for Nutrition, Toxicology and Metabolism, Maastricht University Medical Centre, 2Faculty of Health, Medicine and Life Sciences, Maastricht University, Maastricht, 3DSM Resolve, Geleen, 4Department of Internal Medicine, Division of Gastroenterology and Hepatology, Maastricht University Medical Centre, Maastricht, the Netherlands
Background: The combination of ribavirin (RBV) and pegylated interferon (PEG-IFN) is effective in the treatment of chronic hepatitis C infection. Reducing the frequency of RBV intake from twice to once a day will improve compliance and opens up the opportunity to combine RBV with new and more specific direct-acting agents in one pill. Therefore, the purpose of this study was to evaluate the pharmacokinetic profile of RBV in a once-daily to twice-daily regimen. The secondary aim was to determine tolerability as well as the severity and differences in side effects of both treatment regimens.
Methods: In this randomized open-label crossover study, twelve patients with chronic type 1 hepatitis C infection and weighing more than 75 kg were treated with 180 µg of PEG-IFN weekly and 1,200 mg RBV daily for 24 weeks. The patients received RBV dosed as 1,200 mg once-daily for 12 weeks followed by RBV dosed as 600 mg twice-daily for 12 weeks, or vice versa. In addition to the pharmacokinetic profile, the hematological profile and side effects were recorded. The RBV concentrations in plasma were determined using liquid chromatography-tandem mass spectrometry.
Results: Eight of twelve patients completed the study. Neither the time taken for RBV to reach peak plasma concentration nor the AUC0-last (adjusted for difference in dose) was significantly different between the two groups (P>0.05). Furthermore, the once-daily regimen did not give more side effects than the twice-daily regimen (P>0.05). No significant differences in the hematological profile were observed (P>0.05).
Conclusion: The standard twice-daily RBV regimen is interchangeable with the once-daily regimen. The once-daily regimen will improve compliance and opens the opportunity to combine RBV with other drugs dosed once a day, in a single pill.
Keywords: hepatitis C, dose regimen, ribavirin, pegylated interferon
Introduction
Approximately 3% of the world population, 170 million people, are chronically infected with hepatitis C virus (HCV).1 During the standard treatment with pegylated interferon (PEG-IFN) and in the majority of interferon-free regimens, ribavirin (RBV) is given as part of the therapy.2–5 RBV is administered orally, twice a day, for a period ranging from 12, to 24, to 48 weeks depending on virological factors and patient characteristics.2–4
The dosing regimen is crucial for adherence to successful therapy. Clinical studies indicate that patients who received over 80% of the prescribed dose of interferon and over 80% of the dose of RBV during at least 80% of the treatment period were more likely to obtain a sustained virological response than patients with suboptimal compliance.6,7 In chronic medical conditions, reduction of dosing frequency increases adherence to therapy.8,9 The introduction of PEG-IFN increased compliance to interferon by reducing the required number of injections from three to one a week.10 The relatively low adherence to RBV is partly based on the requirement to take RBV twice a day and will improve by reducing the dosage regimen to once a day.11–13
The sustained virological response for HCV genotype 1 has been improved with the introduction of protease inhibitors, boceprevir and telaprevir.14 New and more specific direct-acting agents such as sofosbuvir, daclatasvir, and simeprevir, will soon be commercially available for HCV treatment regimens in which RBV still plays a therapeutic role.2,14 Many of the new compounds are given once a day. Subsequently, the treatment would become much more convenient by combining all the drugs in one tablet. This is an additional reason to reduce the dosing frequency of RBV from twice to once a day. However, the reduction in frequency should not lead to toxic peak levels or inactivity of RBV. The pharmacokinetics of both dose regimens should be interchangeable, which prompted us to compare the pharmacokinetic profile of RBV in a once-daily regimen with that in a twice-daily regimen in a crossover pilot study.
Further, treatment of HCV with PEG-IFN and RBV is known to have many side effects, including severe anemia.15,16 Therefore, the secondary aim was to determine tolerability as well as the severity and differences in side effects of both treatment regimens.
Patients and methods
Study population
Twelve chronic HCV type 1 patients weighing more than 75 kg were enrolled in the study. The exclusion criteria were similar to those of the standard treatment protocol,17 and patients with human immunodeficiency virus or hepatitis B virus coinfection were also excluded. During 24 weeks, the patients were treated with 180 μg PEG-IFN weekly and 1,200 mg RBV daily. Four patients did not complete the study. Two patients were excluded because the RBV dose was reduced to conform to standard treatment procedure; one because of a fatal car accident; and one because a 2-log10 viral decrease was not achieved at week 12, which is required to continue RBV therapy according to the standard treatment procedure (Figure 1). None of these events were directly related to the study.
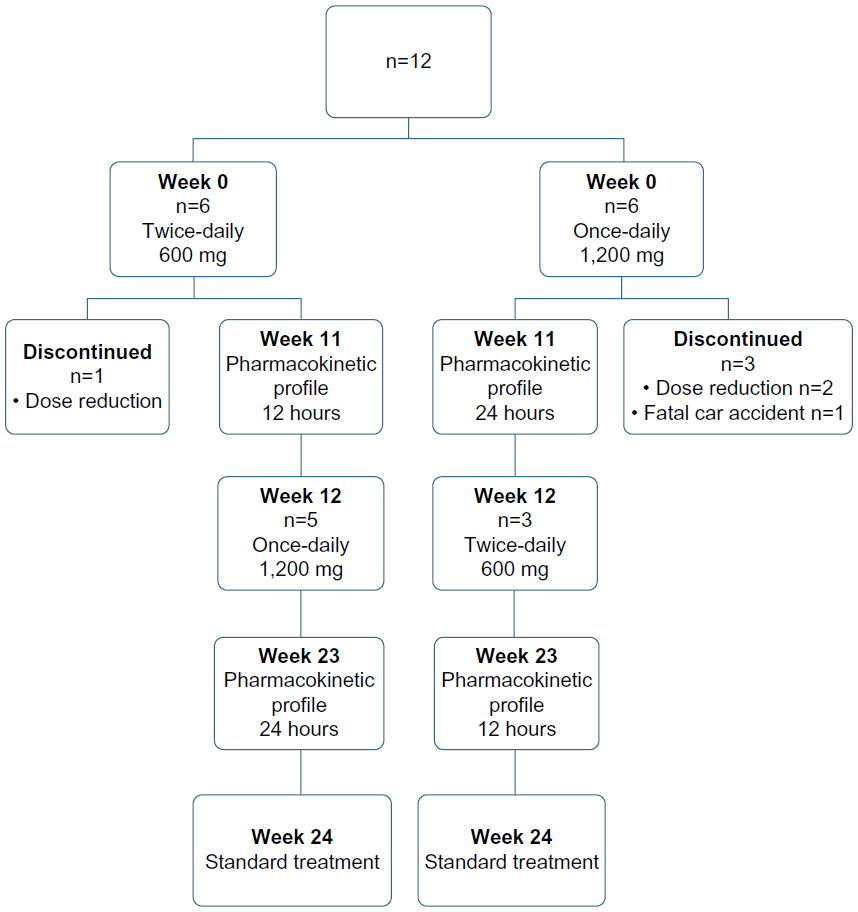 | Figure 1 Flow diagram showing the design of the RIBADOS study, with the number of patients included and reasons for discontinuation of intervention. |
The medical ethics committee of the University Hospital Maastricht approved the study and the patients gave written informed consent. The study, named RIBADOS, is registered at clinicaltrials.gov with the identifier NTC00484328.
Study design
This was a randomized, open-label, crossover study. During 24 weeks, the patients were treated with 180 μg PEG-IFN (Pegasys®) weekly and 1,200 mg RBV (Copegus®, 200 mg) daily. Subsequently, two dosing regimens of RBV were analyzed for their pharmacokinetic parameters, ie, 1,200 mg RBV once-daily and 600 mg RBV twice-daily (standard regimen). RBV was administered as 200 mg controlled-release tablets. The patients were randomly assigned, alternating between the two study groups. In a parallel, crossover design, the patients received one dose regimen for the first 12 weeks. In weeks 13–24, they received the other dose regimen (Figure 1). The physician confirmed compliance with therapy during the standard outpatient visits at weeks 1, 2, and 4, and every 4 weeks thereafter. In weeks 11 and 23, a time profile of RBV plasma concentration was determined during one dosing interval (12 or 24 hours). Blood samples were taken in heparinized tubes before intake of the RBV dose (t=0 minutes), at 30 minutes, 60 minutes, 90 minutes, 120 minutes, 150 minutes, 3 hours, 4 hours, 8 hours, and 12 hours (24 hours on the once-daily dosing regimen) after intake. The blood plasma was stored at −80°C until analysis.
The secondary objective of the study was the tolerability of the once-daily dose regimen. During the 24 weeks of treatment, the hematological profile was determined according to standard procedures and side effects were registered according to good clinical practice. If side effects reached grade 3 of the World Health Organization recommendations for grading of acute and subacute toxicity, the patients were withdrawn from the study and dose reduction of medication according to good clinical practice was performed.
Bioanalysis
RBV (1-β-D-Ribofuranosyl-1,2,4-triazole-3-carboxamide), necessary for the control experiments, and the internal standard (IS) 5-methylcytidine were obtained from Sigma (Schnelldorf, Germany). The other chemicals were of analytical grade.
The IS was added to the plasma sample before extraction; 100 μL of IS solution (6.72 μM in 80:20 acetonitrile to water) was added to 100 μL of plasma. RBV and the IS were extracted by adding 300 μL of acetonitrile, followed by vortexing and centrifugation. The supernatant was injected into a liquid chromatography-tandem mass spectrometer.
The chromatography-mass spectrometry experiments were performed on an Agilent 1290 Infinity high-performance liquid chromatography (Agilent, Waldbronn, Germany), coupled with an Agilent 6460 triple quad mass spectrometer, equipped with an electrospray ionization (ESI) Jet Stream. The mass spectrometer was used in MRM mode: RBV was detected using precursor ion m/z 245.1 (unit resolution) and product ion m/z 113.1 (wide resolution) at a fragmentation voltage of 80 V and a collision energy of 5 V; the IS was detected using precursor ion m/z 258.2 (unit resolution) and product ion m/z 126.1 (wide resolution) at a fragmentation voltage of 80 V and a collision energy of 10 V. The dwell time used was 50 milliseconds. The ESI was used in positive mode with the following settings: peak width 0.07 minute, EMV +400 V, 325°C gas temperature, 8 L N2/min, nebulizer pressure 35 psig, sheath gas temperature, 6 L N2/min sheath gas at 300°C, capillary voltage 3.5 kV cap, and 1 kV nozzle voltage.
The liquid chromatography analysis was performed with a 250×4.6 mm Prevail C18 column (5 μm) at 30°C (Alltech, Deerfield, IL, USA). Mobile phase A consisted of 10 mM ammonium acetate at pH 5 (Merck, Whitehouse Station, TX, USA) in ultrapure water and mobile phase B was methanol (Merck). The gradient was started at t=0 minutes with 1% (v/v) B, stayed there for 1 minute, and changed in 5 minutes to 90% (v/v) B (t=6 minutes), and was maintained until 9 minutes. The total run time was 13 minutes. The flow rate was 1.2 mL/min and the injection volume was 10 μL. A typical example of a chromatogram and MS spectrum for RBV and the IS 5-methylcytidine are shown in Figure 2.
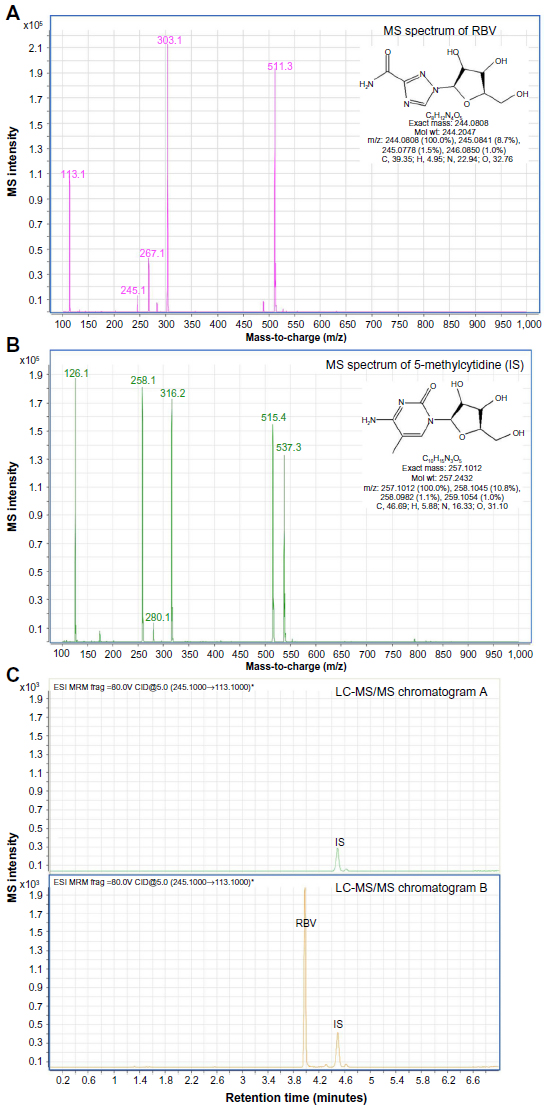 | Figure 2 LC-MS/MS RBV analysis in blood plasma. |
The RBV extraction efficiency of the plasma was 82.1%±3.6%. By using the IS, the corrected efficiency was 99.4%±2.0%. The calibration line of RBV was linear in the range of 0.04–24 μM (R2>0.99), using nine calibrators of RBV. The lowest limit of quantification of RBV was 0.04 μM.
Data analysis
Noncompartmental analysis was used to study the pharmacokinetics of RBV. The following blood sampling schedule was used: before intake of the RBV dose (t=0 minutes), at 30 minutes, 60 minutes, 90 minutes, 120 minutes, 150 minutes, 3 hours, 4 hours, 8 hours, and 12 hours (24 hours for the once-daily dosing regimen) after intake. The area under the plasma concentration-time curve (AUC) of one dosing interval was calculated using the trapezoidal rule and was adjusted for the difference in dose. From the concentration parameters, the maximum concentration (Cmax) and the minimum concentration (Cmin) of RBV in plasma were determined. A Wilcoxon signed-rank test with α=0.05 was used for statistical analysis of RBV concentrations. Due to the small sample size, effects of covariates were not included in the data analysis. Oral clearance was not measured. Liver enzymes, kidney function tests, and hematological profile were performed at every outpatient clinical visit and the results obtained with the two dosing regimens were compared.
Results
Eight of the twelve patients enrolled in the study completed the 24 weeks of treatment. Five patients started with the 600 mg twice-daily dose regimen, and three started with the 1,200 mg once-daily dose regimen (Figure 1).
The patient characteristics are presented in Table 1. All patients had HCV genotype 1 infection confirmed by polymerase chain reaction, with five patients having a high viral load (>800,000 IU/mL). Five patients had elevated alanine amino-transferase, aspartate amino-transferase, and gamma-glutamyl transpeptidase levels during the study. One patient also had elevated alkaline phosphatase levels. During the study, alanine amino-transferase, aspartate amino-transferase, and gamma-glutamyl transpeptidase levels returned to normal in one of these patients, but remained unchanged in the other patients. Three patients started with normal liver function parameters, which remained normal during the study. All patients had normal bilirubin, albumin, and serum creatinine levels during the study.
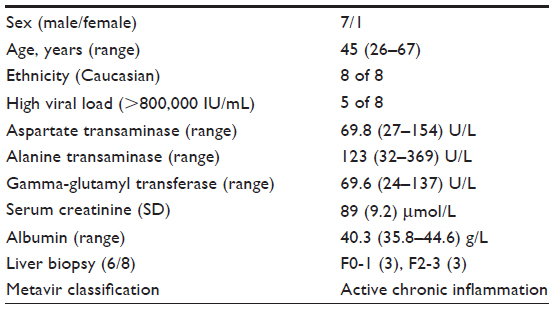 | Table 1 Patient characteristics and hematological parameters at the start of the treatment |
Steady-state RBV plasma concentrations were measured at the time the pharmacokinetic profile was determined. RBV plasma concentrations ranged from 7 to 15 μM (Figure 3). Neither the Cmax nor the Cmin for RBV was significantly different between the two dosing regimens (P>0.05, Figure 4A and B). The total drug exposure, described by the AUC, was not significantly different between the two dosing regimens (P=0.46, Figure 4C).
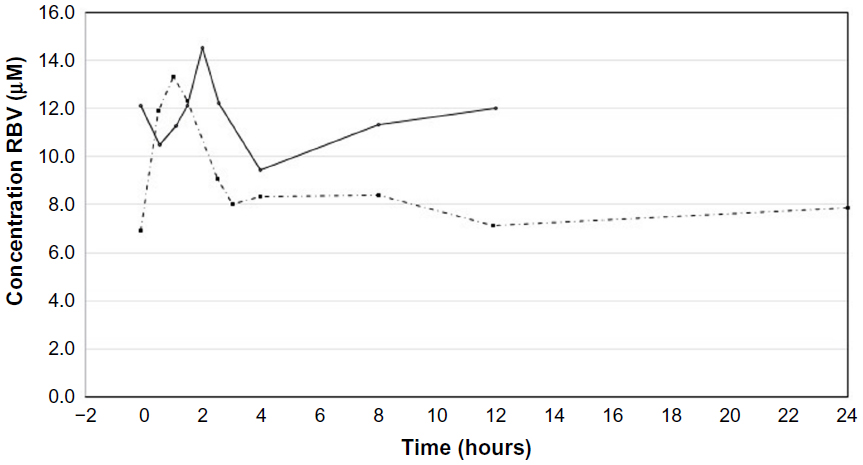 | Figure 3 Typical pharmacokinetic profile for RBV in one patient. The first blood sample was taken before intake of the prescribed dose of RBV. The intake is at time zero. In the twice-daily dose regimen, the RBV plasma concentrations were monitored for 12 hours (solid line). In the once-daily dose regimen, the RBV plasma concentrations were monitored for 24 hours (dashed line). |
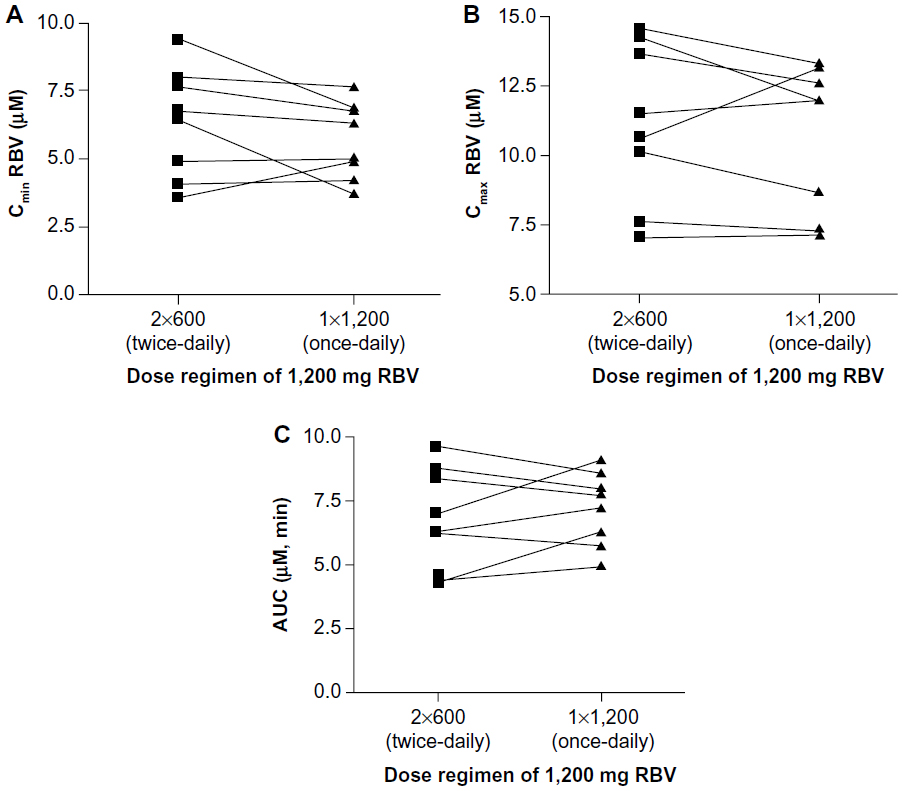 | Figure 4 Pharmacokinetic parameters for RBV. |
Hemoglobin levels in plasma decreased in all patients after initiating treatment. One patient had hemoglobin levels between 5.8 and 6.7 mmol/L (grade 1 toxicity) during the standard dose regimen. The other patients had hemoglobin levels above 6.8 mmol/L (grade 0 toxicity). The average decrease in hemoglobin in week 4 of treatment compared with baseline did not significantly differ between the two groups (P>0.05, Table 2).
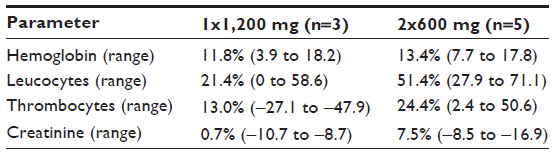 | Table 2 Change in hematological parameters after 4 weeks of treatment |
Five patients had reduced leucocyte levels between 3.0 and 3.9*109/L (grade 1 toxicity) and between 2.4 and 2.9*109/L (grade 2 toxicity), which was not related to the dose regimen. Although the amount of thrombocytes decreased, it remained above 100*109/L (grade 0 toxicity). No significant difference was observed in the number of leucocytes and thrombocytes between the two dose regimens (P>0.05).
Discussion
Adherence to dosing regimen is a concern in the treatment of HCV.18 The introduction of PEG-IFN improved the compliance with IFN.10 Until recently, the standard treatment was addition of RBV twice-daily for 12, 24, and 48 weeks of treatment. Moreover, with the introduction of new oral compounds like sofosbuvir, daclatasvir, and simeprevir, RBV is still part of the therapy.2–5 The disadvantage of RBV is the twice-daily dosing, raising the question if this drug could not be given in a once-daily dose, thereby simplifying treatment and improving adherence. The aim of the present study was to evaluate the interchangeability of a twice-daily dosing with a single dose.
In this crossover study, the once-daily and twice-daily dose regimens were compared. Neither the Cmax, the Cmin, nor the total drug exposure (AUC) for RBV was significantly different between the two dosing regimens. This suggests that the RBV dosing regimen could be simplified to once-daily.
The crossover design was chosen because of the relatively high variation in plasma RBV concentrations between patients.19 In the crossover design, each patient served as their own control and the possibility to compare the pharmacokinetics within each patient drastically increased the power of the study.
RBV has a bioavailability of approximately 60% and is easily taken up by cells via nucleoside transporters. The presence of nucleoside transporters in most cells results in a distribution volume of approximately 2 L and a plasma half-life of approximately 270 hours.20 Steady-state plasma concentrations are reached between 4 and 8 weeks.21 Steady-state plasma concentrations were not different in the choice of once-daily or twice-daily dose regimen. This similarity can be explained by the relative small difference in 12-hour RBV plasma concentration between both dosing regimens compared with the 4–8 weeks needed to reach steady state.
Once inside the cell, RBV is phosphorylated by adenosine kinase into its monophosphate, diphosphate, and triphosphate forms. With the use of phosphate-hydrolyzing enzymes, RBV can be transported back into the plasma for excretion via the kidneys.22,23 In all patients, kidney function was normal at the beginning of the study and did not alter during treatment.
The major side effect of RBV is anemia. In erythrocytes, RBV monophosphates, diphosphates, and triphosphates are formed, but phosphate-hydrolyzing enzymes are absent. Therefore, the RBV phosphates are unable to cross the cell membrane and accumulate in erythrocytes. This accumulation of RBV, resulting in RBV concentrations 60 times higher than the plasma concentration, is accompanied by oxidative membrane damage and subsequently premature removal of erythrocytes from the circulation.24
During standard treatment, anemia is most pronounced in the first weeks of treatment and is the major reason for RBV dose reduction.24–26 The single-dose regimen showed an equal decrease in hemoglobin levels after 4 weeks of treatment when compared with the standard dose regimen.
Additional to the effect of RBV on the amount of erythrocytes, PEG-IFN suppresses hematopoiesis, resulting in an overall decrease in the number of blood cells.27 The amounts of leucocytes and thrombocytes indeed decreased after start of treatment. However, this decrease after 4 weeks was not significantly different between the two dose regimens. Cmax, which has been associated with severity of side effects, also did not differ significantly between the dosing regimens. It should be noted that the impact of the different dosing regimens on side effects was the secondary aim of the study. Although no significant effect on side effects was observed, this should be examined in a larger cohort of patients.
Study limitations
One should realize that this study had a small sample size of eight patients, with only one female. In addition, all eight patients were Caucasian. Therefore, a validation study should confirm that, as previously reported by Wade et al,28 the observed pharmacokinetic results are not race-specific or sex-specific.
Future recommendations
This pharmacokinetic study opens up the route to further assess this change in dose regimen. We would therefore recommend larger clinical studies to assess the change in dose regimen of RBV on pharmacokinetics and tolerability as well as the severity and differences in side effects of both treatment regimens.
Conclusion
In conclusion, the pharmacokinetic parameters of RBV in the once-daily and twice-daily dose regimen indicate that the RBV twice-daily dosing regimen is interchangeable with once-daily dosing. Reduction of the dosing frequency is expected to increase adherence to therapy. For patients, it would be easier to comply with the 80/80/80 rule if RBV has to be taken once-daily. This pharmacokinetic study opens up the route for larger clinical studies to further assess this change in dose regimen of RBV and will probably lead to one combined oral drug therapy in the future treatment of HCV infection and other viral infections.
Disclosure
The authors report no conflicts of interest in this study.
References
Marcellin P. Hepatitis B and hepatitis C in 2009. Liver Int. 2009; 29 Suppl 1:1–8. | |
Alexopoulou A, Karayiannis P. Interferon-based combination treatment for chronic hepatitis C in the era of direct acting antivirals. Ann Gastroenterol. 2015;28:55–65. | |
Cholongitas E, Papatheodoridis GV. Sofosbuvir: a novel oral agent for chronic hepatitis C. Ann Gastroenterol. 2014;27:331–337. | |
Izquierdo L, Helle F, Francois C, Castelain S, Duverlie G, Brochot E. Simeprevir for the treatment of hepatitis C virus infection. Pharmgenomics Pers Med. 2014;7:241–249. | |
Hoofnagle JH, Seeff LB. Peginterferon and ribavirin for chronic hepatitis C. N Engl J Med. 2006;355:2444–2451. | |
Numata M, Kagawa T, Kojima S, et al. Differential impact of adherence to pegylated interferon and ribavirin in the treatment of genotype 1 high viral titer chronic hepatitis C. Hepat Res Treat. 2010;2010:702748. | |
McHutchison JG, Manns M, Patel K, et al. Adherence to combination therapy enhances sustained response in genotype-1-infected patients with chronic hepatitis C. Gastroenterology. 2002;123:1061–1069. | |
Ingersoll KS, Cohen J. The impact of medication regimen factors on adherence to chronic treatment: a review of literature. J Behav Med. 2008;31:213–224. | |
Laliberte F, Nelson WW, Lefebvre P, Schein JR, Rondeau-Leclaire J, Duh MS. Impact of daily dosing frequency on adherence to chronic medications among nonvalvular atrial fibrillation patients. Adv Ther. 2012;29:675–690. | |
Manns MP, McHutchison JG, Gordon SC, et al. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet. 2001;358:958–965. | |
Alam I, Stainbrook T, Cecil B, Kistler KD. Enhanced adherence to HCV therapy with higher dose ribavirin formulation: final analyses from the ADHERE registry. Aliment Pharmacol Ther. 2010;32:535–542. | |
Marcellin P, Chousterman M, Fontanges T, et al. Adherence to treatment and quality of life during hepatitis C therapy: a prospective, real-life, observational study. Liver Int. 2011;31:516–524. | |
Kripalani S, Yao X, Haynes RB. Interventions to enhance medication adherence in chronic medical conditions: a systematic review. Arch Intern Med. 2007;167:540–550. | |
Hayes CN, Chayama K. Emerging treatments for chronic hepatitis C. J Formos Med Assoc. 2015;114:204–215. | |
Hung CH, Lee CM, Lu SN, et al. Anemia associated with antiviral therapy in chronic hepatitis C: incidence, risk factors, and impact on treatment response. Liver Int. 2006;26:1079–1086. | |
Tappero G, Ballare M, Farina M, Negro F. Severe anemia following combined alpha-interferon/ribavirin therapy of chronic hepatitis C. J Hepatol. 1998;29:1033–1034. | |
European Association for Study of Liver. EASL clinical practice guidelines: management of hepatitis C virus infection. J Hepatol. 2014;60:392–420. | |
Evon DM, Esserman DA, Bonner JE, Rao T, Fried MW, Golin CE. Adherence to PEG/ribavirin treatment for chronic hepatitis C: prevalence, patterns, and predictors of missed doses and nonpersistence. J Viral Hepat. 2013;20:536–549. | |
Munoz-de-Rueda P, Ruiz-Extremera A, Candel JM, et al. Plasma ribavirin trough concentrations during treatment of chronic hepatitis C in genotype-1 patients. J Clin Gastroenterol. 2012;46:328–333. | |
Glue P. The clinical pharmacology of ribavirin. Semin Liver Dis. 1999;19 Suppl 1:17–24. | |
Furusyo N, Murata M, Ogawa E, et al. Ribavirin concentration in the later stages of 48 week pegylated interferon-alpha2b plus ribavirin therapy for chronic hepatitis C is useful for predicting virological response. J Antimicrob Chemother. 2011;66:1127–1139. | |
Bruchfeld A, Lindahl K, Schvarcz R, Stahle L. Dosage of ribavirin in patients with hepatitis C should be based on renal function: a population pharmacokinetic analysis. Ther Drug Monit. 2002;24:701–708. | |
Kamar N, Chatelut E, Manolis E, Lafont T, Izopet J, Rostaing L. Ribavirin pharmacokinetics in renal and liver transplant patients: evidence that it depends on renal function. Am J Kidney Dis. 2004;43:140–146. | |
De Franceschi L, Fattovich G, Turrini F, et al. Hemolytic anemia induced by ribavirin therapy in patients with chronic hepatitis C virus infection: role of membrane oxidative damage. Hepatology. 2000;31:997–1004. | |
Krishnan SM, Dixit NM. Ribavirin-induced anemia in hepatitis C virus patients undergoing combination therapy. PLoS Comput Biol. 2011;7:e1001072. | |
Morello J, Rodriguez-Novoa S, Jimenez-Nacher I, Soriano V. Usefulness of monitoring ribavirin plasma concentrations to improve treatment response in patients with chronic hepatitis C. J Antimicrob Chemother. 2008;62:1174–1180. | |
Peck-Radosavljevic M, Wichlas M, Homoncik-Kraml M, et al. Rapid suppression of hematopoiesis by standard or pegylated interferon-alpha. Gastroenterology. 2002;123:141–151. | |
Wade JR, Snoeck E, Duff F, Lamb M, Jorga K. Pharmacokinetics of ribavirin in patients with hepatitis C virus. Br J Clin Pharmacol. 2006;62:710–714. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.