")
Back to Journals » Lung Cancer: Targets and Therapy » Volume 6
Non-small-cell lung carcinoma: role of the Notch signaling pathway
Authors Barse L, Bocchetta M
Received 13 March 2015
Accepted for publication 30 April 2015
Published 26 June 2015 Volume 2015:6 Pages 43—53
DOI https://doi.org/10.2147/LCTT.S60329
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Pan-Chyr Yang
Levi Barse, Maurizio Bocchetta
Department of Pathology, Oncology Institute, Loyola University Chicago, Maywood, IL, USA
Abstract: Notch signaling plays a pivotal role during embryogenesis. It regulates three fundamental processes: lateral inhibition, boundary formation, and lineage specification. During post-natal life, Notch receptors and ligands control critical cell fate decisions both in compartments that are undergoing differentiation and in pluripotent progenitor cells. First recognized as a potent oncogene in certain lymphoblastic leukemias and mesothelium-derived tissue, the role of Notch signaling in epithelial, solid tumors has been far more controversial. The overall consequence of Notch signaling and which form of the Notch receptor drives malignancy in humans is deeply debated. Most likely, this is due to the high degree of context-dependent effects of Notch signaling. More recently, it has been discovered that Notch (especially Notch-1) can exert different, even opposite effects in the same tissue under differing microenvironmental conditions. Further complicating the understanding of Notch receptors is the recently discovered role for non-canonical Notch signaling. Additionally, the most frequent Notch signaling antagonists used in biological systems have been inhibitors of the transmembrane protease complex γ-secretase, which itself processes a plethora of class one transmembrane proteins and thus cannot be considered a Notch-specific upstream regulator. Here we review the available empirical evidence gathered in recent years concerning Notch receptors and ligands in non-small-cell lung carcinoma (NSCLC). Although an overview of the field reveals seemingly contradicting results, we propose that Notch signaling can be exploited as a therapeutic target in NSCLC and represents a promising complement to the current arsenal utilized to combat this malignancy, particularly in targeting NSCLC tissues under specific environmental conditions, such as hypoxia.
Keywords: Notch-1, Notch-3, developmental signaling pathways, Notch inhibitors, NSCLC progenitor cells, CBF-1, MamL
Progenitor cells in NSCLC and embryonic development signaling pathways
Generally, it is assumed that different non-small-cell lung carcinoma (NSCLC) tumor subtypes originate in different sections of the airways. Squamous cell carcinoma (SCC) is commonly located proximal to the trachea and bronchi whereas adenocarcinoma of the lung (ACL) occurs distally. Until recently, large cell carcinoma was a diagnosis of exclusion, but has since been reclassified into several different subclasses.1,2 Currently, our understanding of cancer development is that it most likely derives from a subset of cells in an organ with well-defined characteristics. Such cells should have lived long in order to acquire the large number of genetic insults that characterize any malignancy. Additionally, these cells should not be terminally differentiated in order to present the classic undifferentiated appearance of most malignancies, which often resemble the organization of embryonic tissue. Based on injury models, a minority of cells in the lung epithelia appear resistant to most injuries and repopulate the damaged areas through a production of transit-amplifying cells, which terminally differentiate. Given the histopathological differences of the major types of NSCLC, well-designed studies have identified different progenitor cells giving rise to NSCLC. The Jacks’ group has identified a small number of cells positioned at the bronchioalveolar junction (or BASCs, for bronchioalveolar stem cells) that, under genetic manipulation, can give rise to genuine ACL.3 The progenitor cells of SCC have yet to be clearly defined, but a population of naphthalene-resistant, basal cells of the pseudostratified bronchial epithelium may be necessary for bronchial regeneration.4 More recently, keratin 5/14 positive basal cells located at the submucosal gland junctions and within intracartilaginous sections of the upper airway and main bronchi have been strongly implicated as SCC progenitor cells.5 It appears that in these cells, ablation of the transcription factor Sox2 drastically reduces their capability to repair damage.6
Stem or progenitor cell identity appears to be mediated by a number of factors that are provided within cellular niches. Major developmental pathways (eg, Wnt, Hedgehog, and Notch) all provide critical cues regulating stem and progenitor cell identity and status.7,8 Which signaling pathway has a predominant role appears to be dependent on specialized niches. Solid tumors, due to their overgrowth of the tissue vasculature, have one unique feature in common: all solid tumors contain hypoxic regions. While the hypoxic region of a tumor is deprived of oxygen and nutrients, it appears rich in growth factors.9–11 Since the pioneering study by Gustafsson et al,12 a wealth of data has been gathered establishing the link between hypoxia inducible factors (HIFs), Notch signaling, the maintenance of the undifferentiated state and the survival of NSCLC under hypoxia.10,13,14 Under these principles, it is conceivable to hypothesize a role for Notch signaling in NSCLC pathobiology.
Canonical Notch signaling
Notch receptors and ligands are a group of single pass, transmembrane proteins conserved from Caenorhabditis elegans to humans. The nomenclature of the molecular players, some structural features and the mechanism of activation of the receptor(s) differ among C. elegans, Drosophila melanogaster and mammalian cells. For this reason, we will refer to the Notch signaling pathway in human cells. Notch receptors (Notch-1 to Notch-4) are synthesized as long precursors. Throughout their maturation, Notch receptors undergo a number of post-translational modifications, including extensive glycosylation in the endoplasmic reticulum and in the Golgi apparatus, where they first undergo a proteolytic cleavage at site S1 operated by a Furin-like pro-protein convertase15 (Figure 1). Because of this cleavage, Notch receptors are presented on the plasma membrane as heterodimers of a large, modular, N-terminal portion (or NotchEC), which is exposed to the extracellular environment. NotchEC is non-covalently bound to a C-terminal portion (Notch™) that comprises a short extracellular “stump”, the transmembrane domain and a large, multidomain intracellular portion (or NotchIC). Generally, NotchEC is recognized as inhibitory and the Notch heterodimerization region, which includes the three NotchEC C-terminal negative regulatory regions (NRRs) and the “stump” of Notch™, plays a vital role in preventing improper Notch activation.15–17 To date, preferential binding of any ligands to specific Notch receptors has not been demonstrated. Several publications have indicated that post-translational modifications, operated by Manic and Lunatic Fringe in the Golgi, of both the receptors and the ligands can “slant” the binding toward certain pairs of receptor–ligand complexes.18,19 However, these modifications can temporally and spatially produce differing effects. Humans have five canonical Notch ligands: three Delta-like proteins (Delta-1, -3, and -4) and two Serrate-like polypeptides (Jagged-1 and -2). Following the canonical route of Notch activation, a receiving cell (expressing Notch receptor) makes contact with a signaling cell (expressing Notch ligands). The ligand establishes specific interactions with the modular portion of NotchEC and initiates a transendocytosis process in the signaling cell, which includes the ligand and NotchEC. This event destabilizes the heterodimerization region, rendering the extracellular portion of Notch™ susceptible to cleavage, operated by A-disintegrin and metalloproteinase (ADAM) 10 or 17 (or both) at site S2. In turn, this makes the transmembrane portion of Notch™ available to γ-secretase digestion at site S3. The exact site of γ-secretase cleavage of Notch is not defined; γ-secretases cleave Notch receptors at multiple sites within the transmembrane domain, and the stability of the resulting cleavage products will be determined by the N-terminal rule. Most likely, this event occurs both at the plasma membrane and in the endocytic vesicles. The final product of γ-secretase cleavage is NotchIC, which translocates into the nucleus, and interacts with the transcription factor recombining binding protein suppressor of hairless, known as CBF-1 in humans. This interaction radically changes the composition of CBF-1 binding partners. In the absence of Notch, CBF-1 associates with transcriptional co-repressors like histone de-acetylases and demethylases, Ski interacting protein, CBF-1 interacting co-repressor, silencing mediator for retinoid or thyroid-hormone receptors, the helix-loop-helix transcriptional repressor SHARP and C-terminal-binding protein 1. This complex is a potent repressor of Notch target genes. In the presence of NotchIC, Ski interacting protein facilitates the dissociation of the entire repressor complex20 while Notch recruits CBF-1 transcriptional co-activators such as Mastermind-like (MamL) and histone acetyl-transferases. The net result is transcriptional activation of a family of genes known as Enhancer of split (HES, HEY, and HERP genes in humans), which act as suppressors of tissue-specific differentiation activators.21 Other Notch targets include (in a tissue-dependent fashion) Myc, cyclin D3, and p21WAF/CIP1.22
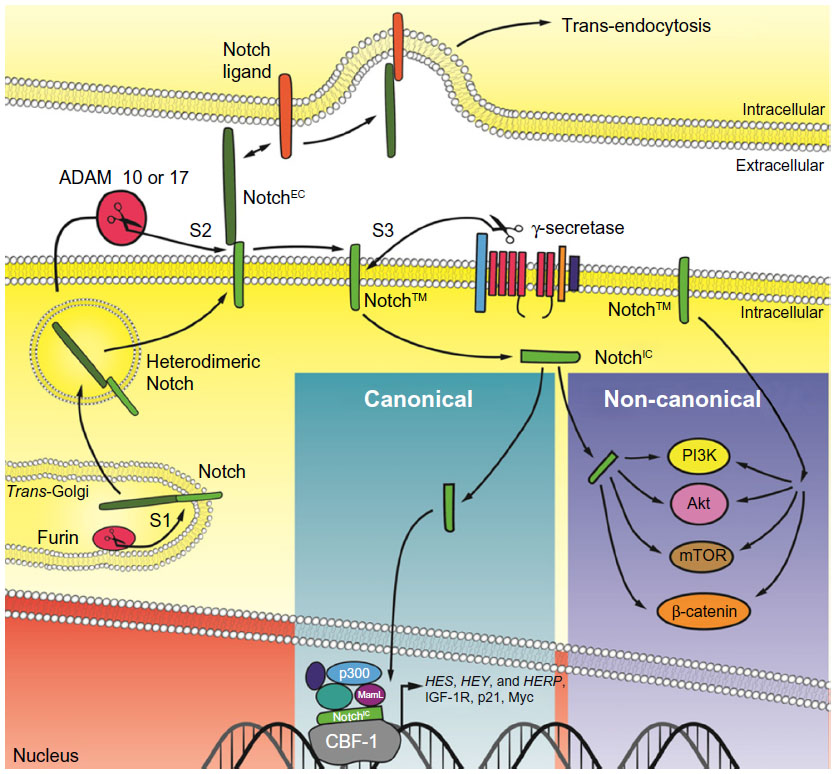 | Figure 1 Simplified schematic of Notch signaling. |
Non-canonical Notch signaling
The recognition that non-canonical Notch signaling triggers a multitude of cellular effects independently of interaction of Notch receptors with ligand has increased among the Notch research community. These non-canonical events partially overlap with other well-known pathways with drive cellular transformation. The following is a summary of distinctive features of non-canonical Notch signaling; a more detailed description of this pathway can be found elsewhere.23
Early anecdotal evidence suggested Notch had oncogenic potential independent of its interaction with CBF-1. In baby rat kidney cells, Notch seemed to synergize with the adenoviral E1A oncogene. However, some experiments indicated that a truncation of NotchIC unable to bind CBF-1 or transcriptional co-activators (missing the RAM and OPA domains) was still active in E1A/Notch cotransformation.24 Studies of conditional CBF-1 knockout mice have revealed that canonical Notch-4 signaling is required for mammary gland development, while non-canonical Notch-4 signaling may be involved in mammary carcinogenesis.25 Recent studies have demonstrated that prevention of nutrient-deprivation induced apoptosis in HeLA cells by non-nuclear, membrane-tethered NotchIC mutants is mediated by mechanistic target of rapamycin complex 2.26 Non-canonical Notch-1 signaling can enhance HIF-1α expression, thereby hypoxic response, by sequestering the HIF-1α negative regulator factor-inhibiting HIF-1α (FIH-1).27 This complicates establishing a hierarchical order between HIF-1α and Notch-1IC under hypoxia. For instance, binding of HIF-1α to Notch-1IC stabilizes and enhances the transcriptional activity of Notch-1IC.12 Alternatively, in hypoxic conditions FIH-1 has a higher affinity to Notch-1IC compared with HIF-1α. FIH-1 then promotes hydroxylation of Notch-1IC at the expense of HIF-1α.27 Due to these conflicting data, further clarification is needed.
Other key feature of non-canonical Notch signaling includes the ability of Notch-1 to directly activate the phosphoinositide 3-kinase/Akt pathway in T-cells.28 In this study, the effects of Notch-1IC on Akt seemed to occur too rapidly to be solely ascribed to transcriptional activation of the insulin-like growth factor 1 receptor (IGF-1R) and its ligands.10 It would be interesting to clarify whether Notch-1 can activate Akt both in a rapid, transient manner and in a more prolonged, transcriptionally dependent fashion. Furthermore, the tissue dependency of these processes must be assessed.
Notch signaling, both canonical and non-canonical, has far-reaching cellular implications in its variety of facets. As the tissue specificity of Notch signaling is well recognized, understanding the prevalence of either canonical or non-canonical signaling in diseases, including cancer, will be important in the development of effective therapeutic strategies.
Notch signaling in NSCLC
Little is known about the role of Notch-2 and Notch-4 in NSCLC. Of the handful of available reports, a very recent study supports a tumor suppressive role for Notch-2 in a Kras(G12D)-driven mouse NSCLC in vivo model.29 Here we will focus on those Notch receptors extensively studied in NSCLC (Notch-3 and Notch-1) due to the large amount of information available.
Notch-3 signaling and NSCLC
First reported in 2000, a t(15;19) somatic translocation was discovered in a poorly differentiated aggressive lung carcinoma implicating that Notch signaling may play a role in lung cancer.30 A detailed analysis of the tumor-derived cell line (HCC2429) exhibited a high expression of Notch-3. Observing no other genetic abnormalities, Dang et al30 assayed a number of cell lines and found elevated expression of the Notch-3 mRNA in 7 of 44 NSCLC cell lines by Northern blot analysis. Peculiarly, all positive cell lines were either ACL or mesotheliomas, and most harbored chromosomal abnormalities involving 19p translocations with different chromosomes. This situation was very similar to the t(7,9) translocation found in about 10% of T cell acute lymphoblastic leukemia (T-ALL) cases. The latter translocation generated a truncated version of Notch-1 (TAN-1) characterized by the expression of a mutated Notch-1 receptor lacking most of the N-terminal domain.16 Since truncated, constitutively active forms of Notch-1 are highly oncogenic in T-cells. This suggested that abnormal Notch-3 expression, which is virtually absent in non-neoplastic lung epithelia, could be a main oncogenic driver in at least some ACLs. In T-cells, constitutively active Notch-1 is a potent oncogene due to its ability to generate an exacerbated positive feedback loop with Myc.30 A similar situation could be hypothesized for Notch-3 in NSCLC.
Subsequent investigations have supported a role for Notch-3 in NSCLC, both in ACL and especially in SCC. In one study, a Notch-3 receptor with a truncated C-terminal domain was generated which acted as a dominant-negative Notch-3. Expression of this mutant in two cell lines (HCC2429 and H460) reduced their growth rate, increased apoptosis and caused increased growth factor dependence.31 Immunochemical analysis of Notch-3 in a large number of lung cancers of different histopathology revealed Notch-3 expression in more than 38% of specimens. Notch-3 expression appeared to be correlated with epidermal growth factor receptor (EGFR) overexpression. The highest frequency of Notch-3 positive lung cancer was found in SCC (45%) and ACL (37%). Interestingly, dominant-negative Notch-3 expression caused increased sensitivity to the EGFR inhibitor AG1478,32 indicating the first evidence of synergism between Notch signaling and receptors tyrosine kinase in NSCLC.32 The interaction between Notch-3 and EGFR in NSCLC seems to converge on the expression of the pro-apoptotic protein Bcl-like protein 11 (or Bim); both EGFR inhibition and siRNA to Notch-3 can elevate Bim expression. However, maximal Bim expression is achieved when both treatments are implemented. According to this model, EGFR and Notch-3 would support NSCLC resistance to apoptosis through a dual mechanism: Akt activation of the transcription factor Forkhead box O3 (Foxo3a), which promotes transcription of Bim, and an Akt-independent, Notch-3 driven Bim activation.33 It is possible that Foxo3a mostly mediates this cross talk. In myosatellite cells, Foxo3a deficiency mimics Notch-1 and Notch-3 attenuated expression. Ultimately, Foxo3a may be necessary for the proper expression of these two Notch receptors to maintain quiescence and prevent differentiation.34
A recent report has indicated that Notch-3 is a driver for the maintenance of the cancer initiating cell phenotype in a subpopulation of sphere-derived, NSCLC cells characterized by high expression of the cell adhesion molecule CD24 and integrin β4 markers. These cells appear to have a very high metastatic potential. Sorting this subpopulation resulted in a drastic reduction of tumor formation upon syngeneic transplantation of cells harboring oncogenic Kras and p53−/−.35 Finally, a 5-year longitudinal survey has associated high Notch-3 expression in NSCLC with poor outcome.36 The available empirical evidence support the possibility that Notch-3 can be a driver oncogene in about 40% of NSCLC and that its functions are closely linked to EGFR signaling.
The controversial role of Notch-1 in NSCLC
The expression of Notch-1 and Jagged-1 during lung development in mice steadily increases from embryonic day E12 until birth. However, Notch-1 and Jagged-1 expression co-localize with PECAM-1, and is found mostly in endothelial cells.37 Notch-1 is also expressed in non-endothelial, mesenchymal cells and lung epithelial cells. This suggests that Notch-1, besides its well-recognized role in angiogenesis, participates in the terminal differentiation of lung epithelia.
Recently, Notch-1 gain-of-function has been often studied by overexpressing Notch-1IC. Overexpression of this portion of Notch-1 in hematopoietic cells caused T-cells leukemias in about 50% of bone marrow transplanted mice.38 Conversely, Notch-1IC overexpression in NSCLC cell lines causes cell growth arrest (or apoptosis) and inhibited tumor formation in vivo.39 This situation is comparable to what has been observed in small cell lung cancer cell lines, where Notch-1 appears to have tumor suppressive roles.40 Importantly, ectopic expression of a constitutively active protein in the context of malignant transformation, where Notch signaling can be already overactivated, may lead to confusing results. Clearer data could be obtained by stimulating Notch-1 in a physiologic fashion in cells withdrawn of Notch-1 signaling. Nonetheless, data have been reported supporting a tumor suppressive role for Notch-1 in NSCLC. For example, a recent report presented a high percentage (75%) of Notch (Notch-1 or Notch-2) loss-of-function mutation in head and neck SCC and, to a lesser extent, in lung SCC. These mutations where heterozygous and yielded truncated, N-terminal fragments of Notch-1 and Notch-2. Alternatively, these mutations affected critical domains, potentially compromising the functionality of the receptors. By utilizing cell-based assays, it was confirmed that these mutations represented Notch loss-of-function mutations.41 It should also be noted that while Notch-1 or Notch-2 knock out mice die at mid gestation in utero, heterozygous knock out mice for either Notch-1 or Notch-2 are viable, fertile, of normal size and do not display noticeable phenotypes. Hence, at least in mice, haploinsufficiency for Notch-1 and Notch-2 do not appear to generate cancer susceptibility. Furthermore, Notch-1 and Notch-2 signaling are not redundant and opposite effects deriving from activation of the two receptors have been described. In cancer, Notch-1 and Notch-2 activation seems to have opposite effects in certain brain tumors, breast cancer, multiple myeloma, and mesothelioma (reviewed in Galluzzo and Bocchetta42). These non-overlapping sometimes opposite effects on the same promoter (phosphatase and tensin homolog [PTEN], or p21WAF/Cip1), may be a consequence of differential post-translational modifications of the intracellular portion of the two receptors20 or may simply be inherent to the structure of the receptors. The fact that heterozygous, inactivating mutations of either Notch-1 or Notch-2 found in a portion of SCCs of the lung may reflect some adaptive responses to certain microenvironmental conditions rather than being the driver of this malignancy as a whole. Imbalances in Notch receptors and ligands may also be related to Notch cis-inhibition. Without delving too deeply into the details of this phenomenon, the ligands of cells that express both Notch receptors and ligands, can still bind Notch receptors but this association prevents activation. Recently, a study demonstrated that endothelial cells, through augmented expression of Delta-4, prevent the proliferation of NSCLC cells by activating Notch signaling. In this specific model, most of the anti-oncogenic functions were mediated by Notch-4 through elevation of Notch-1 expression, hence enhancing PTEN expression.43 In conclusion, there are data supporting anti-oncogenic role of Notch signaling in the lung epithelia that may contradict the previous summary of Notch-3 involvement in lung carcinogenesis.
There is, however, ample experimental evidence supporting a pro-oncogenic role for Notch-1 in NSCLC, which we can only briefly summarize here. In some reports, Notch-1 is presented as a pure oncogene: downregulation of Numb (a Notch-1 negative regulator, which promotes Notch-1IC polyubiquitination and proteasomal degradation44) is found in about one-third of NSCLC specimens.45 Additionally, about one-tenth of NSCLC studied harbored Notch-1 mutations that were confirmed to represent Notch-1 gain-of-function mutations in vitro. These mutations conferred enhanced sensitivity of the cell lines derived from such tumors to the γ-secretase inhibitors (GSIs) DAPT and MRK-003. Notch-1 activation correlated with poor prognosis and seemed present in association with wild-type p53. Also, mutations often occurred in domains considered as “hot spots” for Notch-1 mutations in T-ALL.45 More recently, a transgenic mouse with activated Notch-1 expressed in lung epithelia developed alveolar hyperplasia and adenomas. Notch-1 activation seemed to positively regulate Myc expression, similar to previous observations in some leukemias.46 In this experimental system, Notch-1 overactivation alone was unable to generate adenocarcinomas, while ectopic addition of Myc led to generation of adenocarcinomas. Other Notch-1 regulators have been implicated in the maintenance or the severity of the transformed phenotype in NSCLC. Overexpression of ADAM-10 (one of the proteases required for proteolytical Notch cleavage at S2, rendering Notch receptors susceptible to γ-secretase cleavage) was observed in NSCLC samples when compared to normal lung using immunohistochemistry. Although ADAM-10 staining intensity differed from specimen to specimen, a clear association between the intensity of the signal and the stage of the disease was found, with ADAM-10 maximally expressed in distal metastases.47 ADAM-10 depletion in A549 cells using RNA interference (RNAi) greatly diminished Notch-1 activation. Contrary to other reports, active Notch-1 did not co-operate with β-catenin, but interfered with activation of ERKs and motility (measured as Matrigel invasion). RNAi depletion of both ADAM-10 and γ-secretase exhibited similar effects of halting cell migration.47 This study also revealed a high association of ADAM-10 and Notch-1 overexpression, which was particularly evident in late-stage NSCLC, once again raising the issue that Notch-1 activation is related to tumor progression and metastasis.
Furthermore, Notch-1 has also been implicated in NSCLC cell survival. Artificial downregulation of the protein kinase Tribbles homolog 3 (TRB3), which is induced in response to NFκB activation, led to decreased Notch-1 expression in ACL. Often found in several solid tumors, TRB3 overexpression was correlated with poor prognosis. RNAi-mediated TRB3 depletion in A549 cells caused depressed expression of Notch-1, leading to a 50% reduced viability of A549 cells and a significant reduction in cell motility.48 In summary, there are numerous reports concerning Notch-1 expression, upstream or downstream regulators, the interaction of tumor cells with stroma (or immediately surrounding tissues), prognosis variables, and therapy susceptibility, which support both a tumor suppressive and a pro-oncogenic role for Notch-1 in NSCLC.
While reconciling these apparently conflicting data is difficult, we submit that the most likely explanation is that Notch-1 biological output is highly dependent upon microenvironmental conditions. We found that Notch-1 genetic depletion caused no evident phenotype in ACL cells grown in standard conditions. Notch-1IC overexpression in these cells resulted in significant cell death.13 When performing immunohistochemistry using a highly specific antibody against Notch-1IC in ACL specimens, we found that Notch-1 staining was restricted to pockets within some tumor specimens. When analyzing consecutive slides, we encountered a paradoxical scenario in which a slide would essentially test negative for Notch-1IC, while sections deeper in the tumor specimens had very evident and intense staining. Using orthotropic xenograft models, we determined that Notch-1 expression (and that of its downstream targets) was confined to sections of the tumors expressing the hypoxia markers HIF-1α and glucose transporter 1. However, in non-hypoxic tumor areas, Notch-1IC was undetectable.10 In ACL cells grown under hypoxia, Notch-1 depletion was incompatible with cell survival, and Notch-1IC expression at physiologic levels could rescue GSI-induced apoptosis.13 Notch-1 signaling appears indispensable for ACL survival under hypoxia as it promotes transcription of the IGF-1R and its major ligands IGF-1 and IGF-2.10 Therefore, under hypoxia, Notch-1 promotes an autocrine feedback loop that ultimately results in overactivation of the phosphoinositide 3-kinase/Akt axis (Figure 2). In vivo, where hypoxic tumor tissue is not only deprived of oxygen but also of nutrients, this activation is limited to the anti-apoptotic signals of Akt. In this hypoxic NSCLC environment, mechanistic target of rapamycin complex 1 appears to be under a dominant metabolic checkpoint, resulting in NSCLC cells quiescence.49 Hence, Notch-1 expression and signaling output seem to be determined by microenvironmental conditions (in this case hypoxia) and Notch-1 interplay with HIF-1α.
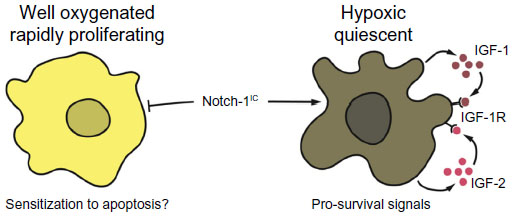 | Figure 2 Possible explanation for opposing Notch-1 effects on NSCLC cells. |
Hypoxia also modulates the expression and the activity of critical Notch-1 activators. For examples, the γ-secretase complex component anterior pharynx-defective 1 transcription is promoted by HIF-1α in a context-dependent fashion.50 Additionally, the γ-secretase component Presenilin-1 (which harbors the proteolytic catalytic site within the γ-secretase complex) can associate with HIF-1α and prevents its proteosomal degradation.51 ADAM 10 expression and activity is also increased under hypoxia, a phenomenon most likely mediated by nitric oxide, which is a key molecule needed to stabilize HIF-1α.52
Hypoxia also upregulates Notch ligands expression. Breast cancer cells in which Notch-1 seems to induce epithelial to mesenchymal transition (EMT) by stimulating Snail expression, demonstrated an increased expression of Delta-1.53 Taking all of this information into consideration, examining the role of Notch-1 in NSCLC cannot be accomplished without accounting for the effects of tumor hypoxia, or the mutational status of central regulatory mediators of the hypoxic response, such as the Von Hippel–Lindau tumor suppressor gene. Furthermore, Notch-1 signaling outcome is highly context-dependent. For this reason, the status of growth factor tyrosine kinase receptors signaling, Ras activation, and other unknown parameters should be taken into consideration when interpreting data concerning Notch-1, its activators, and downstream mediators in NSCLC. Overall, the study of Notch-1 signaling in NSCLC can lead to apparently contrasting results when studied in isolation. In our opinion, the amount of evidence supporting an essential role of Notch-1 signaling for survival under hypoxia, the promotion of EMT and metastasis in NSCLC cells is paramount.
Notch-1 signaling consequences for the treatment of NSCLC
Cisplatin and carboplatin are essential drugs used in combination with taxanes in the standard of care therapy for NSCLC. Although often effective, treatment with platin-based compounds regularly leads to the appearance of cisplatin or carboplatin resistant recurrent disease. The development of this phenotype has been ascribed to upregulation of Notch-1 in a population of prominin-1 (or CD133) positive NSCLC cells. Cisplatin resistance (and the arise of CD133 positive cells) could be reversed using either RNAi to deplete Notch-1 in these cells, or treating them with the GSI DAPT. The effects of GSI treatment could be reversed with Notch-1IC forced expression.54 Additionally, a number of groups have reported enhanced cisplatin cytotoxicity when administered alongside GSI, both in vitro and in vivo, in a number of cells derived from a variety of different solid tumors, including NSCLC.55,56 Furthermore, several reports have indicated NSCLC cases with overactivated Notch-1 signaling were resistant to several types of radio/chemotherapy regimens. For instance, Notch-1 overactivation has been linked with poor prognosis and decreased radiation sensitivity.57
Doxorubicin is an anthracycline antitumor antibiotic that is often used in combination therapies for NSCLC. Recent studies have linked the expression of the microRNA-1 (miR-1) with sensitivity to doxorubicin.58 It appears that miR-1 and Delta-1 expression are negatively correlated in NSCLC. Additionally, miR-1 targets the Delta-1 mRNA directly, hence suggesting that doxorubicin resistance arising from reduced miR-1 expression is ultimately due to Notch-1 enhanced activity.59
Others have shown that Notch-1 participates in the process of acquired resistance to receptor tyrosine kinase inhibition in NSCLC.60 Similarly, in NSCLC, gefitinib-resistant PC9 and H1650 cells upregulated Notch-1 expression exclusively (eg, the expression of other Notch receptors was found unchanged), alongside increased HES-1 expression and expression of the markers of EMT vimentin and Snail.60 Notch-1IC ectopic expression exclusively conferred the previously mentioned properties to gefitinib-sensitive PC9 and H1650 cells, while genetic depletion of Notch-1 using RNAi caused overexpression of the epithelial marker E-cadherin and suppressed vimentin expression. Taken together, these data derived from studies of intrinsic or acquired NSCLC resistance to different therapeutic agents seem to support a pro-oncogenic role for Notch-1 in this malignancy, at least at the level of susceptibility to therapy and emergence of radio/chemotherapy resistance.
Notch receptors as therapeutic targets: strategies to achieve selective inhibition
A summary of various strategies to inhibit Notch signaling is reported in Figure 3.
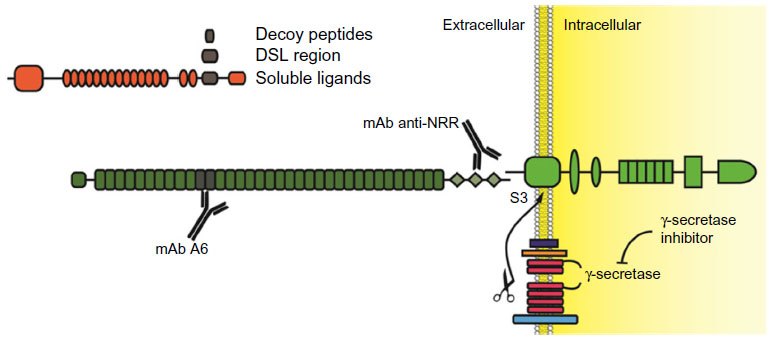 | Figure 3 Experimental strategies to inhibit Notch receptors. |
The traditional, and possibly over-used, Notch inhibitors are GSIs. Principally, this is due to the ample availability of small molecules inhibiting the γ-secretase complex, which were originally developed in order to inhibit amyloid precursor protein cleavage as a potential treatment for Alzheimer’s disease. The problem with GSIs is twofold. First, γ-secretase cleaves more than 90 cellular substrates,61 including Notch ligands. Therefore, they cannot be considered Notch-specific inhibitors. Second, pan-Notch inhibition interferes with enterocyte differentiation, rapidly leading to accumulation in the intestinal epithelia of goblet cells, which cause profuse diarrhea and impaired nutrient uptake.62 This first problem could be bypassed through the development of novel substrate specific GSIs, possibly through the modulation of Nicstrin function as this protein appears to be the main discriminant in the process of γ-secretase substrate selection.63 Currently, GSIs that can discriminate between amyloid precursor protein and Notch cleavage have emerged and a more thorough understanding of the γ-secretase complex function(s) may lead to the development of GSI that are mostly Notch-specific. Several notable GSIs, including RO-4929097, MK0752, PF-03084014, and BMS-708163, have been tested in clinical trials for use in a variety of solid tumors, including NSCLC. Nonetheless, even Notch-specific GSIs could only be pan-Notch inhibitors. ADAM inhibitors are currently under development,64 but these molecules do not promise increased Notch receptor selectivity or decreased undesired off-target effects as compared to GSIs. Moreover, ADAM inhibitors appear to be pan-Notch inhibitors as well.
Other strategies aimed at targeting Notch receptors non-selectively include utilizing soluble Notch ligands or receptors binding domain decoy peptides. A Notch soluble ligand is inhibitory (in vivo, ligand shedding is routinely used as a mechanism to modulate Notch activity) because mammalian Notch receptors, unlike their C. elegans homologs, require trans-endocytosis and physical removal of NotchEC away from Notch™ in order to promote ADAM and γ-secretases sequential cleavage. The binding of a soluble ligand would prevent a membrane-tethered ligand, which is presented on a signaling cell, from associating with Notch receptors on the receiving cell, thus preventing Notch activation. Although these strategies have been attempted in experimental settings with differing degrees of success,65 we are unaware of any drug based on soluble or decoy Notch ligands or receptors that has yet to enter the clinic.
Due to a lack of a Notch receptor “catalytic site”, the development of small inhibitory molecules appears limited to the enzymes that cause Notch receptor activation, or their interaction with CBF-1. Interestingly, a recent report described the generation of soluble, cell permeable alpha-helical peptides that were able to disrupt the association between Notch-1 and MamL.66 Utilizing coimmunoprecipitation techniques, the authors indicated that one such peptide, named SAHM1, disrupted the in vivo association between MamL and Notch-1. This event was paralleled by decreased expression of a reporter gene under a Notch-1-regulated promoter. SAHM1 administered to T-ALL cells caused decreased expression of multiple Notch-1 downstream genes and cell growth arrest in vitro, and reduced leukemic potential in experimental animals. Although conceptually very interesting, SAHM1 is currently sold by a few companies for use only as an investigative compound, though it does appear that SAHM1 and its derivatives are slowly moving into experimentation in human subjects.
Recently, humanized monoclonal antibodies (mAbs) which specifically target individual Notch receptors have been described and tested in vitro and in vivo.67 These antibodies were able to inhibit either Notch-1 or Notch-2 without cross-reactivity. Using phage display techniques, these antibodies were raised against the association domain between NotchEC and Notch™, producing a “clip” which maintains the binding of the Notch NRRs of NotchEC to the extracellular stump of Notch™. The unique property of these antibodies is in the identity of the epitope they recognize; all known point mutations that destabilize NotchEC association to Notch™ in T-ALL are outside of the regions recognized by the paratopes. This feature makes these antibodies particularly useful, because they have the potential of not only interfering with ligand-facilitated activation of Notch, but also interfering with improper activation of Notch due to mutational destabilization of the Notch NRR. Additionally, these antibodies did not display any major detrimental phenotype concerning intestinal cells differentiation. A variety of targets are recognized by other humanized antibodies, trastuzumab, which targets Her2; MK-0646 and several others that target IGF-1R; and bevacizumab that targets vascular-endothelial growth factor A. These humanized antibodies are increasingly utilized in clinical trials with a number of other inhibitory molecules. Quite surprisingly, these Notch-specific antibodies have yet to be deployed in the clinic. Recently, a Phase I using enoticumab (REGN421), a fully humanized anti Delta-4 mAb, has been initiated for the treatment of solid tumors. While the extent of the side effects has made the determination of maximal tolerated dose uncertain, positive therapeutic effects have been identified in patients with ovarian cancer.68
Conclusion
Currently, there is a plethora of evidence linking Notch signaling with NSCLC. For malignancies that appear Notch-3 driven, GSI treatment may be highly beneficial along with standard of care drugs like platinum-based compounds. For the eradication of minimal residual disease and micrometastases in lymph nodes, with much lower oxygen concentration than the bloodstream or well-vascularized tissues, Notch-1 inhibition may be of principal importance. Yet, Notch-1 inhibition should be restricted to this receptor only, and not to other Notch isoforms. The current status of compounds development indicates that specific and sustained Notch-1 inhibition remains a future, exciting perspective.
Acknowledgment
Grant support: NCI – R01 CA134503 to MB.
Disclosure
The authors report no conflicts of interest in this work.
References
Brambilla E, Travis WD, Colby TV, Corrin B, Shimosato Y. The new World Health Organization classification of lung tumours. Eur Respir J. 2001;18(6):1059–1068. | |
Fernandez FG, Battafarano RJ. Large-cell neuroendocrine carcinoma of the lung. Cancer Control. 2006;13(4):270–275. | |
Kim CF, Jackson EL, Woolfenden AE, et al. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell. 2005;121(6):823–835. | |
Hong KU, Reynolds SD, Watkins S, Fuchs E, Stripp BR. Basal cells are a multipotent progenitor capable of renewing the bronchial epithelium. Am J Pathol. 2004;164(2):577–588. | |
Rock JR, Onaitis MW, Rawlins EL, et al. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc Natl Acad Sci U S A. 2009;106(31):12771–12775. | |
Que J, Luo X, Schwartz RJ, Hogan BL. Multiple roles for Sox2 in the developing and adult mouse trachea. Development. 2009;136(11):1899–1907. | |
Joseph NM, Morrison SJ. Toward an understanding of the physiological function of Mammalian stem cells. Dev Cell. 2005;9(2):173–183. | |
Ohlstein B, Kai T, Decotto E, Spradling A. The stem cell niche: theme and variations. Curr Opin Cell Biol. 2004;16(6):693–699. | |
Harris AL. Hypoxia-a key regulatory factor in tumour growth. Nat Rev Cancer. 2002;2:38–47. | |
Eliasz S, Liang S, Chen Y, et al. Notch-1 stimulates survival of lung adenocarcinoma cells during hypoxia by activating the IGF-1R pathway. Oncogene. 2010;29:2488–2498. | |
Lee YH, Morrison BL, Bottaro DP. Synergistic signaling of tumor cell invasiveness by hepatocyte growth factor and hypoxia. J Biol Chem. 2014;289(30):20448–20461. | |
Gustafsson MV, Zheng X, Pereira T, et al. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev Cell. 2005;9(5):617–628. | |
Chen Y, De Marco MA, Graziani I, et al. Oxygen concentration determines the biological effects of NOTCH-1 signaling in adenocarcinoma of the lung. Cancer Res. 2007;67(17):7954–7959. | |
Keith B, Simon MC. Hypoxia-inducible factors, stem cells, and cancer. Cell. 2007;129(3):465–472. | |
Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137(2):216–233. | |
Ellisen LW, Bird J, West DC, et al. TAN-1, the human homolog of the Drosophila notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell. 1991;66(4):649–661. | |
Malecki MJ, Sanchez-Irizarry C, Mitchell JL, et al. Leukemia-associated mutations within the NOTCH1 heterodimerization domain fall into at least two distinct mechanistic classes. Mol Cell Biol. 2006;26(12):4642–4651. | |
Hicks C, Johnston SH, diSibio G, Collazo A, Vogt TF, Weinmaster G. Fringe differentially modulates Jagged1 and Delta1 signalling through Notch1 and Notch2. Nat Cell Biol. 2000;2(8):515–520. | |
Shimizu K, Chiba S, Saito T, Kumano K, Takahashi T, Hirai H. Manic fringe and lunatic fringe modify different sites of the Notch2 extracellular region, resulting in different signaling modulation. J Biol Chem. 2001;276(28):25753–25758. | |
Zhou S, Fujimuro M, Hsieh JJ, et al. SKIP, a CBF1-associated protein, interacts with the ankyrin repeat domain of NotchIC To facilitate NotchIC function. Mol Cell Biol. 2000;20(7):2400–2410. | |
Iso T, Kedes L, Hamamori Y. HES and HERP families: multiple effectors of the Notch signaling pathway. J Cell Physiol. 2003;194(3):237–255. | |
Andersson ER, Sandberg R, Lendahl U. Notch signaling: simplicity in design, versatility in function. Development. 2011;138(17):3593–3612. | |
Ayaz F, Osborne BA. Non-canonical notch signaling in cancer and immunity. Front Oncol. 2014;4:345. | |
Dumont E, Fuchs KP, Bommer G, Christoph B, Kremmer E, Kempkes B. Neoplastic transformation by Notch is independent of transcriptional activation by RBP-J signalling. Oncogene. 2000;19(4):556–561. | |
Raafat A, Lawson S, Bargo S, et al. Rbpj conditional knockout reveals distinct functions of Notch4/Int3 in mammary gland development and tumorigenesis. Oncogene. 2009;28(2):219–230. | |
Lee KS, Wu Z, Song Y, et al. Roles of PINK1, mTORC2, and mitochondria in preserving brain tumor-forming stem cells in a noncanonical Notch signaling pathway. Genes Dev. 2013;27(24):2642–2647. | |
Zheng X, Linke S, Dias JM, et al. Interaction with factor inhibiting HIF-1 defines an additional mode of cross-coupling between the Notch and hypoxia signaling pathways. Proc Natl Acad Sci U S A. 2008;105(9):3368–3373. | |
Sade H, Krishna S, Sarin A. The anti-apoptotic effect of Notch-1 requires p56lck-dependent, Akt/PKB-mediated signaling in T cells. J Biol Chem. 2004;279(4):2937–2944. | |
Baumgart A, Mazur PK, Anton M, et al. Opposing role of Notch1 and Notch2 in a Kras(G12D)-driven murine non-small cell lung cancer model. Oncogene. 2015;34(5):578–588. | |
Dang TP, Gazdar AF, Virmani AK, et al. Chromosome 19 translocation, overexpression of Notch3, and human lung cancer. J Natl Cancer Inst. 2000;92(16):1355–1357. | |
Palomero T, Lim WK, Odom DT, et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc Natl Acad Sci U S A. 2006; 103(48):18261–18266. | |
Haruki N, Kawaguchi KS, Eichenberger S, et al. Dominant-negative Notch3 receptor inhibits mitogen-activated protein kinase pathway and the growth of human lung cancers. Cancer Res. 2005; 65(9):3555–3561. | |
Konishi J, Yi F, Chen X, Vo H, Carbone DP, Dang TP. Notch3 cooperates with the EGFR pathway to modulate apoptosis through the induction of bim. Oncogene. 2010;29(4):589–596. | |
Gopinath SD, Webb AE, Brunet A, Rando TA. FOXO3 promotes quiescence in adult muscle stem cells during the process of self-renewal. Stem Cell Reports. 2014;2(4):414–426. | |
Zheng Y, de la Cruz CC, Sayles LC, et al. A rare population of CD24(+)ITGB4(+)Notch(hi) cells drives tumor propagation in NSCLC and requires Notch3 for self-renewal. Cancer Cell. 2013;24(1):59–74. | |
Ye YZ, Zhang ZH, Fan XY, et al. Notch3 overexpression associates with poor prognosis in human non-small-cell lung cancer. Med Oncol. 2013;30(2):595. | |
Taichman DB, Loomes KM, Schachtner SK, et al. Notch1 and Jagged1 expression by the developing pulmonary vasculature. Dev Dyn. 2002; 225(2):166–175. | |
Pear WS, Aster JC, Scott ML, et al. Exclusive development of T cell neoplasms in mice transplanted with bone marrow expressing activated Notch alleles. J Exp Med. 1996;183(5):2283–2291. | |
Zheng Q, Qin H, Zhang H, et al. Notch signaling inhibits growth of the human lung adenocarcinoma cell line A549. Oncol Rep. 2007; 17(4):847–852. | |
Sriuranpong V, Borges MW, Ravi RK, et al. Notch signaling induces cell cycle arrest in small cell lung cancer cells. Cancer Res. 2001; 61(7):3200–3205. | |
Wang NJ, Sanborn Z, Arnett KL, et al. Loss-of-function mutations in Notch receptors in cutaneous and lung squamous cell carcinoma. Proc Natl Acad Sci U S A. 2011;108(43):17761–17766. | |
Galluzzo P, Bocchetta M. Notch signaling in lung cancer. Expert Rev Anticancer Ther. 2011;11(4):533–540. | |
Ding XY, Ding J, Wu K, et al. Cross-talk between endothelial cells and tumor via delta-like ligand 4/Notch/PTEN signaling inhibits lung cancer growth. Oncogene. 2012;31(23):2899–2906. | |
McGill MA, McGlade CJ. Mammalian numb proteins promote Notch1 receptor ubiquitination and degradation of the Notch1 intracellular domain. J Biol Chem. 2003;278(25):23196–23203. | |
Westhoff B, Colaluca IN, D’Ario G, et al. Alterations of the Notch pathway in lung cancer. Proc Natl Acad Sci U S A. 2009;106(52):22293–22298. | |
Allen TD, Rodriguez EM, Jones KD, Bishop JM. Activated Notch1 induces lung adenomas in mice and cooperates with Myc in the generation of lung adenocarcinoma. Cancer Res. 2011;71(18):6010–6018. | |
Guo J, He L, Yuan P, et al. ADAM10 overexpression in human non-small cell lung cancer correlates with cell migration and invasion through the activation of the Notch1 signaling pathway. Oncol Rep. 2012; 28(5):1709–1718. | |
Zhou H, Luo Y, Chen JH, et al. Knockdown of TRB3 induces apoptosis in human lung adenocarcinoma cells through regulation of Notch 1 expression. Mol Med Rep. 2013;8(1):47–52. | |
Sobol A, Galluzzo P, Liang S, et al. Amyloid precursor protein (APP) affects global protein synthesis in dividing human cells. J Cell Physiol. 2015;230(5):1064–1074. | |
Wang R, Zhang YW, Zhang X, et al. Transcriptional regulation of APH-1A and increased gamma-secretase cleavage of APP and Notch by HIF-1 and hypoxia. FASEB J. 2006;20(8):1275–1277. | |
De Gasperi R, Sosa MA, Dracheva S, Elder GA. Presenilin-1 regulates induction of hypoxia inducible factor-1α: altered activation by a mutation associated with familial Alzheimer’s disease. Mol Neurodegener. 2010;5:38. | |
Barsoum IB, Hamilton TK, Li X, et al. Hypoxia induces escape from innate immunity in cancer cells via increased expression of ADAM10: role of nitric oxide. Cancer Res. 2011;71(24):7433–7441. | |
Guo S, Liu M, Gonzalez-Perez RR. Role of Notch and its oncogenic signaling crosstalk in breast cancer. Biochim Biophys Acta. 2011; 1815(2):197–213. | |
Liu YP, Yang CJ, Huang MS, et al. Cisplatin selects for multidrug-resistant CD133+ cells in lung adenocarcinoma by activating Notch signaling. Cancer Res. 2013;73(1):406–416. | |
Liang S, Galluzzo P, Sobol A, Skucha S, Rambo B, Bocchetta M. Multimodality approaches to treat hypoxic non-small cell lung cancer (NSCLC) microenvironment. Genes Cancer. 2012;3(2):141–151. | |
Yuan X, Wu H, Han N, et al. Notch signaling and EMT in non-small cell lung cancer: biological significance and therapeutic application. J Hematol Oncol. 2014;7(1):87. | |
Theys J, Yahyanejad S, Habets R, et al. High NOTCH activity induces radiation resistance in non small cell lung cancer. Radiother Oncol. 2013;108(3):440–445. | |
Nasser MW, Datta J, Nuovo G, et al. Down-regulation of micro-RNA-1 (miR-1) in lung cancer. Suppression of tumorigenic property of lung cancer cells and their sensitization to doxorubicin-induced apoptosis by miR-1. J Biol Chem. 2008;283(48):33394–33405. | |
Kwon C, Han Z, Olson EN, Srivastava D. MicroRNA1 influences cardiac differentiation in Drosophila and regulates Notch signaling. Proc Natl Acad Sci U S A. 2005;102(52):18986–18991. | |
Xie M, He CS, Wei SH, Zhang L. Notch-1 contributes to epidermal growth factor receptor tyrosine kinase inhibitor acquired resistance in non-small cell lung cancer in vitro and in vivo. Eur J Cancer. 2013; 49(16):3559–3572. | |
Hemming ML, Elias JE, Gygi SP, Selkoe DJ. Proteomic profiling of gamma-secretase substrates and mapping of substrate requirements. PLoS Biol. 2008;6(10):e257. | |
Riccio O, van Gijn ME, Bezdek AC, et al. Loss of intestinal crypt progenitor cells owing to inactivation of both Notch1 and Notch2 is accompanied by derepression of CDK inhibitors p27Kip1 and p57Kip2. EMBO Rep. 2008;9(4):377–383. | |
Xie T, Yan C, Zhou R, et al. Crystal structure of the γ-secretase component nicastrin. Proc Natl Acad Sci U S A. 2014;111(37):13349–13354. | |
Minond D, Cudic M, Bionda N, et al. Discovery of novel inhibitors of a disintegrin and metalloprotease 17 (ADAM17) using glycosylated and non-glycosylated substrates. J Biol Chem. 2012;287(43):36473–36487. | |
Nickoloff BJ, Osborne BA, Miele L. Notch signaling as a therapeutic target in cancer: a new approach to the development of cell fate modifying agents. Oncogene. 2003;22(42):6598–6608. | |
Moellering RE, Cornejo M, Davis TN, et al. Direct inhibition of the NOTCH transcription factor complex. Nature. 2009;462(7270):182–188. | |
Wu Y, Cain-Hom C, Choy L, et al. Therapeutic antibody targeting of individual Notch receptors. Nature. 2010;464(7291):1052–1057. | |
Chiorean EG, LoRusso PM, Strother RM. A phase I first-in-human study of enoticumab (REGN421), a fully-human Delta-like ligand 4 (Dll4) monoclonal antibody in patients with advanced solid tumors. Clin Cancer Res. Epub February 27, 2015. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.